

In this study, Dowen et al. identified a microRNA-regulated developmental timing pathway that coordinates the mobilization of intestinal fat stores to the germline during C. elegans development. Their results show that lin-4 and let-7 microRNAs promotes mTOR signaling, which regulates intestinal fat metabolism, thereby providing insight into a novel function for microRNAs.
Keywords: vitellogenesis, fat metabolism, microRNAs, insulin, mTORC2, pqm-1
Abstract
Animals integrate metabolic, developmental, and environmental information before committing key resources to reproduction. In Caenorhabditis elegans, adult animals transport fat from intestinal cells to the germline to promote reproduction. We identified a microRNA (miRNA)-regulated developmental timing pathway that functions in the hypodermis to nonautonomously coordinate the mobilization of intestinal fat stores to the germline upon initiation of adulthood. This developmental timing pathway, which is controlled by the lin-4 and let-7 miRNAs, engages mTOR signaling in the intestine. The intestinal signaling component is specific to mTORC2 and functions in parallel to the insulin pathway to modulate the activity of the serum/glucocorticoid-regulated kinase (SGK-1). Surprisingly, SGK-1 functions independently of DAF-16/FoxO; instead, SGK-1 promotes the cytoplasmic localization of the PQM-1 transcription factor, which antagonizes intestinal fat mobilization at the transcriptional level when localized to the nucleus. These results revealed that a non-cell-autonomous developmental input regulates intestinal fat metabolism by engaging mTORC2 signaling to promote the intertissue transport of fat reserves from the soma to the germline.
During Caenorhabditis elegans post-embryonic development, the newly hatched L1 larvae, comprised of 556 somatic cells and two germline stem cells, feeds on bacteria to grow ∼100-fold in volume into a reproductive adult containing 959 somatic cells and thousands of germline cells that yield >300 embryos (Kipreos 2005). The larval growth of the animal is precisely choreographed by gene expression programs and requires sufficient nutrient sources to drive developmental switches. For example, the lin-4 and let-7 microRNAs (miRNAs), through repression of their mRNA targets, direct the L1-to-L2 and L4-to-adult developmental transitions, respectively (Chalfie et al. 1981; Ambros and Horvitz 1984; Reinhart et al. 2000). These regulatory events as well as others are used throughout larval development in various somatic blast cell lineages, all of which terminally differentiate at the adult stage, coinciding with the onset of oogenesis.
At adulthood, the nutrients that once licensed larval development now accumulate as fat in the intestine of the animal. This metabolic switch promotes the accumulation of energy reserves that support either somatic longevity or, alternatively, reproductive robustness. The energetic commitment to reproduction at adulthood is epitomized by the initiation of vitellogenesis, a process that reallocates intestinal fat stores to the germline via low-density lipoprotein (LDL)-like particles called vitellogenins (Kimble and Sharrock 1983). The vitellogenin genes (vit-1 to vit-6) encode lipid-binding proteins that share sequence similarity to the mammalian ApoB proteins and are responsible for depositing fats or fat-like substances (i.e., cholesterol) into oocytes, where they provide nutrients for the developing embryo (Spieth et al. 1991). Vitellogenin expression and secretion from the intestine deplete somatic tissues of crucial fat resources and supply the embryos with a dowry of nutrients, thereby supporting the metabolism and development of the animal until it hatches and commences feeding.
Vitellogenesis is tightly regulated by developmental programs that restrict it to the adult stage; however, it is also likely to be dynamically regulated by nutritional conditions. One such regulatory input is insulin/insulin-like growth factor (IGF) signaling, which mediates key aspects of C. elegans growth, development, metabolism, and reproduction but is also required for vitellogenesis (Murphy et al. 2003; DePina et al. 2011). Although insulin-like signaling is required for vitellogenesis, it is unlikely to be the sole regulatory input, and additional pathways or genetic modifiers are likely to exist. Here, we report that a C. elegans developmental timing pathway in the hypodermis, mediated by the lin-4 and let-7 miRNAs, couples to intestinal mTOR (mechanistic target of rapamycin) signaling to regulate fat metabolism and vitellogenesis via the PQM-1 zinc finger transcription factor.
Results
An RNAi screen implicates miRNAs in intestinal fat regulation
The onset of vitellogenesis normally occurs at the L4-to-adult transition and represents a significant metabolic switch that we sought to explore genetically via an RNAi screen. To generate a genetically tractable vitellogenesis reporter, we fused the promoter of the vit-3 gene, which encodes a highly expressed vitellogenin, to GFP (Pvit-3::GFP) and found that this fusion gene is faithfully activated upon the initiation of vitellogenesis at adulthood. The reporter gene is expressed exclusively in the intestines of adult animals and is amenable to high-throughput screening for genetic perturbations that disrupt the normal onset of GFP expression at adulthood. We screened the Pvit-3::GFP reporter strain against a cherry-picked RNAi library composed of ∼1400 genes that are likely to function in cellular signaling: kinases, transcription factors, nuclear hormone receptors, and RNA metabolism genes. We identified 16 gene inactivations that disrupt adult expression of the Pvit-3::GFP reporter gene (Supplemental Table S1). The gene inactivations that most severely disrupted Pvit-3::GFP activation were the miRNA Argonaute cofactor alg-1, the Meis-class homeodomain transcription factor unc-62, the GATA transcription factors elt-1 and elt-2, and the serum/glucocorticoid-regulated kinase (sgk-1) and drl-1 kinase. Importantly, ceh-60 and klf-3, which we also identified in the screen, and unc-62 are all required for full expression of the vit genes, and UNC-62 directly binds the vit promoters (Van Nostrand et al. 2013; Zhang et al. 2013; Van Rompay et al. 2015), further supporting the validity of our screening approach.
Developmental timing genes direct intestinal fat mobilization
The dramatic disruption of Pvit-3::GFP expression caused by inactivation of the miRNA cofactor alg-1 suggested that the normal adult onset of vitellogenesis in the intestine may be coupled to miRNA-dependent regulation of developmental timing. The alg-1 mutants display weak developmental timing defects (heterochrony) in the hypodermis (Ding et al. 2005). The alg-1 gene, along with alg-2, encodes the two miRNA-specific Argonautes in C. elegans, which together are required for viability (Vasquez-Rifo et al. 2012). Simultaneous knockdown of alg-1 and alg-2 (alg-1/2) by RNAi strongly attenuates expression of the Pvit-3::GFP reporter in adult animals, as determined by fluorescence microscopy or Western blot analysis (Fig. 1A,B). Using a different transcriptional reporter, Pvit-2::NLS::GFP, we observed a similar genetic requirement for alg-1/2. To verify that transcriptional down-regulation of vit gene expression results in disruption of vitellogenesis, we treated worms expressing a GFP-tagged vitellogenin protein (VIT-2::GFP) with alg-1/2 RNAi. Indeed, inactivation of alg-1/2 disrupted both the intestinal expression and germline deposition of VIT-2::GFP (Supplemental Fig. S1).
Figure 1.

Successful mobilization of fat stores from the intestine requires miRNAs. (A,B) Overlaid DIC and GFP fluorescence images (A) or Western blot analysis (B) of transgenic Pvit-2::NLS::GFP (vit-2 promoter fused to a nuclear localization sequence fused gfp) and Pvit-3::GFP (vit-3 promoter fused to gfp) day 1 adult animals subjected to control or alg-1/alg-2 double RNAi. (C) Representative images of GFP fluorescence in wild-type, lin-4(e912), and let-7(n2853ts) adult animals grown at 20°C carrying the Pvit-3::GFP transgene. (D) RT-qPCR assay for the endogenous vit genes in mir-71(n4115), lin-4(e912), and let-7(n2853ts) mutants grown at 20°C. Data are represented relative to the wild-type control as the mean ± standard error of the mean (SEM) of three biological replicates. (E) A diagram depicting the timing of expression and the mRNA targets of the lin-4 and let-7 miRNAs during larval development. (F) A qualitative description of Pvit-3::GFP expression in wild-type, lin-4(e912), and let-7(n2853ts) backgrounds after performing the indicated gene inactivations by RNAi.
The observation that miRNA-specific Argonautes are required to initiate vitellogenesis suggests that individual miRNAs modulate intestinal fat mobilization. miRNAs are ∼22-nucleotide noncoding RNAs that post-transcriptionally regulate their target mRNAs by binding to their 3′ untranslated regions (UTRs), causing translational repression and/or degradation of the transcript (Bartel 2004). Inactivation of alg-1/2 would be expected to disrupt the function of all of the ∼150 individual miRNAs encoded in the C. elegans genome; however, a comprehensive analysis of miRNA knockout mutations revealed that loss of only a few individual miRNAs causes developmental defects (Miska et al. 2007). In particular, mutations in the lin-4 and let-7 miRNAs cause a heterochronic phenotype (the reiteration of earlier larval cell fates and failure to implement later cell fates) in the hypodermis of the animal (Chalfie et al. 1981; Ambros and Horvitz 1984; Reinhart et al. 2000). These temporally expressed miRNAs are good candidate regulators of the developmental timing programs that could couple to the intestinal pathways to regulate vitellogenesis. Indeed, crossing the lin-4 or let-7 loss-of-function mutations into the Pvit-3::GFP background abrogates activation of the reporter at adulthood (Fig. 1C). Moreover, the lin-4 and let-7 mutants, but not the unrelated mir-71 mutant, display a marked reduction in the expression of the endogenous vit genes, as determined by RT-qPCR (Fig. 1D). These data are consistent with the previous observation that the vit-1 mRNA is down-regulated in the let-7 mutant (Ding and Grosshans 2009).
The lin-4 and let-7 miRNAs direct hypodermal seam cell divisions at specific developmental stages through negative regulation of their mRNA targets, lin-28/lin-14 and hbl-1/lin-41, respectively (Fig. 1E; Ambros and Horvitz 1984, 1987; Ambros 1989; Ruvkun and Giusto 1989; Moss et al. 1997; Slack et al. 2000; Abrahante et al. 2003; Lin et al. 2003; Grosshans et al. 2005). Thus, it is possible that inappropriate expression of lin-28, lin-14, hbl-1, or lin-41 mediates the failure to express vitellogenin in the intestine of the lin-4 or let-7 mutants. In such a case, genetic inactivation of these target genes may suppress the vitellogenesis defects in the lin-4 or let-7 mutants. Indeed, knockdown of the miRNA target genes by RNAi at least partially restored Pvit-3::GFP expression in the lin-4 or let-7 mutants (Fig. 1F). Genetic inactivation of the nuclear hormone receptor daf-12 in the let-7 mutant background, which suppresses many of the developmental defects caused by the let-7 mutation (Grosshans et al. 2005), did not suppress the Pvit-3::GFP phenotype (Fig. 1F), suggesting that restoration of Pvit-3::GFP expression in the lin-4 or let-7 mutant background is not simply a function of improving the overall health of the animal. These results indicate that the lin-4 and let-7 miRNAs are required for vitellogenin production in the intestine; however, it is unclear from these experiments in which tissue these genes function. It is possible that failure to initiate vitellogenesis in the miRNA mutants is due to disrupted developmental timing in either the intestinal cells that express vitellogenins or the hypodermal cells that are coupled to the intestine via an endocrine system.
Developmental timing genes function in the hypodermis to mediate vitellogenesis
In the hypodermis, sequential expression of developmental timing genes at successive larval transitions culminates in the expression of lin-29, which encodes a zinc finger transcription factor, during the final larval stage (L4). The lin-4 and let-7 mutants fail to express lin-29 in the hypodermis at this stage (Bettinger et al. 1996; Slack et al. 2000), and thus we reasoned that lin-29 mutants may phenocopy the vitellogenesis defects displayed by these heterochronic mutants. Crossing a lin-29-null mutation into the Pvit-3::GFP background strikingly reduced expression of the reporter gene in the intestine (Fig. 2A). Furthermore, systemic knockdown of lin-29 by RNAi dramatically decreased the expression of the endogenous vit genes (Fig. 2B). However, when lin-29 RNAi was performed exclusively in the intestine using a sid-1 mutant strain that is incapable of transporting dsRNAs outside of the intestine, the vit gene expression remained unchanged, indicating that lin-29 functions in a non-cell-autonomous fashion to regulate intestinal fat stores. Consistent with this observation, LIN-29 antibody staining of L4 stage animals demonstrated that LIN-29 is absent in the intestine but is found throughout the hypodermis, a specialized tissue responsible for forming the cuticle (Bettinger et al. 1996). The larval-specific functions of the hypodermis, including collagen production and secretion, are directed at each larval stage by a specialized set of hypodermal blast cells (called the seam cells) and the heterochronic genes that control their divisions. Terminal differentiation of the seam cells requires proper temporal expression of lin-29 (Ambros and Horvitz 1984). We performed tissue-specific lin-29 RNAi in the hypodermis and found that endogenous vit gene expression is markedly reduced (Fig. 2B), suggesting that lin-29 functions in the hypodermis to promote vitellogenin production in the intestine.
Figure 2.
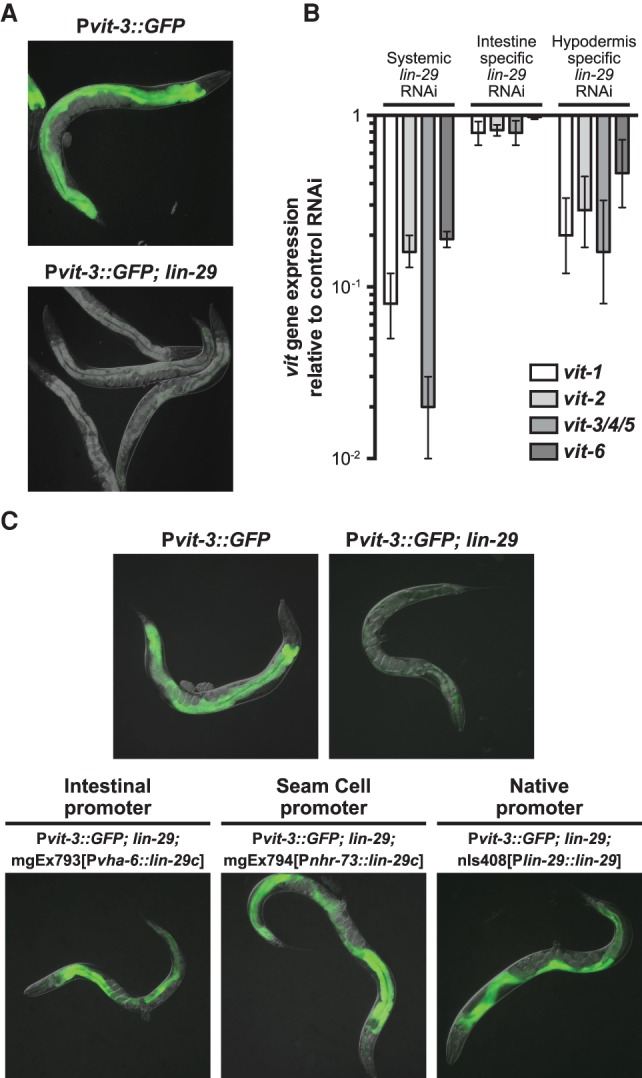
The hypodermal heterochronic gene lin-29 modulates intestinal expression of the vit genes. (A) Images of GFP fluorescence for wild-type and lin-29(n333) adult animals carrying the Pvit-3::GFP transgene. (B) RT-qPCR analysis of vit gene expression after systemic, intestinal-specific, or hypodermal-specific knockdown of lin-29 by RNAi. Data are plotted relative to a control RNAi treatment and represent the mean ± SEM of three biological replicates. (C) Representative GFP fluorescence images of wild-type, lin-29(n333), and lin-29(n333) transgenic animals that express the indicated rescue constructs. All animals also carry the Pvit-3::GFP transgene.
To expand on these observations, we performed tissue-specific rescue experiments while taking advantage of the tractability of the Pvit-3::GFP reporter strain to monitor expression of vitellogenin in the intestine. First, we expressed the let-7 miRNA in the Pvit-3::GFP; let-7 mutant background under control of either an intestinal or a hypodermal promoter. Hypodermal, but not intestinal, expression of let-7 rescues the let-7 mutant defect in vitellogenin production (Supplemental Fig. S2). To test the tissue specificity of lin-29 gene activity, we expressed lin-29 under control of a hypodermal seam cell-specific promoter (Miyabayashi et al. 1999) in the Pvit-3::GFP; lin-29 mutant and found that expression of lin-29 exclusively in the seam cells rescues intestinal Pvit-3::GFP expression in the lin-29 mutant (Fig. 2C). Surprisingly, ectopic expression of lin-29 in the intestine also rescues expression of Pvit-3::GFP in the lin-29 mutant background, suggesting that the LIN-29 transcription factor may direct expression of another gene that can control vitellogenesis in either a cell-autonomous or cell-nonautonomous manner. Together, these data indicate that proper specification of the hypodermal seam cells through the temporal expression of the heterochronic genes is required for mobilization of fat stores from the intestine.
SGK-1 perceives hypodermal development in the intestine to modulate vitellogenesis
In C. elegans, insulin signaling mediates various aspects of the animal's physiology, including its development, metabolism, reproduction, and longevity (Baumeister et al. 2006). Moreover, genetic inactivation of the insulin receptor disrupts vitellogenin expression in adult animals (Murphy et al. 2003; DePina et al. 2011). In C. elegans, the insulin pathway mediates the cellular response to insulin-like peptides via activation of the insulin receptor DAF-2/IGFR and its downstream effector, the phosphoinositide 3-kinase AGE-1/PI3K, which ultimately results in phosphorylation and activation of the AKT and SGK-1 protein kinases (Fig. 3A). To assess the role of insulin signaling in vitellogenesis regulation, we measured vit gene expression in a variety of insulin mutants. Global disruption of insulin signaling via inactivation of daf-2 or pdk-1 (3-phosphoinositide dependent protein kinase-1) as well as individual disruption of akt-1 or sgk-1 strongly reduced vit gene expression (Fig. 3B). Moreover, sgk-1 RNAi severely disrupts expression of the Pvit-3::GFP reporter in the intestines of adult animals (Supplemental Table S1).
Figure 3.
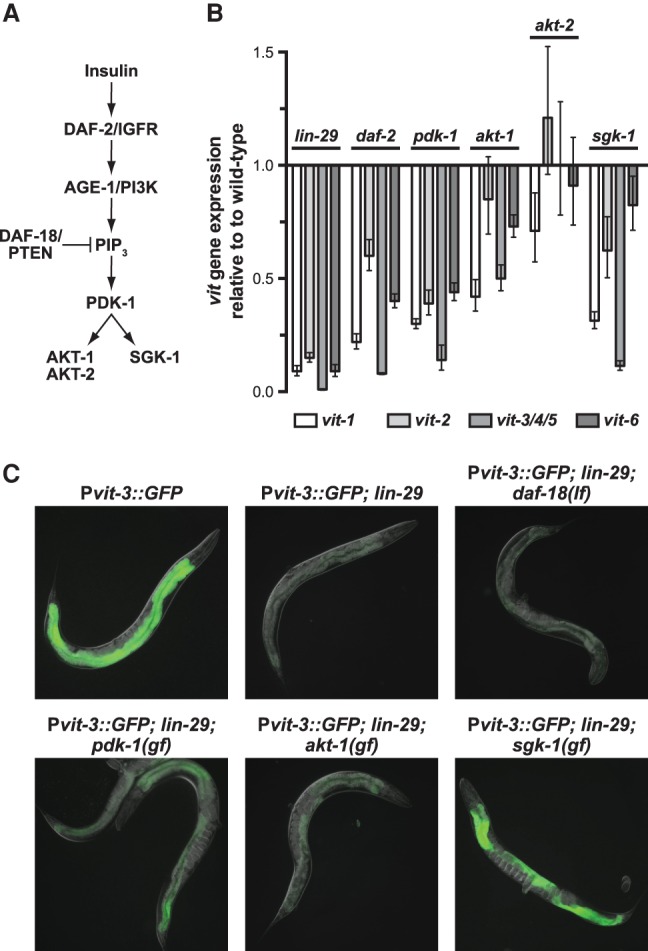
The SGK-1 kinase functions downstream from hypodermal development to regulate intestinal fat mobilization. (A) A diagram of the evolutionarily conserved insulin signaling pathway. (B) A RT-qPCR analysis of endogenous vit gene expression in lin-29(n333), daf-2(e1370ts), pdk-1(sa680ts), akt-1(mg306), akt-2(ok393), and sgk-1(ok538) mutants. Animals were grown to the L4 stage at 15°C before shifting to the nonpermissive temperature (25°C) for 24 h prior to harvesting. The data are plotted relative to a wild-type control as the mean ± SEM of three biological replicates. (C) Representative GFP fluorescence images of adult animals of the indicated genotypes grown at 15°C. The daf-18(mg198) loss-of-function mutant and the pdk-1(mg142), akt-1(mg144), and sgk-1(ft15) gain-of-function mutants were used in this epistasis analysis.
Although pdk-1, akt-1, and sgk-1 are all required for vit gene expression, it is unclear which kinases act downstream from lin-29 to regulate vitellogenesis. Therefore, we introduced pdk-1, akt-1, or sgk-1 gain-of-function mutations into the Pvit-3::GFP; lin-29 mutant background and scored these strains for GFP expression. The sgk-1 gain-of-function mutation robustly suppressed the defect in Pvit-3::GFP expression caused by the lin-29 mutation (Fig. 3C). Surprisingly, a loss-of-function mutation in daf-18/PTEN, which potentiates insulin signaling by increasing PIP3 levels, failed to suppress the Pvit-3::GFP expression defect in the lin-29 mutant, suggesting that LIN-29 does not function exclusively through insulin signaling to control vitellogenesis.
In C. elegans, the insulin pathway interacts with mTOR signaling to mediate the development, metabolism, and life span of the animal (Baumeister et al. 2006). Moreover, the mTOR complex 2 (mTORC2) functions in parallel to insulin signaling to modulate the activity of the AKT and SGK kinases (Hertweck et al. 2004; Kamada et al. 2005; Sarbassov et al. 2005; Jones et al. 2009; Soukas et al. 2009). To determine whether the mTOR kinase (LET-363 in C. elegans) functions in vitellogenesis, we first performed a partial knockdown of let-363/MTOR by RNAi in the Pvit-3::GFP reporter background, which revealed that let-363/MTOR is indeed required for induction of the reporter (Fig. 4A). mTOR assembles into two functionally distinct multiprotein complexes, mTORC1 and mTORC2, which are distinguished by their subunit constituents (Fig. 4B). These include the mTORC1-specific scaffolding protein Raptor (DAF-15 in C. elegans) and the mTORC2-specific scaffold Rictor (RICT-1), which in part differentiate the two mTOR complexes (Hara et al. 2002; Sarbassov et al. 2004). To determine which mTOR complex regulates vitellogenesis, we knocked down individual components of mTORC1 and mTORC2 in the Pvit-3::GFP background as well as their targets, the ribosomal protein S6 kinase (rsks-1) and sgk-1, respectively (Brown et al. 1995; Kamada et al. 2005). Knockdown of the individual components of mTORC2 (sinh-1, rict-1, and sgk-1), but not mTORC1 (daf-15 and rsks-1), disrupted Pvit-3::GFP expression to a degree similar to that of knockdown of let-363/MTOR (Fig. 4C). Consistent with these observations, rict-1 mutants displayed a reduction in endogenous vit gene expression, as determined by RT-qPCR (Supplemental Fig. S3). Together, these data indicate that RICT-1/mTORC2 and its major downstream target, SGK-1, function in parallel to the insulin pathway to modulate vitellogenesis and intestinal fat mass (Jones et al. 2009; Soukas et al. 2009).
Figure 4.
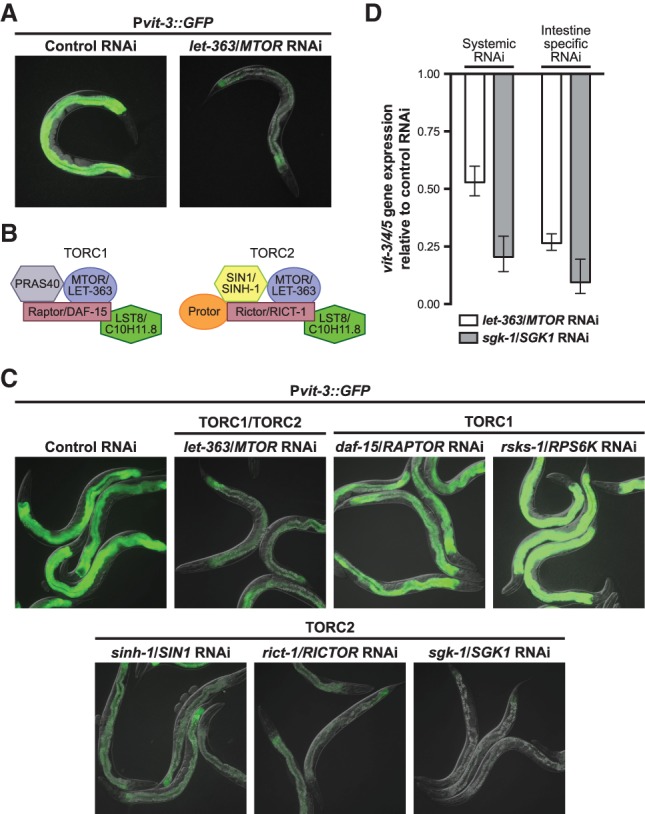
mTORC2 functions cell-autonomously to modulate vitellogenesis through SGK-1. (A) Representative GFP fluorescence images of Pvit-3::GFP transgenic animals after partial knockdown of let-363/MTOR by RNAi. Control animals were fed the empty vector RNAi plasmid. (B) An illustration of the evolutionarily conserved protein subunits of mTORC1 and mTORC2. Both the human and C. elegans protein names are provided. (C) Representative images of Pvit-3::GFP transgenic animals grown on the indicated RNAi strains at 15°C for either one (control, let-363, and daf-15, sgk-1) or two (rsks-1, sinh-1, and rict-1) generations before imaging them as day 1 adults. Animals that are grown on control RNAi for one or two generations display indistinguishable levels of GFP fluorescence. (D) A RT-qPCR analysis of vit-3/4/5 gene expression in animals grown at 15°C after systemic or intestinal-specific knockdown of let-363 or sgk-1 by RNAi. Expression data are plotted relative to a control RNAi treatment and represent the mean ± SEM of three biological replicates.
Although let-363/MTOR is ubiquitously expressed in C. elegans, the SGK-1 protein is expressed only in intestinal cells and neurons at the adult stage (Long et al. 2002; Hertweck et al. 2004), suggesting that mTORC2/SGK-1 may function in the intestine to mediate vitellogenin production. To test this hypothesis, we preformed systemic or intestinal-specific RNAi against let-363/MTOR and sgk-1 and assessed vit-3/4/5 gene expression in adult animals by RT-qPCR (Fig. 4D). Both systemic and intestinal-specific knockdown of let-363/MTOR or sgk-1 caused down-regulation of vit-3/4/5 gene expression, suggesting that mTORC2 signaling autonomously regulates vitellogenesis in the intestine. Moreover, loss of lin-29 or sgk-1 can be rescued by intestinal-specific expression of the gain-of-function or wild-type sgk-1 allele, respectively (Supplemental Fig. S4). Together, these data indicate that insulin and mTORC2 signaling function in parallel to modulate vitellogenesis; however, intestinal SGK-1 activation via mTORC2 is the major lin-29-dependent route of vit gene expression.
SGK-1 represses the PQM-1 transcription factor to control intestinal fat mobilization
The downstream targets of SGK-1, including any putative transcriptional regulators of vitellogenin expression, are unknown in C. elegans. A sgk-1(ok538)-null mutation, like sgk-1 RNAi, strongly disrupts induction of the Pvit-3::GFP reporter in the intestines of adult animals (Fig. 5A; Supplemental Table S1). To investigate which transcriptional regulators act downstream from sgk-1, we performed a large-scale forward genetic screen for ethyl methanesulfonate (EMS)-induced mutations that suppress the defect in Pvit-3::GFP gene expression in the sgk-1(ok538) mutant background. We reasoned that dominant gain-of-function or recessive loss-of-function sgk-1 suppressors could encode mutations in novel transcriptional activators or repressors, respectively. Using this approach, we identified 11 different probable loss-of-function alleles of pqm-1, which encodes a C2H2 zinc finger transcription factor (Supplemental Table S2). Notably, most of the missense mutations in pqm-1 that we identified are located in the DNA-binding domain, including some mutations that specifically alter the cysteine or histidine residues that constitute the zinc finger. To further characterize these mutant alleles, we introduced the previously characterized pqm-1-null mutation (Tepper et al. 2013) into the Pvit-3::GFP; sgk-1 mutant background and found that the pqm-1(ok485)-null mutation phenocopied the pqm-1(mg477) missense mutation (Fig. 5A).
Figure 5.

PQM-1 is a transcriptional effector of mTORC2/SGK-1 and antagonizes vit gene expression. (A) Representative GFP fluorescence images of wild-type, sgk-1(ok538), pqm-1(mg477); sgk-1(ok538), pqm-1(ok485); sgk-1(ok538), and daf-16(mgDf47); sgk-1(ok538) adults carrying the Pvit-3::GFP transgene. (B) Images of wild-type, lin-29(n333), daf-2(e1370ts), rict-1(mg360), and sgk-1(ok538) Pvit-3::GFP transgenic animals subjected to control or pqm-1 RNAi. Animals were grown to the L4 stage at 15°C before shifting to 25°C for 24 h prior to imaging. (C) Images of C-terminally GFP-tagged PQM-1 (PQM-1::GFP) protein localization in the intestines of L3 larvae or day 1 adults. Animals were grown on control, lin-29, or sgk-1 RNAi for 36 h (L3s) or 72 h (adults) at 20°C. The white triangles indicate examples of nuclear PQM-1::GFP. (D) GFP fluorescence images of pqm-1(ok485); sgk-1(ok538) adults carrying the Pvit-3::GFP transgene after being subjected to control or unc-62 RNAi.
The PQM-1 protein was identified using an integrative genome-wide approach to uncover transcription factors that cooperate with DAF-16/FoxO to mediate the transcriptional changes triggered by disruption of insulin-like signaling (Tepper et al. 2013). Active insulin signaling results in AKT-dependent phosphorylation of DAF-16, restricting its localization to the cytoplasm; conversely, low levels of insulin-like ligands or inactivation of daf-2 causes dephosphorylation of DAF-16/FoxO, driving it into the nucleus to modify the expression of its target genes (Lin et al. 2001). Although controversial, SGK-1 may function in part through DAF-16 to modulate C. elegans development, stress resistance, and life span (Hertweck et al. 2004; Chen et al. 2013). To assess whether DAF-16, which represses vitellogenesis in the daf-2 mutant (DePina et al. 2011), also functions downstream from SGK-1 to negatively regulate vitellogenin production, we crossed the daf-16-null mutation into the Pvit-3::GFP; sgk-1 mutant background. Loss of daf-16 failed to suppress the defect in Pvit-3::GFP expression caused by the sgk-1 mutation (Fig. 5A). Consistently, genetic inactivation of daf-16 by RNAi resulted in a modest reactivation of the Pvit-3::GFP reporter in the daf-2 mutant but no reactivation in the lin-29 or sgk-1 mutants (Supplemental Fig. S5). To rule out the possibility that SGK-1 redundantly modulates the repressive activity of both PQM-1 and DAF-16, we crossed the daf-16-null mutation into the Pvit-3::GFP; sgk-1; pqm-1 mutant background and measured gfp mRNA levels by RT-qPCR. Deletion of daf-16 had no additive effect on the expression of the reporter relative to the Pvit-3::GFP; sgk-1; pqm-1 background (Supplemental Fig. S6A). Together, these data indicate that lin-29/mTORC2/sgk-1 act in a daf-16-independent manner to regulate vitellogenesis.
It is possible that PQM-1 represses vitellogenesis upon disruption of either insulin or mTORC2 signaling. To examine this more closely, we knocked down pqm-1 by RNAi in a variety of Pvit-3::GFP mutant strains. Loss of pqm-1 reactivates Pvit-3::GFP expression in the lin-29, rict-1, and sgk-1 mutants but not the daf-2 mutant (Fig. 5B), suggesting that PQM-1-dependent regulation of vitellogenesis is specific to mTORC2 signaling. Surprisingly, introduction of the pqm-1-null allele into the lin-29 mutant background only partially suppressed the defects in endogenous vit gene expression caused by the lin-29 mutation (Supplemental Fig. S6B), suggesting that additional repressors may contribute to these defects (see the Discussion).
In wild-type animals, the PQM-1 protein accumulates in the nucleus of intestinal cells during the larval stages but then largely redistributes into the cytoplasm during adulthood (Fig. 5C; Tepper et al. 2013), which is consistent with our observation that PQM-1 antagonizes vitellogenesis. Thus, we predicted that PQM-1 protein might inappropriately localize to the nucleus of intestinal cells at the adult stage in the lin-29 and sgk-1 mutants to mediate the repression of vitellogenesis. Using a transgenic strain that expresses a PQM-1::GFP fusion protein, we found that PQM-1 does indeed accumulate in the intestinal nuclei of lin-29 and sgk-1 knockdown animals at adulthood (Fig. 5C). PQM-1 and DAF-16 localize to the nuclei of intestinal cells in a mutually exclusive manner to control their target genes (Tepper et al. 2013). Knockdown of daf-2 by RNAi stimulated nuclear translocation of a DAF-16::GFP fusion protein, as expected; however, knockdown of lin-29, rict-1, or sgk-1 had no effect on the localization of DAF-16::GFP (Supplemental Fig. S7A), further supporting the notion that mTORC2/SGK-1 functions independently of DAF-16 to control vitellogenesis. Consistent with these observations, the lin-29 and sgk-1 mutants exhibited an ∼2.5-fold increase in endogenous pqm-1 gene expression without altering daf-16 mRNA levels (Supplemental Fig. S7B). Together, our genetic and cytological analyses suggest that PQM-1 functions downstream from lin-29/mTORC2 and independently of insulin signaling to antagonize vitellogenesis either directly or indirectly.
While accumulation of nuclear PQM-1 in the sgk-1 mutant represses the expression of the Pvit-3::GFP reporter, it remains unclear where PQM-1 functions in the vitellogenesis regulatory pathway relative to known transcriptional activators. To explore this further, we focused on the unc-62 gene, which encodes a direct transcriptional activator of vit gene expression (Van Nostrand et al. 2013) and was a strong hit in our RNAi screen for genes that are required to activate Pvit-3::GFP at the adult stage (Supplemental Table S1). Knockdown of unc-62 by RNAi in the Pvit-3::GFP; sgk-1; pqm-1 mutant background abrogated expression of the Pvit-3::GFP reporter (Fig. 5D), indicating that unc-62 likely functions downstream from pqm-1 to regulate vitellogenin production.
LIN-29 and SGK-1 maintain proper fat mass and body size
The signaling pathways that control vitellogenesis, which are responsible for reallocating key somatic fat resources to the germline, are likely to respond to or control additional aspects of C. elegans fatty acid metabolism. Therefore, we reasoned that fat content in the intestines of lin-29/sgk-1 mutants could be either elevated due to a failure to mobilize fatty acids from the intestine or reduced due to widespread defects in fat metabolism. To examine this more closely, we stained wild-type and mutant animals with Oil Red O, which stains neutral triglycerides and lipids, and then visualized the animals by microscopy and quantified their fat content (Wahlby et al. 2012, 2014). The lin-29 and sgk-1 mutants displayed reduced fat levels as revealed by Oil Red O staining (P < 0.0001) (Fig. 6A,B), suggesting that intestinal fatty acid metabolism is altered in these strains. Fat mass measurements of sgk-1 mutants using Oil Red O have been conflicting (Soukas et al. 2009; Yen et al. 2010). Thus, we expanded on our initial characterization of the sgk-1-null mutant by knocking down sgk-1 by RNAi and staining either L4 larvae or adult animals with Oil Red O. Consistent with our initial observations, we observed a decrease in Oil Red O staining in sgk-1 knockdown animals relative to the controls at both developmental time points (P < 0.0001) (Supplemental Fig. S8A,B). The vitellogenesis defects displayed by sgk-1 mutants can be suppressed by inactivation of pqm-1, and thus it is possible that the decrease in fat mass exhibited by sgk-1 mutant animals may be similarly suppressed. Indeed, introduction of the pqm-1(ok485)-null allele into the sgk-1 mutant background caused a modest but reproducible suppression of the fat storage defects observed in sgk-1 mutants (Fig. 6C).
Figure 6.
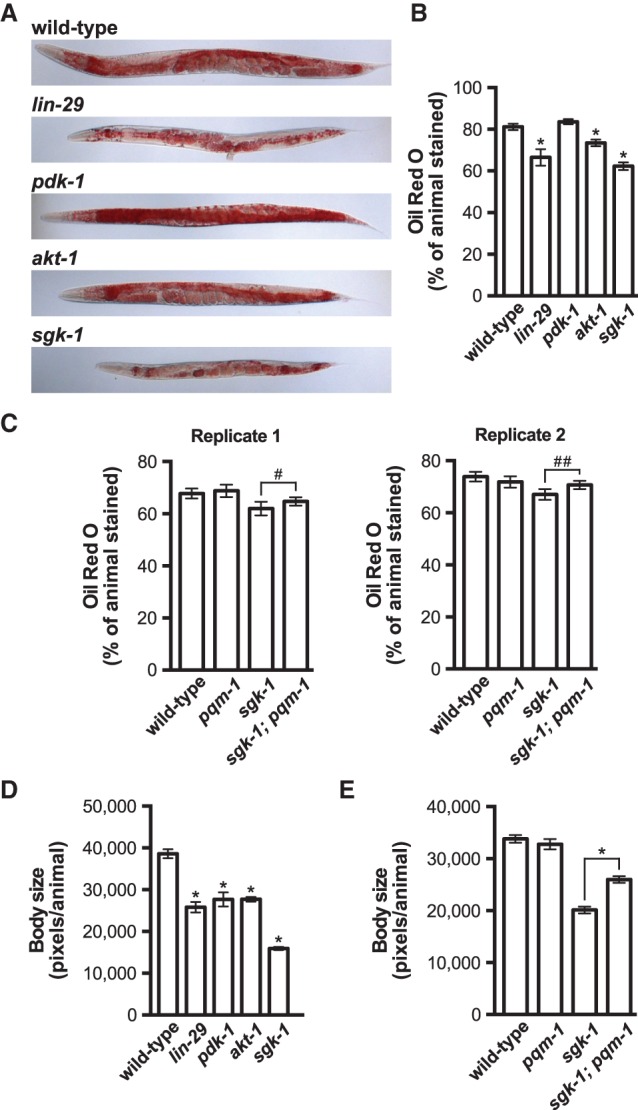
Targets of mTORC2 are required for maintaining fat content and body size. (A,B) Representative images (A) and quantification (B) of wild-type, lin-29(n333), pdk-1(sa680ts), akt-1(mg306), and sgk-1(ok538) adult animals stained with Oil Red O. Animals were grown at 15°C until they reached the L4 stage before shifting to 25°C for 24 h. (C) Quantification of Oil Red O staining for wild-type, pqm-1(ok485), sgk-1(ok538), and pqm-1(ok485); sgk-1(ok538) adult animals grown at 20°C. Two independent replicate experiments are shown. (D,E) Body size measurements were extracted from the corresponding Oil Red O images used in B and C (replicate 1). All Oil Red O quantification data and body size measurements are plotted as the mean and the 95% confidence interval. The P-values were generated by one-way ANOVA using a Bonferroni's multiple comparison test ([*] P < 0.0001) (B,D) or a two-tailed Student's t-test ([*] P < 0.0001; [#] P = 0.072; [##] P = 0.006) (C,E).
The rict-1 and sgk-1 mutants are small, possibly due to defects in intestinal fat metabolism, since the rict-1 body size phenotype can be rescued by intestinal-specific expression of rict-1 (Soukas et al. 2009). We reasoned that inactivation of lin-29, which functions genetically upstream of mTORC2, may phenocopy the body size defects observed in the rict-1 and sgk-1 mutants. Indeed, the lin-29, pdk-1, akt-1, and sgk-1 mutants are all smaller than wild-type animals (Fig. 6D; Supplemental Fig. S8C), supporting previous observations that implicated mTORC2 in maintaining C. elegans body size. Additionally, inactivation of pqm-1 in the sgk-1 mutant background partially suppresses the small body size of the sgk-1 mutant (Fig. 6E). These fat mass and body size analyses support our vitellogenesis data and suggest that LIN-29 is an upstream regulator of mTORC2/SGK-1, which functions in part through modulating the activity of PQM-1.
Discussion
We identified an intertissue signaling axis that couples a miRNA-dependent hypodermal developmental timing pathway to the evolutionarily conserved mTORC2 signaling pathway (Fig. 7), which mediates various aspects of cellular growth and metabolism. We developed a transgenic reporter strain that robustly expresses GFP upon initiation of vitellogenesis. The sensitivity of the reporter enabled us to identify several new regulators of vit gene expression as well as suppressor mutations that restore expression when vitellogenesis is disrupted. This approach revealed that hypodermal timing genes, the insulin and mTORC2 pathway genes, and the largely uncharacterized pqm-1 transcription factor all modulate vitellogenin-dependent fat mobilization. Moreover, our epistasis analysis demonstrated that mTORC2 and its downstream target, SGK-1, couple to hypodermal miRNA pathways to mediate intestinal fat metabolism by controlling the activity of the PQM-1 transcription factor (Fig. 7).
Figure 7.
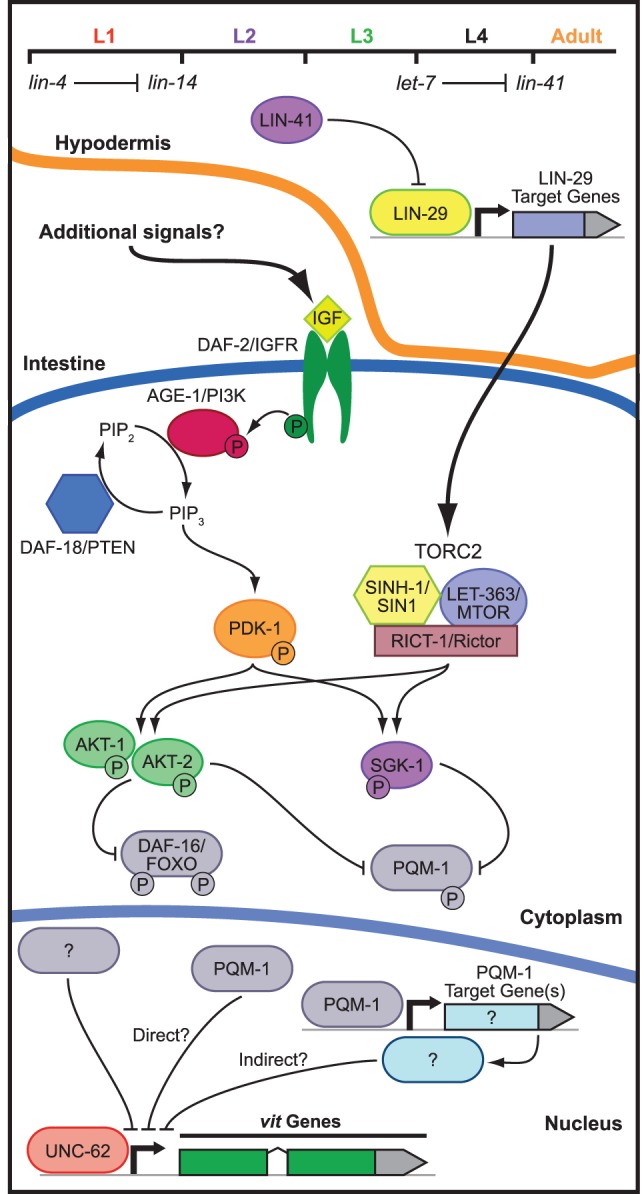
mTOR and insulin signaling collaborate to modulate intestinal fat mobilization. Hypodermal developmental timing genes engage mTORC2 signaling in the intestine to direct the expression of the vit genes. The model illustrates the genetic interactions identified in this study as well as those described elsewhere (Baumeister et al. 2006). Two alternative hypotheses (direct and indirect) are presented for the mechanism by which PQM-1 may regulate vitellogenesis.
The C. elegans heterochronic mutants display defects in cell lineage patterning at specific stages of larval development, often resulting in striking morphological defects. Heterochronic defects are not specific to any one tissue; however, the development of the hypodermis is particularly sensitive to inappropriate hypodermal seam cell divisions, which are mediated in part by the heterochronic genes lin-4 and let-7 (Chalfie et al. 1981; Ambros and Horvitz 1984; Reinhart et al. 2000). We showed that disruption of these miRNAs as well as the downstream heterochronic gene lin-29 causes not only an alteration in hypodermal developmental timing but also a failure to initiate the adult-specific expression of vitellogenin proteins in the intestine (Fig. 7). Interestingly, elt-1, which we identified in our RNAi screen as a regulator of Pvit-3::GFP expression, is required for proper hypodermal development and directly regulates let-7 expression, indicating that additional layers of gene regulation in the hypodermis control vitellogenin production (Cohen et al. 2015). Terminal differentiation of the seam cells at the initiation of adulthood coincides with maximal expression of the LIN-29 transcription factor in the hypodermis, which may be responsible for directing the hypodermal-specific expression of adult-specific signaling molecules that are perceived by surrounding tissues, including the intestine. Indeed, ectopic expression of lin-29 in the intestine rescues the vitellogenesis defects in the lin-29 mutant, suggesting that LIN-29 may directly activate expression of a soluble intertissue signaling molecule that is perceived by intestinal cells. Interestingly, proper migration of the hermaphrodite-specific neurons and arborization of the sensory dendrites during development are mediated by hypodermal expressed cell surface receptors or adhesion molecules, further underscoring the role of the hypodermis in non-cell-autonomous developmental regulation (Pedersen et al. 2013; Salzberg et al. 2013).
Although insulin signaling is required for expression of the Pvit-3::GFP reporter gene, we found that hypodermal development functions predominantly through the mTORC2 pathway to mediate vitellogenesis. While it is possible that insulin regulates the activity of mTORC2 through cross-talk between the two pathways, insulin-mediated signaling is unlikely to be solely responsible for mTORC2 activation, since disruption of daf-18/PTEN, which potentiates insulin signaling via up-regulation of PIP3, fails to suppress the vitellogenesis defects in the lin-29 mutant. Moreover, conclusive evidence that insulin can directly activate mTORC2 is lacking despite the fact that activation of mTORC1 by Akt is well established in mammals (Gingras et al. 1998). A more likely scenario is that insulin and mTORC2 signaling converge on AKT-1, AKT-2, and SGK-1 in the intestine to synergistically promote the activity of these kinases and, consequently, expression of the vitellogenins. In this way, insulin secreted from other tissues (e.g., germline and sensory neurons) could function in parallel to mTORC2 to dictate at least in part whether intestinal resources should be committed to reproduction (Fig. 7). Intriguingly, the homeodomain transcription factor gene ceh-60, which was a hit in our RNAi screen for Pvit-3::GFP regulators, is required for vitellogenesis and is expressed exclusively in a pair of amphid sensory neurons (Van Rompay et al. 2015). Although a link between ceh-60 and insulin signaling has yet to be established, these data do underscore the importance of additional tissues in the regulation of vitellogenesis.
We found that the mTORC2-specific protein RICT-1 is required for vitellogenin expression and that a gain-of-function allele of sgk-1, which encodes the major downstream target of mTORC2, partially suppresses the defects in vitellogenesis caused by the lin-29 mutation. Furthermore, lin-29 mutants display a reduction in fat mass and a small body size, which is phenocopied by loss of sgk-1 (Soukas et al. 2009; Yen et al. 2010). In contrast, we did not observe a detectable reduction in expression of the Pvit-3::GFP reporter after partial knockdown of the mTORC1-specfic protein DAF-15/Raptor or the mTORC1 target RSKS-1/RPS6 kinase, and loss of lin-29 does not phenocopy the arrest phenotype associated with mutations that completely disrupt mTORC1 signaling (i.e., rheb-1, let-363, and daf-15 mutations). These data suggest that lin-29 functions upstream of mTORC2 and not mTORC1; however, we cannot completely rule out that mTORC1 signaling functions at least to some degree to regulate vitellogenin production. Notably, mTORC1 signaling is required for vitellogenin expression in the fat body of the mosquito in response to a blood feeding (Hansen et al. 2004), suggesting that an analogous pathway may exist in C. elegans.
To identify transcriptional regulators that function downstream from mTORC2/SGK-1, we performed a forward genetic screen to identify suppressor mutations that reactivate expression of the vitellogenesis reporter in the sgk-1(ok538) mutant strain. This screen yielded many alleles of pqm-1, a zinc finger transcription factor that is highly conserved in nematodes and parasitic nematodes but not beyond. It is possible that other vitellogenin-producing organisms (e.g., insects, birds, and fish) use different transcriptional regulators to control vitellogenesis, or, alternatively, PQM-1 may be conserved functionally but not at the level of primary structure. Our cytological analysis of PQM-1::GFP indicates that SGK-1 promotes the cytoplasmic localization of PQM-1, presumably through phosphorylation (Fig. 7). This phosphorylation-dependent mechanism is analogous to how AKT regulates the localization and activity of DAF-16 (Lin et al. 2001). Indeed, PQM-1 possesses a SGK consensus phosphorylation motif (RERTSTI) in a small conserved region near the C terminus of the protein (Kobayashi et al. 1999); however, mutation of this threonine residue to alanine by CRISPR/Cas9 fails to constitutively repress expression of the Pvit-3::GFP reporter (data not shown). It is possible that additional phosphorylation sites may compensate when this site is lost, including a nearby conserved, although weaker, putative SGK phosphorylation site (KPRSCTL).
Alternatively, additional factors downstream from SGK-1 may collaborate with PQM-1 to repress vitellogenin expression. In support of this hypothesis, inactivation of pqm-1 only partially suppresses the defects in endogenous vit gene expression caused by the lin-29 mutation. Indeed, the transcriptional repressor MAFR-1/Maf1 negatively regulates vit gene expression in the intestines of hermaphrodites, and MAB-3 represses vitellogenesis in males (Yi and Zarkower 1999; Khanna et al. 2014). It is possible that these transcriptional repressors or other unidentified negative regulators could also function downstream from lin-29/sgk-1 to control intestinal fat metabolism. Moreover, PQM-1 does not directly bind the promoters of the vit genes at the L3 larval stage when vitellogenin expression is repressed, suggesting that PQM-1 may indirectly control vitellogenesis by directing the expression of additional regulators of fatty acid metabolism (Fig. 7; Niu et al. 2011).
We demonstrated that PQM-1 functions downstream from mTORC2/SGK-1, but not DAF-2, to control intestinal fat mobilization. Conversely, disruption of insulin signaling restricts vitellogenin expression through the repressive action of DAF-16 and not PQM-1 (Fig. 5B; Supplemental Fig. S5; DePina et al. 2011), suggesting that insulin and mTORC2 may regulate fat metabolism thorough distinct transcriptional regulators. Indeed, daf-2 and pdk-1 mutants display a daf-16-dependent increase in fat content, likely due to elevated de novo lipogenesis (Fig. 6A; Ashrafi et al. 2003; Perez and Van Gilst 2008), while mutation of sgk-1 causes a decrease in fat mass (Fig. 6A,B; Supplemental Fig. S8; Yen et al. 2010), which can be partially suppressed by inactivation of pqm-1. This is consistent with the observation that SGK-1 opposes AKT-1 in the regulation of C. elegans life span and stress resistance, and, in many cases, loss of sgk-1 alters the expression of DAF-16 target genes differently than loss of akt-1 (Chen et al. 2013). It will be important to determine whether the decrease in fat mass caused by mutation of lin-29 or sgk-1 is a function of increased fat utilization through up-regulation of fatty acid oxidation pathways or whether these mutant animals are defective in nutrient absorption or lipogenesis. Nonetheless, our results suggest that insulin and mTORC2 signaling mediate different aspects of fat metabolism despite the fact that they are both individually required for vitellogenesis.
The vitellogenin proteins are highly expressed in the C. elegans reproductive adult and likely require several layers of regulation, since they deplete the animal of key somatic fat stores. We identified a miRNA-based developmental signaling component in this regulatory network, which originates in the hypodermis and engages mTORC2 in the intestine. Interestingly, almost all of the gene inactivations or genetic mutations that disrupt this developmental pathway do not phenocopy the sterility associated with loss of rme-2, which encodes the sole vitellogenin receptor in the germline (Grant and Hirsh 1999), suggesting that multiple pathways act in parallel to maximally activate vitellogenin expression and that very little vitellogenin is actually necessary to support some level of fertility. The identity of the hypodermal signal is of particular interest, as it may modulate other aspects of the animal's physiology, including its longevity, which is often coupled to metabolic pathways. Importantly, the upstream regulators of mTORC2 are largely unknown, and our vitellogenesis reporter provides a unique screening tool to genetically interrogate this signaling pathway. It is likely that further characterization of the pathways that arbitrate the trade-off between maintaining somatic resources and committing them to reproduction will identify new genes that function in fundamental aspects of energy homeostasis.
Materials and methods
C. elegans strains
The C. elegans wild-type strain used in this study is N2 Bristol. All strains were cultured using standard methods (Brenner 1974). A complete list of the strains used in this study is in the Supplemental Material.
RNAi strains
RNAi sublibraries composed of genes encoding transcriptional regulators (397), nuclear hormone receptors (219), kinases (410), and RNA metabolic proteins (417) were cherry-picked from the Ahringer and Vidal RNAi libraries (Kamath et al. 2003; Rual et al. 2004). RNAi clones for individual experiments were obtained from these sublibraries or the full genome Ahringer library and confirmed by Sanger sequencing. Some “homemade” RNAi clones (daf-15, daf-2, and daf-16) were constructed by amplifying coding sequences from N2 worm genomic DNA and cloning them into the empty L4440 vector (Addgene) using standard cloning techniques. The L4440 empty vector was used in all experiments as the RNAi control. All strains were grown overnight in Luria-Bertani medium with 50 µg/mL ampicillin, concentrated 20×–25× by centrifugation, seeded onto NGM plates containing 5 mM isopropyl-β-D-thiogalactoside (IPTG), and incubated at room temperature overnight to induce dsRNA expression. Adult worms were bleached to liberate embryos, and L1 larvae were synchronized in M9 buffer before seeding them on the seeded RNAi plates. The animals were incubated for 72 or 120 h at 15°C or 20°C, respectively, until they reached the first day of adulthood before they were scored, imaged, or harvested for downstream applications. Growing animals in the presence of dsRNA for 120 h at 15°C enhanced silencing for less effective RNAi clones.
Generation and imaging of transgenic strains
The vit-3 transcriptional reporter was generated by fusing ∼500 base pairs (bp) of sequence upstream of the vit-3 transcriptional start site to GFP by fusion PCR. The PCR product was microinjected into animals at 10 ng/µL with 90 ng/µL sheared salmon sperm DNA. The extrachromosomal array spontaneously integrated into the genome, and the resulting strain was backcrossed to N2 six times.
The lin-29 rescue constructs were generated by fusing the intestinal-specific vha-6 promoter (chromosome II: 11,438,422–11,439,355; WS251) or the seam cell-specific nhr-73 promoter (chromosome I: 11,435,874–11,436,781; WS251) (Miyabayashi et al. 1999) to a mCherry::his-58::SL2 cassette fused to the lin-29 genomic locus (isoform c; chromosome II: 11,917,642–11,921,438; WS251). The SL2-specific transsplice site was used to verify the tissue specificity of lin-29 expression, which can be inferred based on mCherry::his-58 expression. The nIs408[Plin-29::lin-29::mCherry] transgene has been described previously (Harris and Horvitz 2011). The individual DNA fragments (promoters, mCherry::his-58::SL2 cassette, and lin-29) were amplified by PCR (Phusion, New England Biolabs), assembled into the pCFJ151 plasmid by Gibson assembly, and verified by Sanger sequencing. The Pvha-6::mCherry::his-58::SL2::lin-29 construct was microinjected into the wild-type Pvit-3::GFP strain at 2.5 ng/µL, and the resulting strain was crossed to the lin-29(n333) mutant to give GR2126. The Pnhr-73::mCherry::his-58::SL2::lin-29 construct was microinjected directly into lin-29(n333); mgIs70 animals at 20 ng/µL to generate GR2127.
All transgenic worms carrying either the Pvit-3::GFP, the Pvit-2::NLS::GFP, or the VIT-2::GFP reporter were imaged with a 5× or 10× objective on a Zeiss Axio Imager Z1 microscope running AxioVision 4.6 software. Transgenic animals expressing PQM-1::GFP were imaged at 40× or 63×.
Western blot analyses
Animals carrying the Pvit-3::GFP or Pvit-2::NLS::GFP transgene were individually picked and transferred directly to Laemmli buffer (one worm per microliter). Embryos were liberated from VIT-2::GFP animals by bleaching and then washed three times with M9 buffer, counted, and resuspended at equal concentrations in Laemmli buffer. Protein samples were boiled, resolved on a NuPAGE 10% Bis-Tris SDS–polyacrylamide gel (Thermo Fisher), and transferred to a nitrocellulose membrane. Blots were probed with either GFP (Thermo Fisher, A-11122) or Actin (Abcam, ab3280) antibodies.
RT-qPCR assays
Synchronized L1 animals were grown for ∼72 h until they reached the first day of adulthood. Generally, we hand-picked 100–200 gravid adults for RNA preparations, since many of the mutants used in this study display asynchronous growth rates within the population. In the cases where all of the experimental conditions yielded similar growth rates, the gravid animals were simply washed from plates. The total RNA was isolated from adult worms using Trizol (Thermo Fisher) followed by chloroform extraction and precipitation with isopropanol. Residual DNA was removed using the TURBO DNA-free kit per the manufacturer's instructions (Thermo Fisher). The cDNA was synthesized from total RNA primed with oligo(dT) primers using the SuperScript III kit according to the manufacturer's instructions (Thermo Fisher). qPCR was performed on a Bio-Rad real-time PCR detection system using iQ SYBR Green according to the manufacturer's recommendations (Bio-Rad). The qPCR primer sequences are listed in Supplemental Table S3. For a given experimental sample, the CT values for each target gene were first normalized to act-1 before normalization to a wild-type control sample. The resulting ΔΔCT value was averaged across three independently performed experiments, and the standard error of the mean (SEM) was calculated. The mean and SEM values were then transformed using 2−ΔΔCT.
Forward genetic screen and identification of causative mutations
Mutagenesis of sgk-1(ok538); mgIs70 (GR2140) was performed using EMS (Sigma-Aldrich). Briefly, synchronized L4 animals were washed three times with M9 buffer containing 0.01% Tween-20 before resuspending them in 47 mM EMS in M9 buffer. Animals were rotated for 4 h at 20°C, washed three times with M9 buffer containing 0.01% Tween-20, dropped on NGM plates seeded with OP50 bacteria, and grown for 24 h before isolating the embryos by bleach preparation. Synchronized F1 larvae were grown on NGM OP50 plates in nine pools for ∼72 h at 20°C, and any GFP-positive suppressors were picked (dominant alleles or haploinsufficiency). Each pool of F1 gravid adults was individually bleach-prepared and synchronized in M9 buffer. The F2 animals were grown for ∼72 h at 20°C on NGM OP50 plates, and the GFP-positive suppressors were picked (recessive alleles). A total of 90,000 haploid genomes was screened.
Strains carrying the sgk-1 suppressor alleles were backcrossed to GR2140 twice, and ∼50 F2 recombinants arising from the second backcross were picked to individual plates (Doitsidou et al. 2010). The plates were allowed to starve, the animals were pooled, and genomic DNA was isolated using the Gentra Puregene tissue kit (Qiagen). Whole-genome DNA sequencing libraries were prepared using the NEBNext DNA library preparation kit per the manufacturer's instructions (New England Biolabs, E6040). The multiplexed library, consisting of 24 individual libraries, was sequenced on an Illumina HiSeq instrument according to the manufacturer's instructions. Finally, the candidate variants were identified using CloudMap software (Minevich et al. 2012).
Oil Red O staining and quantification
Gravid day 1 adult animals were fixed with 60% isopropanol and stained with Oil Red O as previously described (Wahlby et al. 2014). Animals were mounted on an agarose pad and imaged with a 5× objective on a Zeiss Axio Imager Z1 microscope fitted with a Zeiss AxioCam HRm color camera running AxioVision 4.6 software. Quantification of Oil Red O staining was performed with the WormToolbox component of CellProfiler software (Wahlby et al. 2012). For each worm, the fraction of the animal stained (number of pixels stained/number of total pixels) was calculated, and the data were averaged across a population of worms. Similarly, body size was calculated as the total number of pixels per animal, and the data were averaged across the population. Experiments were performed three times with similar results.
Supplementary Material
Acknowledgments
Some of the strains used in this work were provided by the Caenorhabditis Genetics Center, which is supported by the National Institutes of Health Office of Research Infrastructure Programs (P40 OD010440). The Pvit-3::GFP reporter strain (GR2122) was generated by Justine Melo (Massachusetts General Hospital), and the intestinal-specific RNAi strain (MGH171) was generously provided by Alex Soukas (Massachusetts General Hospital). The transcription factor RNAi sublibrary was constructed by Sean Curran (University of Southern California) and Dave Simon (Josh Kaplan Laboratory, Massachusetts General Hospital), the nuclear hormone receptor RNAi sublibrary was constructed by Ho Yi Mak (Hong Kong University of Science and Technology), the kinase RNAi sublibrary was constructed by Javier Irazoqui (Massachusetts General Hospital), the RNA metabolism RNAi sublibrary was constructed by Sylvia Fischer (Massachusetts General Hospital) and John Kim (Johns Hopkins), and the gfp RNAi clone was generated by Dorian Anderson (Josh Kaplan laboratory, Massachusetts General Hospital). This work was funded in part by a grant to G.R. from the National Institute of Health (GM44619). R.H.D. is an American Cancer Society Postdoctoral Fellow (122240-PF-12-078-01-RMC).
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.283895.116.
References
- Abrahante JE, Daul AL, Li M, Volk ML, Tennessen JM, Miller EA, Rougvie AE. 2003. The Caenorhabditis elegans hunchback-like gene lin-57/hbl-1 controls developmental time and is regulated by microRNAs. Dev Cell 4: 625–637. [DOI] [PubMed] [Google Scholar]
- Ambros V. 1989. A hierarchy of regulatory genes controls a larva-to-adult developmental switch in C. elegans. Cell 57: 49–57. [DOI] [PubMed] [Google Scholar]
- Ambros V, Horvitz HR. 1984. Heterochronic mutants of the nematode Caenorhabditis elegans. Science 226: 409–416. [DOI] [PubMed] [Google Scholar]
- Ambros V, Horvitz HR. 1987. The lin-14 locus of Caenorhabditis elegans controls the time of expression of specific postembryonic developmental events. Genes Dev 1: 398–414. [DOI] [PubMed] [Google Scholar]
- Ashrafi K, Chang FY, Watts JL, Fraser AG, Kamath RS, Ahringer J, Ruvkun G. 2003. Genome-wide RNAi analysis of Caenorhabditis elegans fat regulatory genes. Nature 421: 268–272. [DOI] [PubMed] [Google Scholar]
- Bartel DP. 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281–297. [DOI] [PubMed] [Google Scholar]
- Baumeister R, Schaffitzel E, Hertweck M. 2006. Endocrine signaling in Caenorhabditis elegans controls stress response and longevity. J Endocrinol 190: 191–202. [DOI] [PubMed] [Google Scholar]
- Bettinger JC, Lee K, Rougvie AE. 1996. Stage-specific accumulation of the terminal differentiation factor LIN-29 during Caenorhabditis elegans development. Development 122: 2517–2527. [DOI] [PubMed] [Google Scholar]
- Brenner S. 1974. The genetics of Caenorhabditis elegans. Genetics 77: 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EJ, Beal PA, Keith CT, Chen J, Shin TB, Schreiber SL. 1995. Control of p70 s6 kinase by kinase activity of FRAP in vivo. Nature 377: 441–446. [DOI] [PubMed] [Google Scholar]
- Chalfie M, Horvitz HR, Sulston JE. 1981. Mutations that lead to reiterations in the cell lineages of C. elegans. Cell 24: 59–69. [DOI] [PubMed] [Google Scholar]
- Chen AT, Guo C, Dumas KJ, Ashrafi K, Hu PJ. 2013. Effects of Caenorhabditis elegans sgk-1 mutations on lifespan, stress resistance, and DAF-16/FoxO regulation. Aging Cell 12: 932–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen ML, Kim S, Morita K, Kim SH, Han M. 2015. The GATA factor elt-1 regulates C. elegans developmental timing by promoting expression of the let-7 family microRNAs. PLoS Genet 11: e1005099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePina AS, Iser WB, Park SS, Maudsley S, Wilson MA, Wolkow CA. 2011. Regulation of Caenorhabditis elegans vitellogenesis by DAF-2/IIS through separable transcriptional and posttranscriptional mechanisms. BMC Physiol 11: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding XC, Grosshans H. 2009. Repression of C. elegans microRNA targets at the initiation level of translation requires GW182 proteins. EMBO J 28: 213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Spencer A, Morita K, Han M. 2005. The developmental timing regulator AIN-1 interacts with miRISCs and may target the argonaute protein ALG-1 to cytoplasmic P bodies in C. elegans. Mol Cell 19: 437–447. [DOI] [PubMed] [Google Scholar]
- Doitsidou M, Poole RJ, Sarin S, Bigelow H, Hobert O. 2010. C. elegans mutant identification with a one-step whole-genome-sequencing and SNP mapping strategy. PLoS One 5: e15435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras AC, Kennedy SG, O'Leary MA, Sonenberg N, Hay N. 1998. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev 12: 502–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant B, Hirsh D. 1999. Receptor-mediated endocytosis in the Caenorhabditis elegans oocyte. Mol Biol Cell 10: 4311–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosshans H, Johnson T, Reinert KL, Gerstein M, Slack FJ. 2005. The temporal patterning microRNA let-7 regulates several transcription factors at the larval to adult transition in C. elegans. Dev Cell 8: 321–330. [DOI] [PubMed] [Google Scholar]
- Hansen IA, Attardo GM, Park JH, Peng Q, Raikhel AS. 2004. Target of rapamycin-mediated amino acid signaling in mosquito anautogeny. Proc Natl Acad Sci 101: 10626–10631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K. 2002. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110: 177–189. [DOI] [PubMed] [Google Scholar]
- Harris DT, Horvitz HR. 2011. MAB-10/NAB acts with LIN-29/EGR to regulate terminal differentiation and the transition from larva to adult in C. elegans. Development 138: 4051–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertweck M, Gobel C, Baumeister R. 2004. C. elegans SGK-1 is the critical component in the Akt/PKB kinase complex to control stress response and life span. Dev Cell 6: 577–588. [DOI] [PubMed] [Google Scholar]
- Jones KT, Greer ER, Pearce D, Ashrafi K. 2009. Rictor/TORC2 regulates Caenorhabditis elegans fat storage, body size, and development through sgk-1. PLoS Biol 7: e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada Y, Fujioka Y, Suzuki NN, Inagaki F, Wullschleger S, Loewith R, Hall MN, Ohsumi Y. 2005. Tor2 directly phosphorylates the AGC kinase Ypk2 to regulate actin polarization. Mol Cell Biol 25: 7239–7248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, et al. 2003. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421: 231–237. [DOI] [PubMed] [Google Scholar]
- Khanna A, Johnson DL, Curran SP. 2014. Physiological roles for mafr-1 in reproduction and lipid homeostasis. Cell Rep 9: 2180–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimble J, Sharrock WJ. 1983. Tissue-specific synthesis of yolk proteins in Caenorhabditis elegans. Dev Biol 96: 189–196. [DOI] [PubMed] [Google Scholar]
- Kipreos ET. 2005. C. elegans cell cycles: invariance and stem cell divisions. Nat Rev Mol Cell Biol 6: 766–776. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Deak M, Morrice N, Cohen P. 1999. Characterization of the structure and regulation of two novel isoforms of serum- and glucocorticoid-induced protein kinase. Biochem J 344: 189–197. [PMC free article] [PubMed] [Google Scholar]
- Lin K, Hsin H, Libina N, Kenyon C. 2001. Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat Genet 28: 139–145. [DOI] [PubMed] [Google Scholar]
- Lin SY, Johnson SM, Abraham M, Vella MC, Pasquinelli A, Gamberi C, Gottlieb E, Slack FJ. 2003. The C. elegans hunchback homolog, hbl-1, controls temporal patterning and is a probable microRNA target. Dev Cell 4: 639–650. [DOI] [PubMed] [Google Scholar]
- Long X, Spycher C, Han ZS, Rose AM, Muller F, Avruch J. 2002. TOR deficiency in C. elegans causes developmental arrest and intestinal atrophy by inhibition of mRNA translation. Curr Biol 12: 1448–1461. [DOI] [PubMed] [Google Scholar]
- Minevich G, Park DS, Blankenberg D, Poole RJ, Hobert O. 2012. CloudMap: a cloud-based pipeline for analysis of mutant genome sequences. Genetics 192: 1249–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miska EA, Alvarez-Saavedra E, Abbott AL, Lau NC, Hellman AB, McGonagle SM, Bartel DP, Ambros VR, Horvitz HR. 2007. Most Caenorhabditis elegans microRNAs are individually not essential for development or viability. PLoS Genet 3: e215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyabayashi T, Palfreyman MT, Sluder AE, Slack F, Sengupta P. 1999. Expression and function of members of a divergent nuclear receptor family in Caenorhabditis elegans. Dev Biol 215: 314–331. [DOI] [PubMed] [Google Scholar]
- Moss EG, Lee RC, Ambros V. 1997. The cold shock domain protein LIN-28 controls developmental timing in C. elegans and is regulated by the lin-4 RNA. Cell 88: 637–646. [DOI] [PubMed] [Google Scholar]
- Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C. 2003. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424: 277–283. [DOI] [PubMed] [Google Scholar]
- Niu W, Lu ZJ, Zhong M, Sarov M, Murray JI, Brdlik CM, Janette J, Chen C, Alves P, Preston E, et al. 2011. Diverse transcription factor binding features revealed by genome-wide ChIP-seq in C. elegans. Genome Res 21: 245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen ME, Snieckute G, Kagias K, Nehammer C, Multhaupt HA, Couchman JR, Pocock R. 2013. An epidermal microRNA regulates neuronal migration through control of the cellular glycosylation state. Science 341: 1404–1408. [DOI] [PubMed] [Google Scholar]
- Perez CL, Van Gilst MR. 2008. A 13C isotope labeling strategy reveals the influence of insulin signaling on lipogenesis in C. elegans. Cell Metab 8: 266–274. [DOI] [PubMed] [Google Scholar]
- Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G. 2000. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403: 901–906. [DOI] [PubMed] [Google Scholar]
- Rual JF, Ceron J, Koreth J, Hao T, Nicot AS, Hirozane-Kishikawa T, Vandenhaute J, Orkin SH, Hill DE, van den Heuvel S, et al. 2004. Toward improving Caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome Res 14: 2162–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruvkun G, Giusto J. 1989. The Caenorhabditis elegans heterochronic gene lin-14 encodes a nuclear protein that forms a temporal developmental switch. Nature 338: 313–319. [DOI] [PubMed] [Google Scholar]
- Salzberg Y, Diaz-Balzac CA, Ramirez-Suarez NJ, Attreed M, Tecle E, Desbois M, Kaprielian Z, Bulow HE. 2013. Skin-derived cues control arborization of sensory dendrites in Caenorhabditis elegans. Cell 155: 308–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. 2004. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol 14: 1296–1302. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. 2005. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307: 1098–1101. [DOI] [PubMed] [Google Scholar]
- Slack FJ, Basson M, Liu Z, Ambros V, Horvitz HR, Ruvkun G. 2000. The lin-41 RBCC gene acts in the C. elegans heterochronic pathway between the let-7 regulatory RNA and the LIN-29 transcription factor. Mol Cell 5: 659–669. [DOI] [PubMed] [Google Scholar]
- Soukas AA, Kane EA, Carr CE, Melo JA, Ruvkun G. 2009. Rictor/TORC2 regulates fat metabolism, feeding, growth, and life span in Caenorhabditis elegans. Genes Dev 23: 496–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spieth J, Nettleton M, Zucker-Aprison E, Lea K, Blumenthal T. 1991. Vitellogenin motifs conserved in nematodes and vertebrates. J Mol Evol 32: 429–438. [DOI] [PubMed] [Google Scholar]
- Tepper RG, Ashraf J, Kaletsky R, Kleemann G, Murphy CT, Bussemaker HJ. 2013. PQM-1 complements DAF-16 as a key transcriptional regulator of DAF-2-mediated development and longevity. Cell 154: 676–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand EL, Sanchez-Blanco A, Wu B, Nguyen A, Kim SK. 2013. Roles of the developmental regulator unc-62/Homothorax in limiting longevity in Caenorhabditis elegans. PLoS Genet 9: e1003325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Rompay L, Borghgraef C, Beets I, Caers J, Temmerman L. 2015. New genetic regulators question relevance of abundant yolk protein production in C. elegans. Sci Rep 5: 16381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez-Rifo A, Jannot G, Armisen J, Labouesse M, Bukhari SI, Rondeau EL, Miska EA, Simard MJ. 2012. Developmental characterization of the microRNA-specific C. elegans Argonautes alg-1 and alg-2. PLoS One 7: e33750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlby C, Kamentsky L, Liu ZH, Riklin-Raviv T, Conery AL, O'Rourke EJ, Sokolnicki KL, Visvikis O, Ljosa V, Irazoqui JE, et al. 2012. An image analysis toolbox for high-throughput C. elegans assays. Nat Methods 9: 714–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlby C, Conery AL, Bray MA, Kamentsky L, Larkins-Ford J, Sokolnicki KL, Veneskey M, Michaels K, Carpenter AE, O'Rourke EJ. 2014. High- and low-throughput scoring of fat mass and body fat distribution in C. elegans. Methods 68: 492–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen K, Le TT, Bansal A, Narasimhan SD, Cheng JX, Tissenbaum HA. 2010. A comparative study of fat storage quantitation in nematode Caenorhabditis elegans using label and label-free methods. PLoS One 5: e0012810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi W, Zarkower D. 1999. Similarity of DNA binding and transcriptional regulation by Caenorhabditis elegans MAB-3 and Drosophila melanogaster DSX suggests conservation of sex determining mechanisms. Development 126: 873–881. [DOI] [PubMed] [Google Scholar]
- Zhang J, Hashmi S, Cheema F, Al-Nasser N, Bakheet R, Parhar RS, Al-Mohanna F, Gaugler R, Hussain MM, Hashmi S. 2013. Regulation of lipoprotein assembly, secretion and fatty acid β-oxidation by Kruppel-like transcription factor, klf-3. J Mol Biol 425: 2641–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.