

The normal functions of amyloid-β peptides are poorly understood, as is the early vulnerability of central cholinergic pathways in Alzheimer’s disease. Kumar et al. report that amyloid-β forms complexes with cholinesterases, and thereby increases their ACh-degrading efficacy, and that high apolipoprotein-E alters this by facilitating abnormal accumulation of these complexes.
Keywords: Alzheimer’s disease, apolipoprotein E, beta-amyloid, astrocyte neurodegeneration, inflammation

The normal functions of amyloid-β peptides are poorly understood, as is the early vulnerability of central cholinergic pathways in Alzheimer’s disease. Kumar et al. report that amyloid-β forms complexes with cholinesterases, and thereby increases their ACh-degrading efficacy, and that high apolipoprotein-E alters this by facilitating abnormal accumulation of these complexes.
Abstract
Amyloid-β peptides, through highly sophisticated enzymatic machinery, are universally produced and released in an action potential synchronized manner into the interstitial fluids in the brain. Yet no native functions are attributed to amyloid-β. The amyloid-β hypothesis ascribes just neurotoxicity properties through build-up of soluble homomeric amyloid-β oligomers or fibrillar deposits. Apolipoprotein-ε4 (APOE4) allele is the only confirmed genetic risk factor of sporadic Alzheimer’s disease; once more it is unclear how it increases the risk of Alzheimer’s disease. Similarly, central cholinergic signalling is affected selectively and early in the Alzheimer’s disease brain, again why cholinergic neurons show this sensitivity is still unclear. However, the three main known Alzheimer’s disease risk factors, advancing age, female gender and APOE4, have been linked to a high apolipoprotein-E and accumulation of the acetylcholine degrading enzyme, butyrylcholinesterase in cerebrospinal fluids of patients. Furthermore, numerous reports indicate that amyloid-β interacts with butyrylcholinesterase and apolipoprotein-E. We have proposed that this interaction leads to formation of soluble ultrareactive acetylcholine-hydrolyzing complexes termed BAβACs, to adjust at demand both synaptic and extracellular acetylcholine signalling. This hypothesis predicted presence of acetylcholine-synthesizing enzyme, choline acetyltransferase in extracellular fluids to allow maintenance of equilibrium between breakdown and synthesis of acetylcholine through continuous in situ syntheses. A recent proof-of-concept study led to the discovery of this enzyme in the human extracellular fluids. We report here that apolipoprotein-E, in particular ε4 isoprotein acts as one of the strongest endogenous anti-amyloid-β fibrillization agents reported in the literature. At biological concentrations, apolipoprotein-E prevented amyloid-β fibrillization for at least 65 h. We show that amyloid-β interacts readily in an apolipoprotein-facilitated manner with butyrylcholinesterase, forming highly stable and soluble complexes, BAβACs, which can be separated in their native states by sucrose density gradient technique. Enzymological analyses further evinced that amyloid-β concentration dependently increased the acetylcholine-hydrolyzing capacity of cholinesterases. In silico biomolecular analysis further deciphered the allosteric amino acid fingerprint of the amyloid-β-cholinesterase molecular interaction in formation of BAβACs. In the case of butyrylcholinesterase, the results indicated that amyloid-β interacts with a putative activation site at the mouth of its catalytic tunnel, most likely leading to increased acetylcholine influx into the catalytic site, and thereby increasing the intrinsic catalytic rate of butyrylcholinesterase. In conclusion, at least one of the native physiological functions of amyloid-β is allosteric modulation of the intrinsic catalytic efficiency of cholinesterases, and thereby regulation of synaptic and extrasynaptic cholinergic signalling. High apolipoprotein-E may pathologically alter the biodynamics of this amyloid-β function.
Introduction
The most common type of dementia is sporadic Alzheimer’s disease, with advanced age and apolipoprotein E ( APOE ) ε4 allele (APOE4) as its major risk factors. Although APOE4 is most confirmed genetic risk factor for developing Alzheimer’s disease, its pathological role in Alzheimer’s disease or its involvement with the pathological processes in Alzheimer’s disease, such as fibrillization or oligomerization of amyloid-β peptides and hyperphosphorylation of tau protein is highly obscure.
Some in vitro studies suggest that synthetic amyloid-β peptides readily form complexes with human APOE protein isoforms, in particular the ε4 isoprotein ( Strittmatter et al. , 1993 a , b ; Naiki et al. , 1997 ; Sweeney et al. , 2004 ; Hone et al. , 2005 ), which is found in amyloid-β deposits in the Alzheimer’s disease brain ( Thal et al. , 2005 ). Other well documented proteins, which are found in association with amyloid-β deposits such as amyloid-β plaques and cerebral amyloid angiopathy, and with neurofibrillary tangles of tau protein in the Alzheimer’s disease brain are the cholinergic enzymes, acetylcholinesterase (ACHE, encoded by ACHE ) and butyrylcholinesterase (BuChE, encoded by BCHE ) ( Carson et al. , 1991 ; Mesulam et al. , 1992 ; Wright et al. , 1993 ; Geula et al. , 1994 ; Mesulam and Geula, 1994 ). Nonetheless, these observations have been reported from post-mortem brain studies, which most likely reflect the endpoint outcome of a gradual and subtle long-term interaction between these proteins rather than a fast phenomenon. In vitro studies using purified or recombinant proteins have further suggested that ACHE accelerates but BuChE attenuates aggregation of amyloid-β peptides into fibrils ( Inestrosa et al. , 1996 a , b ; Diamant et al. , 2006 ; Berson et al. , 2008 ; Podoly et al. , 2008 ). However, these in vitro studies were done under APOE protein-free conditions, and because of methodological necessities, the concentrations used were greater than the estimated physiological levels. Hence, it is difficult to extrapolate these findings to their ultimate in vivo outcomes.
Nonetheless, a recent in vivo study on BuCHE-knockout mice show contrasting findings of a decrease in the plaque burden in the brain of mice lacking BuChE ( Reid and Darvesh, 2015 ). While the outcome of ACHE–amyloid-β interaction has been confirmed by studies on double transgenic mice expressing both human ACHE and APPswe genes, suggesting higher level of plaques in these animals compared to single transgenic APP swe mice ( Rees et al. , 2003 , 2005 ) and that the amyloid-β burden is most tightly correlated with memory impairment in the double transgenics ( Rees et al. , 2005 ).
Studies on CSF of patients with Alzheimer’s disease have provided further evidence for accumulation of soluble ACHE–amyloid-β and BuChE–amyloid-β complexes in CSF ( Darreh-Shori et al. , 2011 b , 2012 , 2014 ). In addition, the ACHE–amyloid-β interaction seems to cause a strong inactivation of ACHE in CSF, which could be recovered by donepezil, most likely by displacing amyloid-β from peripheral anionic sites on ACHE ( Darreh-Shori et al. , 2014 ). In addition, intracerebral ventricular injections of the amyloid-β peptide in the brain of rats suggest that amyloid-β may alter the molecular forms of ACHE in the CSF and brains of rats ( Saez-Valero et al. , 2002 ).
Further studies on CSF samples indicated that APOE is also involved in the formation of BuChE/ACHE-amyloid-β complexes leading us to propose the term BAβACs (BuChE/ACHE-amyloid-β-APOE complexes) ( Darreh-Shori et al. , 2011 a , b , 2012 ; Vijayaraghavan et al. , 2013 ). The main characteristic of BAβACs is that they can oscillate between a slow and ultrafast state of acetylcholine hydrolysis, depending on access to amyloid-β peptides ( Darreh-Shori et al. , 2011 a , b ). Intriguingly, cholinergic neurons seem to be the main neuronal populations in the brain that contain intraneuronal amyloid-β in both healthy and diseased brain ( Baker-Nigh et al. , 2015 ; Norvin et al. , 2015 ). We have thus hypothesized that an amyloid-β induced allosteric hyperactivation of these enzymes represents a native physiological function for the universal production and nerve activity synchronized amyloid-β release ( Cirrito et al. , 2005 ). In other words, the physiological action of amyloid-β can include the tuning of cholinergic action at synapses and in interstitial fluid, thereby regulating the functional status of cholinoceptive neuronal and non-neuronal non-excitable cells, which are abundant in the brain and include microglia, astrocytes, oligodendrocytes, endothelia, and vascular smooth muscles ( Darreh-Shori et al. , 2011 a ; Vijayaraghavan et al. , 2013 ). Nonetheless, this hypothesis also explains why cholinergic neurons are particularly vulnerable to the actions of amyloid-β ( Kar et al. , 1998 ). Furthermore, BAβACs seems to be accumulated in the presence of high APOE protein (a condition readily fulfilled in APOE4 carriers) ( Darreh-Shori et al. , 2011 a , b ), explaining how APOE protein may contribute to the pathological events in Alzheimer’s disease.
In the current study we aimed to decipher the details of molecular interaction and formation of BAβACs. We used the well-established thioflavine T assay and investigated the changes in the rate of amyloid-β peptides fibrillization in the presence of APOE, BuChE or ACHE in vitro using purified human proteins at various physiological and non-physiological concentrations. We also used sucrose density gradient (SDG) analyses coupled with quantitative assays as an optimal procedure to separate the spontaneously formed soluble complexes, and to measure their levels and activities. In addition we performed in silico molecular docking studies to decipher further the molecular fingerprint of the interactions between amyloid-β peptides and the cholinesterases. We here provide compelling evidence for the formation of soluble BAβACs. The in silico studies also revealed important insights into the molecular binding and the activation mechanism of the BuChE/ACHE-amyloid-β complexes.
Materials and methods
Peptides and purified proteins
APOE protein
To minimize misinterpretation based on post-transcriptional processing of different recombinant APOE protein we used three different purified APOEs: (i) human APOE protein highly purified from non-frozen human plasma (>95%, catalogue number A-2001, rPeptide), which is mixed isoforms of APOE according to the allelic frequency of APOE genotypes among the human population; (ii) recombinant human APOE ε2, ε3 and ε4 isoforms expressed in Escherichia coli (>90%, catalogue number BV-4660-500, BV-4696-500, BV-4699-500, respectively, purchased from BioVision Inc.); and (iii) recombinant human APOE ε2, ε3 and ε4 (>95%, catalogue number P2002, P2003 and PV2004, respectively, from Invitrogen), which are expressed in insect cells ( Gretch et al. , 1991 ).
We used three different concentrations of APOE proteins [1.0, 0.5 and 0.1 µM in phosphate-buffered saline (PBS)], which represent the mean CSF APOE level and the ranging values, respectively, measured in the CSF of the patients with Alzheimer’s disease included in our previous reports ( Darreh-Shori et al. , 2011 b ).
BuChE protein
Highly purified BuChE from human serum (>99.7%, Sigma-Aldrich, catalogue number B4186) was resolved in PBS to the concentration of 1.0 g/l (17.12 µM based on a molecular weight of 58.42 kDa for its monomeric subunit according to previous report) ( Diamant et al. , 2006 ). BuChE was used in three different concentrations/activity, one at high concentration (0.2 µM; ∼10-fold over the CSF BuChE activity level), and two physiologically relevant concentrations (0.02 µM and 0.004 µM, simulating the CSF BuChE activity level >7.0 and <5.0 nmol/min/ml, in Alzheimer’s disease patients with high- and low-CSF BuChE activity, respectively) ( Darreh-Shori et al. , 2006 a ).
The BuChE protein was incubated with or without APOE proteins in a 37°C water bath and BuChE activity measured at intervals. The BuChE activity in the samples immediately before incubation was used as the initial activity (t 0 ).
Amyloid-β peptides
Ultrapure, hexafluoroisopropanol (HFIP)-treated, recombinant amyloid-β 1–40 (amyloid-β 40, catalogue number A-1153 from rPeptide) and 1–42 peptides (amyloid-β 42 , catalogue number A-1002, rPeptide) were used. The amyloid-β peptide stock solutions were prepared in DMSO (dimethyl sulphoxide, D2650, Sigma) by rigorous vortexing, and sonication 10 times for 3 s on ice, and finally filtering the solution through a 0.2 -μm filter unit (Spartan 3/0.2PA, Whatman Group). The stock solutions were freshly prepared at 850 µM (of amyloid-β 40 ) and 400 µM (amyloid-β 42 ) stock concentrations, based on the molecular weight of the peptides provided by the manufacturer.
Thioflavin T fluorescence assay
The overall experimental workflow is depicted schematically in Supplementary Fig. 1 . The thioflavin T assay was carried out as described previously ( Darreh-Shori et al. , 2014 ). Briefly, the assay was run (i) in PBS (pH = 7.4) using a predefined amount of purified human ACHE and BuChE together with certain amounts of other ‘additives’ (amyloid-β peptides and APOE proteins); and (ii) in 25% pooled CSF samples in PBS together with the ‘additives’. For analysis on pooled CSF samples, the final concentrations of added amyloid-β 42 and amyloid-β 40 peptides in the wells were 2.17 μM and 20 μM, respectively (molar ratio amyloid-β 42/40 of 0.11, based on physiological proportions observed in CSF of patients with Alzheimer’s disease). All samples were applied in triplicate in the wells of a microtitre plate [(NUN96fb_LumiNunc FluoroNunc) - Nunclon 96 Flat Black, DK]. The final volume was adjusted to 200 μl per well with a freshly prepared solution of thioflavin T (#T3516, Sigma) in PBS. The final concentration of thioflavin T in the wells was 1.0 μM. The plates were then sealed with tape (Nunc, #236366, DK) with the lock on, and covered carefully with Parafilm® to avoid evaporation. Continuous incubation and reading took place in a Tecan spectrophotometer (Infinite 1000) at 37°C. The kinetic bottom-reading function was set at 15-min intervals with 5 s orbital shaking just prior to each reading at 450 nm excitation wavelength and 490 nm as the emission wavelength.
Sucrose gradient sedimentation analysis
SDG analysis was used to investigate the presence of stable complexes resulting from molecular interactions between BuChE, amyloid-β and APOE. Immediately after the thioflavin T assay (∼70 h of incubation), the replicate wells of the plates containing certain concentrations of amyloid-β and/or APOE and/or BuChE were collected and pooled. Then 140 µl of these pooled samples were carefully applied on the surface of the gradients in ultracentrifuge tubes (Ultra-Clear tube, catalogue number 344059, Beckman) containing 10 ml of 5–20% sucrose gradients. The gradient was prepared by adding first 5 ml of a 5% sucrose solution (wt/vol) in Tris-buffered saline–MgCl 2 (TBS; 10 mM Tris-HCl, pH 7.4, 0.9% NaCl and 50 mM MgCl 2 ) into the ultracentrifuge tube. Then 5 ml of a 20% sucrose solution was added by inserting the tip of a Pasteur pipette into the bottom of the ultracentrifuge tube and pipetting the 20% sucrose solution into the upper end of the Pasteur pipette. The ultracentrifuge tube was then capped, gently rolled on a 45° angled surface, and brought to vertical position in a tube holder (for details see http://www.science-projects.com/GradCent.htm !). We did not use any detergents such as Brij96 or Triton™ X-100 in the gradient buffer as is commonly used in some SDG analyses to avoid interfering with non-covalent binding forces involved in the molecular interactions between the proteins and peptides used in the experiments.
Immediately after addition of the samples on the top of each SDG-tube, we also added two standard proteins/enzymes with known sedimentation coefficient (S), i.e. bovine liver catalase (11.4 S; and molecular weight of ∼250 kDa, Sigma) and calf intestinal alkaline phosphatase (6.1 S; molecular weight ∼140 kDa, Sigma).
The ultracentrifugation was run for 18 h at 4°C at 165 000 g (36 600 rpm) in a Beckman ultracentrifuge (XL100, rotor SW 41 Ti). Approximately 50 fractions were then collected from the bottom of each tube in 96-well plates. The two enzymes, catalase and alkaline phosphatase, served partly as standard proteins for calculation of the sedimentation coefficient of the SDG fractions, and partly as irrelevant control proteins for the molecular interaction between ACHE or BuChE and amyloid-β and/or APOE. We did not observe any findings that could indicate interaction between amyloid-β or APOE and these two control enzymes.
The mixtures of purified proteins and peptides in PBS buffer had, by necessity, a simple microenvironment fluid. Therefore we also performed similar SDG analyses on CSF samples, as a surrogate for the highly complex parenchymal fluids in the brain, to avoid misinterpretations and to further challenge the specificity of the interactions observed in experiments on purified proteins. These SDG analyses were done on nine distinct pooled Alzheimer’s disease CSF samples, which had been prepared based on the patients APOE4 and BCHE-K genotypes. Each CSF SDG tube was loaded with 500 μl of pooled CSF sample.
Detection and measurement of additives in the SDG fractions
ACHE and BuChE activity measurements after thioflavin T assay and SDG analysis
Ellman’s colorimetric assay was used to determine enzyme activities from both experiments on purified proteins and pooled CSF samples. The protein level of these enzymes in each SDG fraction was determined with enzyme-linked immunosorbent assay as described previously ( Darreh-Shori et al. , 2006 b ).
When purified BuChE was used, a master mix (MMB) that contained the substrate butyrylthiocholine iodide (BTC, 5.0 mM final concentration, Sigma) and the Ellman’s reagent DTNB (0.4 μM final concentration, Sigma) in sodium potassium phosphate buffer (50 mM, pH 7.4) was used for BuChE activity measurements. For ACHE activity, the master mix (MMA) consisted of acetylthiocholine iodide as the substrate (ATC, 0.5 mM, final concentration, Sigma) instead of BTC. When pooled CSF samples was analysed, the MMB and MMA also contained selective ACHE (BW280C50, 1.0 μM final concentration, Sigma) or BuChE (ethopropazine, 0.1 μM final concentration, Sigma) inhibitors, respectively.
Wells containing the buffer, or just the amyloid-β and/or APOE were used as the negative controls. Depending on the experiment, the samples could contain thioflavin T, sodium azide (or thimerosal) and/or DMSO, therefore prior optimization analyses were required to control for, and reduce their possible effect on Ellman’s assay. Thioflavin T and/or DMSO increased the background slightly but did not interfere with kinetic readings of the enzymatic rates. The preservative, sodium azide (or thimerosal) strongly interfered with Ellman’s assay due to direct nucleophilic attack to ATC, BTC and/or DTNB. In our study, a concentration of 0.005% in the wells of thioflavin T assay plates was enough to prevent microorganism growth (up to 120 h). This gave a final concentration of 0.0001% sodium azide in the wells of the replicate plates for enzymatic assay, and did not interfere with kinetic readings of cholinesterase activities by Ellman’s assay as long as the enzyme activity rate was >10 nmol/min/ml. Endpoint readings should however be avoided if changes in absorbance cannot be calculated.
APOE and amyloid-β levels in SDG fractions
Presence and relative levels of the amyloid-β peptides and APOE protein in each SDG fraction were assessed by dot-blot analysis on nitrocellulose membrane (Hybond P, Invitrogen), when the experiment was run on purified proteins in PBS. The blots were prepared by applying 2 µl of each fraction on the membrane. All fractions were applied on the same blot. The optical density of each dot was calculated by the ImageQuant™ TL software (v5, Invitrogen).
APOE protein was detected by the A1.4 antibody (1/8000, sc-13521, Santa Cruz Biotech.) and the secondary HRP-conjugated anti-mouse antibody (1/1000, sc-2005, Santa Cruz Biotech.) in 5% M-TBS-T 0.3% blocking solution (10 mM TBS containing 5% non-fat dry milk, 170-6404, Bio-Rad and 0.3% Tween-20). Amyloid-β peptides in the fractions were detected by the 6E10 mouse monoclonal antibody (1/1000, SIG-39320, Covance) and the secondary HRP-conjugated antibody (sc-2005, Santa Cruz Biotech.).
Biomolecular docking analysis
Crystal structure of BuChE (PDB id: 3BDS) ( Nachon et al. , 2013 ) and ACHE (PDB id: 4EY7) ( Cheung et al. , 2012 ) were downloaded from the PDB database along with solution structures of amyloid-β 42 (PDB id: 1IYT) ( Crescenzi et al. , 2002 ) and amyloid-β 40 (PDB id: 1BA4) ( Coles et al. , 1998 ) and used for molecular docking studies. The obtained structure was thoroughly checked for any artefacts and prepared using the structure preparation tool of SYBYL-X molecular modelling suite installed on Linux based Dell Precision T7610 workstation (Intel® Xeon® E5-2643 CPU @ 3.3 GHz; 16 GB RAM, 2 TB hard disk). Protein preparation steps involved addition of any missing hydrogen, removal of water molecules, and minimization of energy using the Powell method ( Wang et al. , 2003 ) with Tripos force field.
Protein–protein docking was performed with Cluspro 2.0 ( Comeau et al. , 2004 a , b ; Kozakov et al. , 2006 , 2013 ) which is a fully automated web server for prediction of protein–protein interaction and has three main steps. The first step involves running PIPER, a Fast Fourier Transform correlation-based docking method. The second step involves clustering based on a hierarchical pairwise RMSD (root mean squared deviation) algorithm to retain near native conformations and discard the unstable clusters. The third step involves judging the stability of these clusters using Monte Carlo approach and refinement using medium range optimization method. The best models were selected on the basis of cluster size and the parameters generated by the ‘Balanced’ scoring function, which is defined by a balance between the electrostatic, hydrophobic, and van der Waals forces.
Results
High APOE levels strongly prevent amyloid-β fibrillization
In the absence of purified human serum APOE protein, fibrillization of the recombinant amyloid-β 40 or amyloid-β 42 readily occurred within 6 h of incubation at 37°C ( Fig. 1 A and B).
Figure 1.

Apolipoprotein E protein strongly prolongs the lag time and attenuates the maximum fibrillization levels of amyloid-β peptides in a concentration-dependent manner. ( A ) Fibrillization kinetic curve determined by thioflavin T assay for recombinant amyloid-β 40 peptides alone, and together with three physiological concentrations of purified human APOE protein ( Darreh-Shori et al. , 2011 b ). At the highest APOE protein of 1.0 μM, corresponding to the levels observed in CSF of APOE4 homozygotes Alzheimer’s disease patients, no fibrillization of amyloid-β 40 occurred for at least 67 h. ( B ) The effect of APOE protein concentrations on fibrillization kinetics of amyloid-β 42 peptides. Thus, APOE concentration-dependently attenuated the amyloid-β peptides, in particular the amyloid-β 40 peptides. The graphs are per cent of maximum fibrillization (F max ) of the corresponding amyloid-β peptides at the last 2 h of the assay.
Addition of APOE protein at all of the physiologically relevant concentrations corresponding to the levels observed in the CSF of Alzheimer’s disease patients (1.0–0.1 µM) ( Darreh-Shori et al. , 2011 b ) strongly prolonged the lag time (F LT, see inset in Fig. 1 ), reduced the fibrillization rate (F Rate ) and the fibrillization plateau (F max ) of the recombinant amyloid-β peptides ( Fig. 1 A and B). Nonetheless, the anti-amyloid-β fibrillization of APOE was strongly concentration-dependent ( Fig. 1 A and B).
The anti-amyloid-β fibrillization effect of APOE protein was highly isoprotein-dependent
Next we examined whether the effect of APOE was also isoform-dependent by using recombinant human APOE ε2, ε3 and ε4 isoproteins. We found that the anti-amyloid-β fibrillization effect of APOE protein was also isoprotein-dependent, particularly when amyloid-β 42 peptide was involved ( Supplementary Fig. 2A and Supplementary Data ).
The APOE ε3 and ε4 isoproteins had similar effects and increased the F LT of amyloid-β 40 peptide aggregation from 6 h to >30 h, whereas the APOE ε2 isoform prolonged the F LT of amyloid-β 40 to 16 h ( Supplementary Fig. 2A ).
When amyloid-β 42 peptide was used, the APOE ε4 protein exerted the strongest effect and completely abolished the fibrillization of amyloid-β 42 peptides for at least 70 h ( Supplementary Fig. 2B ). In contrast, the fibrillization dynamic of amyloid-β 42 peptides were essentially the same as observed for the amyloid-β 40 peptides due to presence of the APOE ε3 isoprotein i.e. an increased F LT ∼30 h ( Supplementary Fig. 2A ).
Amyloid-β 42 and amyloid-β 40 behave differently in the presence of APOE ε3 and ε4
We also compared the effect of different concentrations of APOE isoproteins on the fibrillization properties of recombinant amyloid-β 40 and amyloid-β 42 peptides ( Supplementary Fig. 3 ). The findings indicated that both amyloid-β 42 and amyloid-β 40 underwent quite similar kinetic changes in their fibrillization properties particularly at the APOE concentration range of 0.5–1.0 µM ( Supplementary Fig. 3A and Supplementary Data ), corresponding to the CSF levels of Alzheimer’s disease patients with moderate-to-high APOE protein ( Darreh-Shori et al. , 2011 b ).
In the presence of APOE ε4, in contrast to ε3 protein, we observed strong concentration-dependent changes in the fibrillization kinetics of amyloid-β 42 , which differed with that observed for the amyloid-β 40 peptides ( Supplementary Fig. 3C and D ). At 1.0 µM concentration of APOE ε4 protein, no significant fibrillization of amyloid-β 42 was observed for at least 70 h ( Supplementary Fig. 3D ). However, a reduction in the APOE ε4 concentration from 1.0 µM to 0.5 and 0.1 µM caused dramatic kinetic changes in the fibrillization of amyloid-β 42 peptides, namely F LT was reduced from at least 65 h to 15 and 5 h, respectively. The F Rates and F max were also increased 200–300% ( Supplementary Fig. 3D ).
Thus, the fluctuations in the concentration of APOE protein were accompanied with the strongest changes in the kinetic of amyloid-β 42 fibrillization, particularly when APOE ε4 isoprotein was included in the experimental setups.
BuChE alone does not significantly change amyloid-β fibrillization
We and others have shown that BuChE may play a role in Alzheimer’s disease, particularly in relation to the presence or absence of APOE ε4 allele ( Diamant et al. , 2006 ; Podoly et al. , 2008 ; Darreh-Shori et al. , 2011 b , 2014 ). For instance, in vitro analyses have shown that BuChE attenuates the kinetics of amyloid-β fibrillization at a concentration of 0.2 µM of BuChE protein ( Diamant et al. , 2006 ), which is at least 10-fold higher than the CSF BuChE levels in patients with Alzheimer’s disease (0.02 to 0.004 µM, based on molecular weight of 65 kDa for its monomeric subunits) ( Darreh-Shori et al. , 2006 a , 2011 b ).
At the high BuChE concentration (0.2 μM) of the highly purified human serum BuChE, the kinetics of amyloid-β fibrillization in the current study were essentially identical to those reported by Diamant et al. (2006) and Podoly et al. (2008) ( Fig. 2 A and B).
Figure 2.
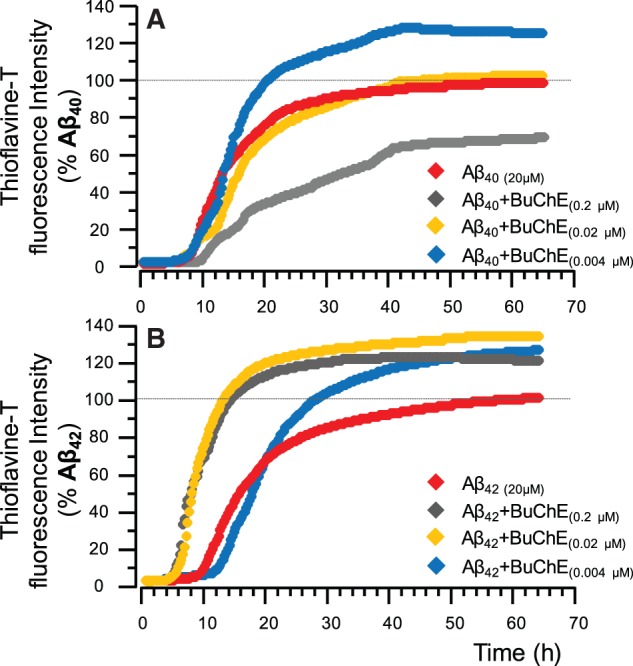
Kinetic changes in the fibrillization of the amyloid-β 40 and amyloid-β 42 peptides in the presence of BuChE. ( A ) Fibrillization kinetic curve determined by thioflavin T assay for recombinant amyloid-β 40 peptides alone, and together with two physiological concentrations of purified human BuChE protein, namely 0.02 and 0.004 μM corresponding to high and low CSF BuChE levels in patients with Alzheimer’s disease ( Darreh-Shori et al. , 2006 a ). In close agreement with a previous report ( Diamant et al. , 2006 ), at the highest BuChE protein of 0.2 μM, which is not physiological, the BuChE increased the amyloid-β 40 lag-time (L t ∼ 4 h), and reduced the fibrillization rate (F Rate ) and maximum (F max ∼ 30%). However, at the two physiologically relevant BuChE concentrations only slight changes in the fibrillization kinetics of amyloid-β 40 are apparent. ( B ) The effect of BuChE protein concentrations on the fibrillization kinetics of amyloid-β 42 peptides. In contrast to the observed changes in amyloid-β 40 , BuChE, particularly at its physiological concentrations seems to actually accelerate the fibrillization of amyloid-β 42 peptides. The graphs are per cent of maximum fibrillization (F max ) of the corresponding amyloid-β peptides at the last 2 h of the assay.
However, we also monitored the fibrillization of amyloid-β peptides at the more relevant concentrations of BuChE protein corresponding to the observed BuChE levels in the CSF of ε4 non-carrier and carrier Alzheimer’s disease patients (0.02 to 0.004 µM, respectively) ( Darreh-Shori et al. , 2006 a , 2011 b ). Within this concentration range, BuChE protein increased the fibrillization properties of the recombinant amyloid-β peptides, particularly when amyloid-β 42 peptide was used ( Fig. 2 A and B). Indeed, presence of BuChE protein at all tested concentrations increased the F max of amyloid-β 42 peptides by 20–30% ( Fig. 2 B).
APOE protein changes the effect of BuChE on amyloid-β fibrillization
Obviously, the composition of the brain parenchymal fluid is far more complex than that of the in vitro experimental paradigms, and thereby many of these endogenous proteins are likely to affect the kinetics of amyloid-β peptides ( Darreh-Shori et al. , 2014 ). As a partial remedy to this lacking, we performed the analyses by combining both APOE and BuChE proteins to the amyloid-β peptides ( Fig. 3 ), since numerous studies indicate a genetic or molecular interaction between APOE, BuChE, or both with amyloid-β peptides in Alzheimer’s disease ( Mesulam and Geula, 1994 ; Guillozet et al. , 1997 ; Lehmann et al. , 1997 ; Diamant et al. , 2006 ; Lane et al. , 2008 ; Podoly et al. , 2008 ; Darreh-Shori et al. , 2011 b , 2012 , 2014 ).
Figure 3.

Kinetic changes in the fibrillization of the amyloid-β 40 peptides in the presence of apolipoprotein E and BuChE proteins. ( A ) Fibrillization kinetics of amyloid-β 40 peptides alone, and together with physiological concentration of 0.5 μM APOE protein, corresponding to the moderate levels observed in CSF of APOE4 heterozygotes Alzheimer’s disease patients ( Darreh-Shori et al. , 2011 b ). In the presences of 0.5 μM APOE protein, BuChE protein, particularly at 0.02 and 0.2 μM concentrations augmented the anti-amyloid-β fibrillization effect of APOE protein. ( B ) The effect of BuChE protein concentrations on the fibrillization kinetics of amyloid-β 40 peptides together with 0.1 μM APOE protein, that corresponds to a low CSF APOE level observed in APOE4 non-carriers ( Darreh-Shori et al. , 2011 b ). At this APOE concentration, BuChE, particularly at low (0.004 μM) and high (0.02 μM) concentrations merely increased the lag-time of amyloid-β 40 fibrillization but seems to actually accelerate the fibrilization rate of amyloid-β 42 peptides. Fibrillization kinetic curves determined by thioflavin T assay as described in the ‘Material and methods’ section using recombinant amyloid-β peptides and purified human APOE and BuChE proteins.
Indeed, the inclusion of both APOE and BuChE protein seemed to synergistically change the aggregation dynamics of the interaction between BuChE and the amyloid-β 40 and amyloid-β 42 peptides, particularly relative to that observed with low concentrations of BuChE ( cf . Figs 2 and 3 ).
SDG analysis reveals formation of molecular complexes
These results apparently indicate that APOE, BuChE and amyloid-β peptides interact and form soluble and/or non-soluble complexes. We hence investigated this notion by subsequent SDG and enzymatic analysis. After the completion of the thioflavin T assay of amyloid-β fibrillization (usually ∼65 h of incubation), the samples were carefully collected from the microplate wells and analysed by the SDG procedure. The rationale of analysis of molecular interactions using SDG techniques is as follows. Monomeric subunits of amyloid-β peptides, APOE and BuChE have a molecular weight of ∼3.5 kDa, ∼34 kDa and ∼65 kDa, respectively. In other words, they have distinct sedimentation coefficients (S). The relative ranking order of S-values is S amyloid-β << S APOE < S BuChE . For instance, when only amyloid-β is included in the tubes the SDG diagram will show quantitatively peaks at S-values corresponding to amyloid-β monomers, dimers etc. Similarly in the case of APOE, we expect to see two main peaks with S corresponding to monomeric (∼34 kDa) and dimeric (∼68 kDa) forms of APOE since it is known that APOE can dimerize. BuChE alone is expected to show peaks with S corresponding to its globular subunit (G 1 for monomeric 65–70 kDa), G 2 (dimeric) and G 4 (tetrameric) forms of BuChE. Thus, in the absence of molecular interaction and formation of stable complexes between these biopolymers, they will end up in fractions with the matching S-values following SDG fractionation and analysis. However when there are stable molecular interactions between these biopolymers, their SDG diagram will show additional peaks at higher S-values than their individual S. In other words, there will be shift in the S-values depending on the number of interacting molecules in each complex.
Amyloid-β peptides produces soluble complexes with APOE protein
The results of the SDG analyses are summarized in Fig. 4 . In the absence of any additives, the SDG diagram for amyloid-β alone (black line in Fig. 4 A) illustrates several light peaks corresponding to monomeric and small multimeric forms of amyloid-β peptides with sedimentation coefficients (S) of ∼0.8, and 1.4, 2.6 and 3.1, consistently reflecting the presence of monomeric, dimeric, trimeric and tetrameric forms of amyloid-β peptide assemblies, respectively. There are also several heavier amyloid-β peaks, for instance between 11.4S and 13.2S, most likely representing soluble amyloid-β protofibrills ( Fig. 4 A, black line).
Figure 4.

SDG analyses of molecular interactions between recombinant amyloid-β peptides and purified human serum APOE and BuChE proteins. The molecular interactions between recombinant human amyloid-β peptides alone or together with purified human APOE and/or BuChE were analysed using the SDG technique. These samples were those that had been recollected after ∼70 h of incubation from the wells of the microtitre plates at the end of the thioflavin T assay as is depicted in the schematic Supplementary Fig. 1 . Linear sucrose gradient solutions were prepared in ultracentrifuge tubes. Samples were then loaded on the top of the gradient, together with two enzymes, alkaline phosphatase (AP) and catalase ( C ) with known sedimentation coefficients (S) of 6.1 and 11.4, respectively, as indicated by the vertical arrows in the SDG diagrams. These two enzymes also served as irrelevant control proteins for the interaction between amyloid-β, APOE and BuChE proteins. After overnight ultracentrifugation, the content of each SDG tube was carefully fractionated into ∼50 equal fractions from the bottom of the tubes, and the levels of amyloid-β, APOE and BuChE were quantified in all fractions, as described in the ‘Material and methods section. ( A ) Presence and relative levels of amyloid-β peptides in the fractions are plotted versus the corresponding S-values of the fractions. The denoted peaks a, b, and c from the SDG tube containing only amyloid-β (black line) exhibit S-values that closely correspond to the molecular weight of amyloid-β monomers (∼4 kDa), dimers (∼8 kDa) and tetramers or hexamers (∼16–24 kDa), respectively. In the presence of APOE protein (blue line), these peaks exhibit strong shifts in their S-values, indicating that all amyloid-β peptides were incorporated in stable amyloid-β-APOE complexes (the light blue-shaded area). In the presence of the BuChE protein (red line), the intensity of these amyloid-β peaks show partial reduction, while new amyloid-β peaks appear with much larger S-values, corresponding to stable amyloid-β-BuChE complexes (the light pink-shaded area). When both APOE and BuChE proteins were present (green line), the peaks’ intensities (relative levels) but not their S-values are altered, indicating competitive displacement of amyloid-β peptides between APOE and BuChE and/or rearrangements of the complexes. (B) Relative levels of APOE protein are plotted against S-values of the fractions. In the absence of amyloid-β peptides, the SDG procedure separated the purified serum APOE protein (black line) in three main peaks, corresponding to a major APOE fragment with ∼24 kDa (a*) and full-length monomers (b* ∼34 kDa) and dimers (c*∼68 kDa) of APOE. In the presence of amyloid-β (blue line), these peaks are completely shifted to the right, evincing that APOE is in complexes with amyloid-β, as was predicted in A (amyloid-β-APOE complexes as is highlighted by the light blue-shaded area). ( C and D ) Relative levels of purified serum BuChE protein ( C ) and activity ( D ) in the corresponding SDG fractions are plotted versus the S-values of the SDG fractions from the specified tubes. Similar to the amyloid-β and APOE SDG diagrams, BuChE-containing peaks show a shift in their S-value when amyloid-β (red line) or both amyloid-β and APOE (green line) were present compared to BuChE alone (orange line in C and D ). Note that in D , the y -axis is cut-off at 1% of overall SDG fractions from each tube to be able to illustrate the presence of lighter globular ( G ) forms of BuChE as the G4 BuChE and/or BuChE-amyloid-β-APOE (BAβA) complexes exhibited ∼99% of the overall BuChE in the fractions, although the relative protein levels of theses heavy BuChE containing peaks protein were ∼10–12% of the total fractions the light-pink and green shaded area in C , which is reasonably comparable with the relative levels of the G 1 (∼1%) or G 2 peaks (∼4%).
Similarly, the corresponding SDG diagram for APOE alone ( Fig. 4 B, black line) shows a broad double peak at ∼2.5S and ∼4S, most likely corresponding to the truncated and full-length monomeric (∼30–34 kDa) forms of APOE proteins. In addition, the overlapping shoulder peak between 4.5–5.2S regions represents the dimeric (∼68 kDa) form of APOE protein. However, when amyloid-β was incubated together with APOE protein (the APOE+amyloid-β condition), the SDG diagram (blue line in Fig 4 A) indicates that all the lighter amyloid-β peaks at 0.8–3.1S are strongly diminished. Instead, a new broad multi-peak is formed between 6S to 10S (compare the black and blue lines in Fig. 4 A). In addition, the heavier amyloid-β peaks corresponding to the amyloid-β protofibrills shifted to the right to S12 and S13. Consistently, the corresponding SDG APOE+amyloid-β diagrams based on APOE detection in the fractions (the blue line in Fig. 4 B) reveals that the twin peak of APOE (at 2.5–5.2S region) is equally shifted to the right, producing a triple peak between 6–10S, as well as between 10–13S (compare the blue and black lines in Fig. 4 B). In other words, the precise co-shifts in S-values of the peaks in Fig. 4 A and B suggest that the major part of the added amyloid-β peptides were co-migrated in the sucrose gradient as stable complexes with APOE protein (denoted amyloid-β-APOE complexes in Fig. 4 A and B). This conclusion is strongly supported by the results from the thioflavin T assay (illustrated in the Fig. 1 ), as by definition thioflavine T exhibits high fluorescent signals when it binds to amyloid-β fibrills/protofibrills [see the weak thioflavin T signals shown as the black graphs corresponding to the amyloid-β+APOE (1.0μM) setups in Fig. 1 A and 1 B].
BuChE interacts with amyloid-β peptides and forms molecular complexes
Similar to amyloid-β alone, the corresponding SDG diagram of the amyloid-β+BuChE setup (the red line in Fig. 4 A) shows the presence of the lighter amyloid-β peaks (at 0.8–3.1S) but with moderated intensities of the peak’s height as compared to the SDG diagram for amyloid-β alone (the black line in Fig. 4 A). Nonetheless, there are new peaks with moderate to high intensities at 9–11S and 12–13.5S regions that are not present in the SDG diagram for amyloid-β alone (compare these S regions in the black with the red lines in Fig. 4 A and C). This finding evinces that amyloid-β peptides also interacted and formed stable molecular complexes with human BuChE protein.
APOE protein facilitates the formation of molecular complexes between BuChE and amyloid-β
Finally, when amyloid-β peptides were incubated together with both APOE and BuChE proteins (amyloid-β+APOE+BuChE setup), the light amyloid-β peaks reappear, in particular the 0.8S peak corresponding to monomeric amyloid-β (compare the black and the green lines in Fig. 4 A), while the 6–10S peaks of the amyloid-β-APOE complexes show strong reduction in their intensities in both Fig. 4 A and B ( cf . green versus blue line). This indicates that BuChE either displaced a substantial part of APOE-bound amyloid-β peptides or prevented the formation of APOE-amyloid-β complexes. Consistent with this notion, the SDG diagram in Fig. 4 B (the green versus blue and black lines) reveals the partial reappearance of free APOE protein in the 2–5S region of the green line, and a reduction of peak intensities of the amyloid-β-APOE in the 6-12S regions. This confirms the above notion that the presence of BuChE indeed displaced amyloid-β-bound APOE protein. Most importantly, the examination of SDG diagrams in Fig. 4 A–C (the green line) show in addition a twin peak at the 12S/13S region, which contains relatively high amounts of amyloid-β peptides ( Fig. 4 A), APOE ( Fig. 4 B) and BuChE ( Fig. 4 C) proteins, indicating formation of a stable molecular complex between BuChE, amyloid-β and APOE. Thus, we have termed these twin 12S/13S peaks as the BuChE-amyloid-β-APOE complexes or BAβACs.
Note the reappearance of the intense 0.8S peak of amyloid-β and the twin peaks of the truncated and full-length monomeric (∼30–34 kDa) forms of APOE proteins, which were absent in the amyloid-β+APOE experiment. Thus, these observations strongly indicate occurrence of mutual displacement reaction and rearrangement of a significant amount of amyloid-β peptides and APOE protein in the process of the formation of the heavy BAβACs.
SDG analysis on CSF samples
The aforementioned SDG analyses were employed on highly purified human proteins, which due to the simplicity of the microenvironment in such assay fluid may not be representing the much more complex microenvironment in the brain parenchymal fluids. Therefore we used nine different pooled Alzheimer’s disease CSF samples as surrogate of the parenchymal fluids in the brain, and performed similar SDG analyses but without any other additives. The results are shown in Fig. 5 , which shows a spectral pattern that is almost identical to the SDG spectrums from the purified human proteins and recombinant amyloid-β peptides ( Fig. 4 ). The major difference is that APOE- or BuChE-containing peaks in the CSF SDG analyses generally exhibit larger sedimentation coefficient than those from the SDG analyses on purified proteins and peptides ( Fig. 4 ).
Figure 5.

SDG analyses of pooled CSF samples evince the molecular interactions between CSF amyloid-β peptides, APOE and BuChE proteins. Nine different pooled CSF samples were prepared and analysed by SDG. After overnight ultracentrifugation, each SDG tube was fractionated into ∼50 fractions with approximately equal volumes. The levels of amyloid-β, APOE, BuChE and ACHE were quantified in each fraction and their relative levels are plotted against the sedimentation coefficient (S), as is described in the ‘Material and methods’ section. The red line depicts the spectrum of amyloid-β-containing peaks detected in the pooled CSF samples, and is the average of SDG diagrams of nine different SDG tubes. The blue and green lines are the corresponding spectrums for APOE protein and BuChE activity in the SDG fractions of the pooled CSF samples. The graph with dashed-black line represents the SDG spectrum corresponding to ACHE activity in the SDG fractions of the CSF samples. The grey-shaded area highlights the amyloid-β region with small S-values. The first two amyloid-β peaks most likely represent smaller amyloid-β fragments (with molecular weight <4 kDa) in CSF. The denoted peaks a, b, and c exhibit S-values that closely correspond to the molecular weight of monomers (∼4 kDa), dimers (∼8 kDa) and tetrameric-to-hexameric homomers of amyloid-β peptides (∼16–24 kDa), respectively. The amyloid-β peaks between 3–6S (the blue-shaded area) are most likely representing various heteromeric amyloid-β complexes with APOE (as is denoted as amyloid-β-APOE complexes in the Fig. 4 A and B). The amyloid-β peaks of 6–9S may represent heteromeric complexes of amyloid-β with APOE and G 1 /G 2 forms of BuChE. Similarly, the heaviest amyloid-β peaks between 9-17S region (the green-shaded area) are representing heteromeric amyloid-β complexes with cholinesterases, in particular G 4 BuChE (amyloid-β-BuChE- and BAβA-complexes). The BuChE SDG spectra (the green lines here and in the Fig. 4 ) support this notion through the closely superimposing multiple peaks of high BuChE activities with the heavy amyloid-β peaks in this region. In contrast, the SDG spectrum of the ACHE activity shows a main relatively G4 ACHE peaks at 9S, where there are no amyloid-β present. Nonetheless, this peak exhibits a shoulder at 10.2S (arrowhead), where there are high amyloid-β and BuChE, most likely representing hybrid BuChE/ACHE-amyloid-β complexes. The scales are 0–2 for amyloid-β; 0–12 for APOE; 0–4 for BuChE and 0–16 for ACHE and are percentages of total fractions. Aβ = amyloid-β.
Nonetheless, these CSF SDG analyses reveal that there might be a range of BAβACs in CSF (denoted in Fig. 5 as light, heavy, and ultra-heavy BAβACs). The light BAβACs may represent an intermediate state of interaction, when the APOE molecules are not yet completely displaced by BuChE as it seems in the case for the heavy and ultra-heavy BAβACs. This is inferred from the very low (or absence) of APOE protein in the fractions in the 10S to 17S region (compare this region with the 6–8.5S region in the blue line in Fig. 5 ).
It is noteworthy that the peaks of amyloid-β in 1–3S region are similar in both CSF ( Fig. 5 ) and purified protein ( Fig. 4 A) SDG spectrums, suggesting that the SDG technique could properly identify two major homomeric amyloid-β peptides, namely an amyloid-β dimer (peak b in Figs 4 and 5 ), and a peak corresponding to an tetrameric or hexameric form of amyloid-β (peak c in Figs 4 and 5 ). To the best of our knowledge, these are the first data that show the presence of these amyloid-β oligomers in CSF without any major manipulation of CSF samples. The amyloid-β peaks in <1S region in the CSF amyloid-β spectrum most likely represent amyloid-β fragments expected to be present in CSF.
BAβACs constitute reactive soluble amyloid-β complexes
We also measured the enzymatic activity of BuChE in both following the thioflavin T and the SDG analyses. The SDG analyses indicated that the BuChE activity had a major peak ∼12S when in the BuChE-alone or BuChE-APOE setups (the orange and blue lines in Fig. 4 D). Nonetheless, the peaks became broader in the BuChE-amyloid-β or the BuChE-amyloid-β-APOE setups, indicating a shift in the heavier part of the peak to an S-value of ∼13S. Two conclusions can be drawn: (i) consistent with Fig. 4 A–C, the shift in S-value of the major peak from ∼12S to ∼13S is in agreement with the molecular interaction in BAβACs; and (ii) the larger area under the curve suggests that BuChE in BAβACs exhibited abnormally high activity. Indeed, the very broad overlapping multiple peaks of high BuChE activity in the 10–17S region of the CSF SDG spectrum (the green line in Fig. 5 ), confirms these notions.
To further challenge the latter conclusion, we performed new analyses using three physiological levels of human BuChE activities which were then mixed with increasing concentrations of amyloid-β 40 peptides ranging from 71 pg/ml to 71 ng/ml, which covers the pathophysiological ranges of amyloid-β 40 in CSF ( Fig. 6 A–C). The CSF levels of amyloid-β 40 and amyloid-β 42 are between 1–10 ng/ml and 200–800 pg/ml (based on our own database).
Figure 6.

Amyloid-beta peptides act as allosteric activator of human BuChE and ACHE . Highly purified human plasma BuChE, at three different physiological concentrations ( A–C ) was incubated with increasing concentrations of amyloid-β peptides. Note that the amyloid-β concentrations that needed (>71.4 pg/ml) to activate BuChE are physiologically relevant considering that CSF levels of amyloid-β are ∼4–10 ng/ml. Similarly, the concentrations of BuChE used in these experiments are also highly relevant since they correspond to activity ranges of 11.7 ± 0.2 ( A ) to 6.7 ± 0.2 nmol/min/ml ( C ). This is the range of BuChE activity in human CSF. However, when BuChE was incubated with amyloid-β the activity of the enzyme reached 32 ± 1 in ( A ), 16 ± 0.5 in ( B ), 10.5 ± 0.4 nmol/min/ml in ( C ). We then repeated these analyses using the lowest BuChE concentration but the measurements were done at different incubation times to investigate the steady-state effect of amyloid-β on BuChE ( D–F ). Comparison between D and E or F shows that the reaction between amyloid-β and BuChE is reasonably fast and reached within 30 min at amyloid-β > 700 pg/ml and within 60 min at amyloid-β < 71.4 pg/ml. ( G–I ) Corresponding changes in ACHE activity by amyloid-β peptides ( G ) are much more pronounced than the altered BuChE activity (shown in A ). However in contrast to its effect on BuChE activity ( A–C ), this amyloid-β-derived hyperactivity of ACHE diminishes with time ( cfG with H and I ), and gradually inhibits the enzyme by 80%, as shown previously ( Darreh-Shori et al. , 2014 ). In the current analyses, the average ACHE activity prior to incubation with amyloid-β was 47 ± 11 nmol/min/ml (in G–I).
This analysis confirmed that amyloid-β peptides at concentrations >700 pg/ml boosted the BuChE activity by up to 2.6-fold ( Fig. 6 A–C). In addition, we found that the maximum activation levels of BuChE at an amyloid-β concentration of 700 pg/ml was reached only at the highest BuChE concentration under this experimental setup.
We therefore performed the experiment using the lowest BuChE concentration but increasing incubation time of BuChE with amyloid-β peptides at 37°C ( Fig. 6 D–F). These analyses confirmed that the interaction and activation of BuChE by amyloid-β peptides was both concentration and time-dependent. In addition the reaction, particularly at lowest amyloid-β levels (70–700 pg/ml), seems to reach certain equilibrium state with regard to activation levels of BuChE by amyloid-β.
We also repeated these analyses on recombinant human ACHE protein ( Fig. 6 G–I). Initially (within the first 15 min), amyloid-β boosted the activity of ACHE more pronouncedly (∼5-fold, Fig. 6 G) than BuChE activity (2.5-fold, Fig 6 A). Nonetheless, with longer incubation time the initial increase was significantly reduced ( Fig. 6 H and I), indicating that some of the ACHE become inactivated by prolonged exposure to amyloid-β, in close agreement with previous observations in CSF samples or on purified ACHE proteins ( Darreh-Shori et al. , 2014 ).
Taken together, these observation suggested that the interaction of APOE and amyloid-β with BuChE (and/or ACHE) boosted the catalytic activities of these enzymes either by an increase in their structural stability or by inducing a structural change that improves the enzymes’ intrinsic catalytic properties or both. In any case, amyloid-β peptides seem to act as allosteric activator of cholinesterases, in particular the BuChE.
In silico modelling docking studies
To deduce insight into the nature of molecular interactions between amyloid-β peptides, BuChE and ACHE, we performed protein–protein docking studies. These docking studies were performed with Cluspro, a highly efficient protein-protein docking server which carried out cluster analysis after docking and identified nearly 28 clusters. The best cluster for BuChE- amyloid-β 42 complex was Cluster 00 with maximum members (184) and lowest energy (−928.5 kcal/mol) and for BuChE-amyloid-β 40 complex was Cluster 00 with maximum members (108) and lowest energy (−902.5 kcal/mol). In the case of ACHE-amyloid-β 42 and ACHE-amyloid-β 40 complexes the best clusters were 00 with cluster size of 112 (lowest energy: −876.2 kcal/mol) and Cluster 00 with cluster size of 108 (lowest energy: −953.3 kcal/mol), respectively. The predicted models were quite good in terms of electrostatic, van der Waals and cluster size. Details of the clusters obtained after docking along with the important interacting amino acid residues are given in Supplementary Table 1 .
Results from the docking of amyloid-β peptides with BuChE revealed that amyloid-β 42 , as well as amyloid-β 40 , preferably bind in the vicinity of the putative activation site, which comprises the amino acid residues A277–Y282 ( Chiou et al. , 2015 ) ( Fig. 7 ). The important interactions of the amyloid-β peptide with binding site amino acid residues are shown in 2D-ligand interaction diagram in Fig. 7 A and B for amyloid-β 42 and amyloid-β 40 , respectively. The important amino acids involved in interaction are also listed in Supplementary Table 1 and it is quite clear that amino acid Y288, which is an integral part of putative activation site, is very frequently involved in the interaction of these peptides with BuChE enzyme. This binding comprises of mainly hydrophobic interactions. This putative activation site is situated at the entrance of the tunnel going to catalytic site of the enzyme. The cave around the active site gorge contained two hydrophobic regions in opposite direction and is surrounded by several hydrophilic amino acid residues.
Figure 7.

Molecular docking of amyloid-β peptides on butyrylcholinesterase. Molecular complex of BuChE with amyloid-β 42 ( A ) and amyloid-β 40 ( B ) obtained after in silico docking. The 2D ligand interaction insets show the most important amino acid residues that seems to be involved in the binding of amyloid-β peptides with the mouth of the catalytic tunnel of BuChE.
The results from docking studies of ACHE and amyloid-β peptides indicated that amyloid-β 42 and amyloid-β 40 accommodate themselves in the vicinity of Y282, and Y280 amino acid residues. The docked complex along with 2D-ligand interaction diagram showing important amino acid interactions are given in Supplementary Fig. 4 .
The cartoon representation of docked complexes from each cluster representing ACHE-amyloid-β 40, ACHE-amyloid-β 42 , BuChE-amyloid-β 40 and BuChE-amyloid-β 42 complexes are shown in Supplementary Fig. 5 and gives a general idea of respective binding position of amyloid-β peptides to the ACHE and BuChE enzymes.
Discussion
In the current study, we report strong evidence regarding mutual physical interaction between APOE, BuChE, amyloid-β 40 and amyloid-β 42 . We found that this interaction alters the pathophysiological properties of the interacting partners. The findings indicate that APOE binds to and strongly prevents the fibrillization of amyloid-β peptides at its physiological concentrations. We also report compelling evidence that amyloid-β and APOE interact with BuChE, producing a reasonably stable heteromeric molecular complex, which we termed BAβACs (BuChE–amyloid-β–APOE complexes) ( Darreh-Shori et al. , 2011 a ). The enzymological analyses indicate that through the formation of BAβACs, the amyloid-β peptide concentrations dependently act as allosteric modulators of the acetylcholine-hydrolyzing capacity of BuChE. We have previously provided some evidence for this interaction and hyperactivation of BuChE is very specific as it also occurs in a complex biological matrix such as in CSF samples or in human embryonic-derived neurosphere cultures ( Darreh-Shori et al. , 2011 a , b , 2014 ; Malmsten et al. , 2014 ). Here, we extended these results by performing SDG analyses on nine different pooled Alzheimer’s disease CSF samples as surrogates of the in vivo parenchymal fluids in the brain. The interaction between amyloid-β and both ACHE and BuChE has also been shown by others through both in vitro analysis using purified proteins ( Inestrosa et al. , 1996 b ; Diamant et al. , 2006 ; Podoly et al. , 2008 ) as well as in numerous reports as components of the amyloid-β deposits in the Alzheimer’s disease brain ( Mesulam et al. , 1992 ; Mesulam and Geula, 1994 ; Guillozet et al. , 1997 ; Lehmann et al. , 2000 ).
Using molecular modelling we further deciphered the possible molecular fingerprint of the amino acid residues of the interaction sites of amyloid-β on the BuChE protein. The amino acid residues A277-Y282 comprise a site at the mouth of the catalytic tunnel of BuChE. Intriguingly, these interacting amino acid residues turned out to be those of the activation sites that had been proposed as the putative activating sites, and targeted for designing small activator molecules of BuChE ( Chiou et al. , 2015 ). Thus, in agreement with this previous report, our molecular modelling analysis seems to further suggest that the interaction of amyloid-β peptides with this putative activation site may cause widening of the mouth of the catalytic tunnel and/or provide a wider hydrophobic environment for entrance of the substrate. Such a facilitated influx of the substrates to the catalytic site ( Chiou et al. , 2015 ) may then explain the increased intrinsic catalytic rate of BuChE, which was determined by the extensive enzymatic analyses reported here and elsewhere ( Darreh-Shori et al. , 2011 a , b , 2014 ; Malmsten et al. , 2014 ).
One of the most important questions that we have aimed to decipher concerns the native biological function of amyloid-β peptides. Indeed, it is highly unlikely that such a sophisticated production of amyloid-β peptides, which consists of a highly precise multi-enzymatic and multi-sites cleavage machinery of APP, evolved with no biological function but to be highly cytotoxic. This notion is further appreciated by the reports indicating that amyloid-β release occurs by synapses in synchrony with action potential. Intriguingly, another recent study indicates that amyloid-β 40 may facilitate glutamate release by acting as a ligand for APP homodimer as its presynaptic receptors ( Fogel et al. , 2014 ).
Overall, our current findings accumulate additional evidence supporting our hypothesis ( Fig. 8 ) that at least one of the native biological functions of amyloid-β peptides is its involvement in the regulation of an extrasynaptic acetylcholine homeostasis in the brain ( Vijayaraghavan et al. , 2013 ). Another essential piece of the puzzle comes from our recent report of the presence of soluble acetylcholine-synthesizing enzyme, choline acetyltransferase (CHAT) in the extracellular fluids, including CSF and plasma ( Vijayaraghavan et al. , 2013 ; Karami et al. , 2015 ). The hypothetical pathway is that extracellular CHAT and formation of BAβACs is implicated in the fine-tuning of an extrasynaptic acetylcholine equilibrium involved in regulation of astroglial function and/or endothelia, mediated through activation of nicotinic α7-ACh receptors ( Parrish et al. , 2008 ). We have proposed a model in which continuous synthesis of acetylcholine by CHAT maintains a certain extrasynaptic acetylcholine concentration ( Vijayaraghavan et al. , 2013 ). When needed, neuronal circuitries can influence the extrasynaptic acetylcholine tone through action potential-mediated production and release of amyloid-β peptides into the interstitial fluid ( Cirrito et al. , 2005 ). In this way amyloid-β peptides interact with ACHE and BuChE, and temporarily form BAβACs with ultrafast acetylcholine-catalytic activity, which will then effectively shift the equilibrium state towards lower acetylcholine. This allows a heightened astroglial status to perform effectively their functional task in the brain. Reuptake of amyloid-β peptides will then result in disintegration of BAβACs, the return of acetylcholine levels to the initial steady-state balance, and the initial astroglial status. Indeed very recent reports show that cholinergic neurons are the main neuronal population in the brain that contains intracellular amyloid-β peptides ( Baker-Nigh et al. , 2015 ; Norvin et al. , 2015 ). However, in the Alzheimer’s disease brain, particularly in the presence of APOE4 gene, a high APOE protein ( Darreh-Shori et al. , 2011 b ) disturbs the formation, the disintegration or both by causing dysfunctional amyloid-β reuptake (clearance) by neurons or astroglial cells. Alternatively or additionally, high APOE protein may, by priming and prolonging the interaction between cholinesterases and amyloid-β, cause gradual accumulation and deposition of BAβACs in the brain in the amyloid-β plaques and cerebral amyloid angiopathies, explaining the extensively documented presence of BuChE, ACHE and APOE in these amyloid-β deposits ( Mesulam and Geula, 1994 ). Indeed, these post-mortem studies indicate that the cholinesterases found in the amyloid-β deposits exhibited altered enzymatic properties ( Geula and Mesulam, 1995 ), and that BuChE reactivity in the amyloid-β deposits in the brain is a characteristic of dementia ( Mesulam and Geula, 1994 ).
Figure 8.

Hypothetical regulatory pathway of extrasynaptic cholinergic signalling by amyloid-β peptides. ( 1 ) Neuronal activity demanding activation of cholinoceptives astroglia, initiate an action potential synchronized release of the intraneuronal amyloid-β peptides ( Cirrito et al. , 2005 ), e.g. from cholinergic neurons ( Baker-Nigh et al. , 2015 ) into the interstitial fluid. ( 2 ) Simultaneous release of APOE by astroglia may facilitate the interaction of the released amyloid-β peptides with the soluble cholinesterases (ChEs) ( 3 ), leading to the formation of BAβACs. Binding of amyloid-β peptides to the ChEs leads to increased intrinsic catalytic rate of these enzymes, plausibly through allosteric modulation of their tertiary structures. ( 4 ) The hyperfunctional BAβACs will then effectively lower the extrasynaptic acetylcholine (ACh) and thus shifting its equilibrium state which is maintained by the soluble ACh-synthesizing enzyme choline acetyl-transferase (ChAT) ( Vijayaraghavan et al. , 2013 ). The lowered ACh will result in a heightened functional status of astroglial cells, allowing them to perform the intended task. ( 5 ) Cellular reuptake of amyloid-β peptides will then reduce the levels of amyloid-β peptides, leading to the disintegration of BAβACs ( 6 ). The enzymatic activity of ChEs will then return to a relatively dormant state ( Darreh-Shori et al. , 2011 a ). ( 7 ) This will allow soluble ChAT to normalize the steady state balance of ACh levels, and thereby the initial astroglial activity status ( Malmsten et al. , 2014 ).
Another important question concerns the still highly obscure role of APOE protein in pathological events of Alzheimer’s disease despite the ε4 allele of the APOE being the most consistent genetic risk factor for sporadic Alzheimer’s disease, while the ε2 allele of this gene seems to be protective. In the current study, we showed that APOE protein affected—both isoform-dependently and concentration-dependently—the fibrillization kinetics of the recombinant amyloid-β 40 and amyloid-β 42 peptides in a highly unexpected way. We found that at high-to-moderate APOE concentration (1–0.5 µM) representative of the CSF levels in ε4-carrier patients with Alzheimer’s disease, APOE protein interacted with and changed the lag-time of the fibrillization of amyloid-β 40 and amyloid-β 42 peptides from 5–10 h to at least 30–60 h and reduced the maximum level of amyloid-β fibrillization to <30% of amyloid-β alone. These findings are unexpected because they suggest that APOE proteins exert a strong anti-amyloid-β fibrillization effect rather than the pro-fibrillogenic effect that has been suggested based on post-mortem studies ( Strittmatter et al. , 1993 a , b ) or associations with amyloid-β load in the brain as assessed by Pittsburgh compound B–PET ( Darreh-Shori et al. , 2011 b ; Ramanan et al. , 2014 ; Vijayaraghavan et al. , 2014 ).
Another important APOE-related observation was the differential effect of APOE isoproteins on the in vitro aggregation kinetic of amyloid-β 42 but not amyloid-β 40 peptides. Namely, all the APOE isoproteins showed a similar attenuating effect on the aggregation kinetics of the amyloid-β 40 peptides. In contrast, it was the APOE ε4 isoprotein that exhibited the strongest anti-fibrillization property on the amyloid-β 42 peptides. At 1.0 µM concentration level, the analyses indicated that for at least 65 h of incubation <1% of amyloid-β 42 peptides were fibrillized compared to the kinetic data from amyloid-β 42 alone. The same APOE ε4 protein concentration extended the lag-time of amyloid-β 40 fibrillization to just ∼35 h. APOE ε3 isoprotein (the most common variant) however increased the lag-time and reduced the F max for fibrillization of amyloid-β 42 and amyloid-β 40 peptides to a similar extent. These observations suggest that the product of the major genetic risk factor (APOE ε4 allele) of the sporadic Alzheimer’s disease might exert a stronger fibrillization-attenuating effect on the most fibrillogenic form of amyloid-β peptides.
Another noteworthy observation concerns an APOE concentration fluctuation effect on amyloid-β fibrillization properties, as we found that reduction of APOE ε4 protein from 1.0 µM to 0.5–0.1 µM caused dramatic changes on the fibrillization properties of amyloid-β 42 peptides. At 0.5 µM or less, the ε4 isoprotein reversed the rate and maximum level of the amyloid-β 42 fibrillization from <1% at 1.0 µM ε4 isoprotein to 200–300% of that observed when the amyloid-β 42 was used alone. Indeed by using a photo-induced cross-linking technique, it has been shown that structural stability of amyloid-β 42 oligomers may differ with that of amyloid-β 40 peptides ( Ono et al. , 2009 ).These observations are therefore important and may highlight a fundamentally distinct biochemical behaviour between amyloid-β 40 and amyloid-β 42 peptides and their interaction with APOE protein, and suggest that these peptides might biochemically behave differently depending on whether the interacting partner is the APOE ε4 or ε3 isoprotein. It also suggests that a possible physiological/pathological fluctuation in levels of the APOE protein may have a clinically relevant significance in rendering the ε4 carriers more vulnerable and/or susceptible to Alzheimer’s disease, which deserves further investigation.
Amyloid-β-derived diffusible ligands (ADDLs), or simply soluble amyloid-β oligomers, rather than fibrillar deposits of amyloid-β peptides are now recognized as the main route for amyloid-β toxicity in Alzheimer’s disease ( Catalano et al. , 2006 ). However, this new revision of the amyloid-β hypothesis once again seems to imply that ADDLs are solely made of homomeric oligomers of amyloid-β peptides. In addition, it is still unclear how these amyloid-β oligomers are formed and/or stabilized in the Alzheimer’s disease brain. Nonetheless, the strong anti-amyloid-β fibrillization of APOE protein is a highly relevant bit of the puzzle concerning its pathological contribution in Alzheimer’s disease. Our findings describe a prominent role of APOE protein in formation and accumulation of reactive soluble amyloid-β oligomers (homomeric and heteromeric forms). Thus, the current findings together with both clinical and paraclinical, and numerous other well-established observations by us or other groups ( Mesulam and Geula, 1994 ; Guillozet et al. , 1997 ; Lehmann et al. , 2000 ; Perry et al. , 2003 ; Diamant et al. , 2006 ; Podoly et al. , 2008 ; Darreh-Shori et al. , 2011 b , 2014 ; Vijayaraghavan et al. , 2013 , 2014 ; Malmsten et al. , 2014 ; Ramanan et al. , 2014 ; Tai et al. , 2014 ), evince a more plausible hypothesis in which APOE (in particular ε4) protein acts as a major driving force for the in vivo formation of heteromeric forms of ADDLs in the brain. BAβACs constitute a solid example of such highly functional and reactive forms of ADDLs. This hypothesis therefore merges the main pathophysiological findings in the main dementia disorders. It proposes a plausible native function for amyloid-β peptides, explains why the central cholinergic activity is one of the first neuronal networks that becomes heavily affected in Alzheimer’s disease ( Davies and Maloney, 1976 ) and dementia with Lewy bodies ( Bohnen et al. , 2003 ; Ziabreva et al. , 2006 ). It also provides support to other substantial mechanistic details described in our previous report ( Vijayaraghavan et al. , 2013 ).
Post-mortem brain studies suggest that several proteins, such as APOE, ACHE and BuChE interact with amyloid-β peptides and become trapped in the amyloid-β deposit in the Alzheimer’s disease brain. In vitro studies support these observations and suggest that APOE ( Strittmatter et al. , 1993 a , b ) , and cholinesterases (in particular ACHE) may have pro-fibrillogenic effect on amyloid-β peptides ( Diamant et al. , 2006 ). However, there are also evidences that high concentration or certain genetic variant of BuChE protein ( Diamant et al. , 2006 ; Podoly et al. , 2008 ) may have an attenuating effect on amyloid-β fibrillization. At BuChE concentration as high and non-physiological as was used in these previous reports, our findings confirmed the attenuating effect of BuChE, but it was marginally less so than the effect of physiological APOE protein.
In vitro analyses on ACHE splice variant proteins suggest that the read-through ACHE-R variant protein concentration-dependently decreases amount of insoluble amyloid-β oligomers and fibrils, and reduces amyloid-β toxicity in cell cultures, while the synaptic ACHE-S variant accelerate amyloid-β fibrillization ( Berson et al. , 2008 ). In CSF, two-thirds of ACHE protein is in the form of ACHE-R protein ( Darreh-Shori et al. , 2006 b , 2008 ). Using SDG analysis, the ACHE-R is separated as part of a light G 1 -G 2 peak of ACHE, in which the majority of ACHEs are in an inactive state ( Darreh-Shori et al. , 2004 ), and in complex with amyloid-β peptides ( Darreh-Shori et al. , 2014 ), indicating prolonged exposure of ACHE to amyloid-β peptides inactivated the enzyme. Here we showed that amyloid-β initially hyperactivated ACHE more strongly than BuChE, but also that the hyperactivation of ACHE, but not BuChE, by amyloid-β was quite transient since even within an hour of exposure to amyloid-β the ACHE activity began exhibiting dwindling signs. Indeed, prolonged incubation of CSF with recombinant amyloid-β has been shown to lead to ∼80% inactivation of the CSF ACHE proteins ( Darreh-Shori et al. , 2014 ). This differential and time-dependent allosteric modulation of amyloid-β on BuChE versus ACHE, and/or between ACHE splice variants may point as a self-regulatory mechanism for adjustment of extracellular acetylcholine signalling. For instance, a physiological consequence of amyloid-β-inactivated ACHE-R is a reduced degradation of extrasynaptic acetylcholine, and decrease astroglial hyperactivity due to suppressive action of acetylcholine on these cholinoceptive cells. In addition, amyloid-β-ACHE-R complexes are expected to remain soluble, and thereby to reduce amount of insoluble amyloid-β forms. Indeed, in vivo study on double transgenic APP swe /ACHE-R mice indicates reduced plaque burden and reactive astrocytes in the brain of these animals ( Berson et al. , 2008 ). Other regulatory mechanisms through specific microRNAs, which are termed as ‘CholinomiRs’ ( Soreq, 2015 ), may also be involved. For instance, miR-289 may be of particular relevance to the concept and biological roles of BAβACs. For instance, exposure of human embryonic stem (hES) cells to physiologically relevant amount of amyloid-β peptides for 20–30 days did not merely hyperactivate BuChE, but in addition altered both protein expression and release of ChAT and BuChE in an apparently synchronized manner favouring low acetylcholine signalling, and differentiation of the human embryonic stem cells toward gliogenesis ( Malmsten et al. , 2014 ).
In vitro studies suggest that BuChE protein exerts a concentration-dependent attenuating role on the amyloid-β fibril formation under APOE protein-free condition ( Diamant et al. , 2006 ; Podoly et al. , 2008 ). Indeed, we were able to reproduce the same findings in the current study when we used the same high 0.2 µM concentration of BuChE. However, even at this relatively high and non-physiological concentration, the anti-amyloid-β fibrillogenic effect of BuChE was negligible compared to the effect of APOE protein at physiological ranges representing in vivo in CSF. Nonetheless, BuChE in CSF ( Darreh-Shori et al. , 2006 a ), exhibited high pro-fibrillogenic properties on amyloid-β peptide assembly, in particular with amyloid-β 42 peptides. In contrast, in the presence of APOE protein, BuChE protein independent to its concentrations augmented the anti-fibrillar effect of APOE on amyloid-β peptides. These observations are consistent with those based on SDG data that indicated that APOE facilitated the formation of BAβACs and are in line with the close but inverse interrelationship between CSF levels of APOE and BuChE ( Darreh-Shori et al. , 2006 a ) and a complex APOE-dependent alteration in the stability and phenotypic display of activity of BuChE ( Darreh-Shori et al. , 2011 b , 2012 ). However, the CSF SDG spectrum in the current study also indicated that the heavy and ultra-heavy BAβACs contained much less (if any) APOE protein than the light BAβACs. The large sedimentation coefficient of ultra-heavy BAβACs, corresponding to a molecular weight of 600–750 kDa, which suggests that at least these ultra-heavy complexes in the CSF samples were about to become deposited in the brain. In the long-term, eliminating the accumulation of BAβACs is hence expected to reduce the amount of amyloid-β deposits in the brain. Indeed, BCHE-knockout mice exhibit a reduced amyloid-β deposition in the brain ( Reid and Darvesh, 2015 ).
Detection and quantification of amyloid-β oligomers has been very problematic and to date the evidence of their existence in vivo is mainly hypothetical and in most cases substantiated based on methods that inherently are not suitable for their detection. For instance, western blotting using conventional Laemmli loading buffer that contains high SDS, a strong anionic detergent capable of eliminating hydrophobic interactions responsible for assembly of homomeric amyloid-β oligomers or their heteromeric assembly with other proteins. There are also reasonable risks for SDS producing artefacts because it has been used to create globulomers following a particular protocol ( Gellermann et al. , 2008 ). In the current study, we showed that SDG analyses easily separated the spontaneously formed homomeric dimers and tetramers as well as heteromeric heavier forms of amyloid-β peptides’ assemblies into distinct fractions. This could be done without any substantial manipulation of the samples, preventing artefacts. Then the quantification can be done accurately by any appropriate quantitative assay.
Funding
This study was supported by grants from Loo and Hans Osterman Foundation; KI Foundations; Olle Engkvist Byggmästare Foundation; Åke Wibergs Foundation; Åhlén-Foundation; Gunvor and Josef Anérs Foundation; Magnus Bergvalls Foundation; Demens Foundation (Demensfonden); Gun and Bertil Stohnes Foundation; Ragnhild and Einar Lundströms Foundation; Foundation for Sigurd and Elsa Goljes Memory; Tore Nilsson Foundation; the Foundation for Old Servants; the Swedish Research Council (project no 05817); Karolinska Institutet Strategic Neuroscience programme; the Stockholm County Council-Karolinska Institutet regional agreement on medical training and clinical research (ALF grant); Swedish Brain Power; the Swedish Brain Foundation; the Alzheimer Foundation in Sweden; and EU FP7 large scale integrating project INMiND ( http://www.uni-muenster.de/InMind ).
Supplementary material
Supplementary material is available at Brain online.
Supplementary Material
Glossary
Abbreviations
- BAβAC
BuChE/ACHE–amyloid-β–APOE complex
- BuChE
butyrylcholinesterase
- SDG
sucrose density gradient
References
- Baker-Nigh A, Vahedi S, Davis EG, Weintraub S, Bigio EH, Klein WL, et al. . Neuronal amyloid-beta accumulation within cholinergic basal forebrain in ageing and Alzheimer's disease . Brain 2015. ; 138 ( Pt 6 ): 1722 – 37 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berson A, Knobloch M, Hanan M, Diamant S, Sharoni M, Schuppli D, et al. . Changes in readthrough acetylcholinesterase expression modulate amyloid-beta pathology . Brain 2008. ; 131 ( Pt 1 ): 109 – 19 . [DOI] [PubMed] [Google Scholar]
- Bohnen NI, Kaufer DI, Ivanco LS, Lopresti B, Koeppe RA, Davis JG, et al. . Cortical cholinergic function is more severely affected in parkinsonian dementia than in Alzheimer disease: an in vivo positron emission tomographic study . Arch Neurol 2003. ; 60 : 1745 – 8 . [DOI] [PubMed] [Google Scholar]
- Carson KA, Geula C, Mesulam MM . Electron microscopic localization of cholinesterase activity in Alzheimer brain tissue . Brain Res 1991. ; 540 : 204 – 8 . [DOI] [PubMed] [Google Scholar]
- Catalano SM, Dodson EC, Henze DA, Joyce JG, Krafft GA, Kinney GG . The role of amyloid-beta derived diffusible ligands (ADDLs) in Alzheimer's disease . Curr Topics Med Chem 2006. ; 6 : 597 – 608 . [DOI] [PubMed] [Google Scholar]
- Cheung J, Rudolph MJ, Burshteyn F, Cassidy MS, Gary EN, Love J, et al. . Structures of human acetylcholinesterase in complex with pharmacologically important ligands . J Med Chem 2012. ; 55 : 10282 – 6 . [DOI] [PubMed] [Google Scholar]
- Chiou SY, Weng TT, Lin GZ, Lu RJ, Jian SY, Lin G . Molecular docking of different inhibitors and activators to butyrylcholinesterase . J Biomol Struct Dyn 2015. ; 33 : 563 – 72 . [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, et al. . Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo . Neuron 2005. ; 48 : 913 – 22 . [DOI] [PubMed] [Google Scholar]
- Coles M, Bicknell W, Watson AA, Fairlie DP, Craik DJ . Solution structure of amyloid beta-peptide(1-40) in a water-micelle environment. Is the membrane-spanning domain where we think it is? Biochemistry 1998. ; 37 : 11064 – 77 . [DOI] [PubMed] [Google Scholar]
- Comeau SR, Gatchell DW, Vajda S, Camacho CJ . ClusPro: a fully automated algorithm for protein-protein docking . Nucleic Acids Res 2004a. ; 32 : W96 – 9 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeau SR, Gatchell DW, Vajda S, Camacho CJ . ClusPro: an automated docking and discrimination method for the prediction of protein complexes . Bioinformatics 2004b. ; 20 : 45 – 50 . [DOI] [PubMed] [Google Scholar]
- Crescenzi O, Tomaselli S, Guerrini R, Salvadori S, D'Ursi AM, Temussi PA, et al. . Solution structure of the Alzheimer amyloid beta-peptide (1-42) in an apolar microenvironment. Similarity with a virus fusion domain . Eur J Biochem 2002. ; 269 : 5642 – 8 . [DOI] [PubMed] [Google Scholar]
- Darreh-Shori T, Hellstrom-Lindahl E, Flores-Flores C, Guan ZZ, Soreq H, Nordberg A . Long-lasting acetylcholinesterase splice variations in anticholinesterase-treated Alzheimer's disease patients . J Neurochem 2004. ; 88 : 1102 – 13 . [DOI] [PubMed] [Google Scholar]
- Darreh-Shori T, Brimijoin S, Kadir A, Almkvist O, Nordberg A . Differential CSF butyrylcholinesterase levels in Alzheimer's disease patients with the ApoE epsilon4 allele, in relation to cognitive function and cerebral glucose metabolism . Neurobiol Dis 2006a. ; 24 : 326 – 33 . [DOI] [PubMed] [Google Scholar]
- Darreh-Shori T, Meurling L, Pettersson T, Hugosson K, Hellstrom-Lindahl E, Andreasen N, et al. . Changes in the activity and protein levels of CSF acetylcholinesterases in relation to cognitive function of patients with mild Alzheimer's disease following chronic donepezil treatment . J Neural Transm 2006b. ; 113 : 1791 – 801 . [DOI] [PubMed] [Google Scholar]
- Darreh-Shori T, Kadir A, Almkvist O, Grut M, Wall A, Blomquist G, et al. . Inhibition of acetylcholinesterase in CSF versus brain assessed by 11C-PMP PET in AD patients treated with galantamine . Neurobiol Aging 2008. ; 29 : 168 – 84 . [DOI] [PubMed] [Google Scholar]
- Darreh-Shori T, Forsberg A, Modiri N, Andreasen N, Blennow K, Kamil C, et al. . Differential levels of apolipoprotein E and butyrylcholinesterase show strong association with pathological signs of Alzheimer's disease in the brain in vivo . Neurobiol Aging 2011a. ; 32 : 2320 e15 – 32 . [DOI] [PubMed] [Google Scholar]
- Darreh-Shori T, Modiri N, Blennow K, Baza S, Kamil C, Ahmed H, et al. . The apolipoprotein E epsilon4 allele plays pathological roles in AD through high protein expression and interaction with butyrylcholinesterase . Neurobiol Aging 2011b. ; 32 : 1236 – 48 . [DOI] [PubMed] [Google Scholar]
- Darreh-Shori T, Siawesh M, Mousavi M, Andreasen N, Nordberg A . Apolipoprotein epsilon4 modulates phenotype of butyrylcholinesterase in CSF of patients with Alzheimer's disease . J Alzheimers Dis 2012. ; 28 : 443 – 58 . [DOI] [PubMed] [Google Scholar]
- Darreh-Shori T, Hosseini SM, Nordberg A . Pharmacodynamics of cholinesterase inhibitors suggests add-on therapy with a low-dose carbamylating inhibitor in patients on long-term treatment with rapidly reversible inhibitors . J Alzheimers Dis 2014. ; 39 : 423 – 40 . [DOI] [PubMed] [Google Scholar]
- Davies P, Maloney AJ . Selective loss of central cholinergic neurons in Alzheimer's disease . Lancet 1976. ; 2 : 1403 . [DOI] [PubMed] [Google Scholar]
- Diamant S, Podoly E, Friedler A, Ligumsky H, Livnah O, Soreq H . Butyrylcholinesterase attenuates amyloid fibril formation in vitro . Proc Natl Acad Sci USA 2006. ; 103 : 8628 – 33 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel H, Frere S, Segev O, Bharill S, Shapira I, Gazit N, et al. . APP homodimers transduce an amyloid-beta-mediated increase in release probability at excitatory synapses . Cell Rep 2014. ; 7 : 1560 – 76 . [DOI] [PubMed] [Google Scholar]
- Gellermann GP, Byrnes H, Striebinger A, Ullrich K, Mueller R, Hillen H, et al. . Abeta-globulomers are formed independently of the fibril pathway . Neurobiol Dis 2008. ; 30 : 212 – 20 . [DOI] [PubMed] [Google Scholar]
- Geula C, Greenberg BD, Mesulam MM . Cholinesterase activity in the plaques, tangles and angiopathy of Alzheimer's disease does not emanate from amyloid . Brain Res 1994. ; 644 : 327 – 30 . [DOI] [PubMed] [Google Scholar]
- Geula C, Mesulam MM . Cholinesterases and the pathology of Alzheimer disease . Alzheimer Dis Assoc Disord 1995. ; 9 ( Suppl 2 ): 23 – 8 . [DOI] [PubMed] [Google Scholar]
- Gretch DG, Sturley SL, Friesen PD, Beckage NE, Attie AD . Baculovirus-mediated expression of human apolipoprotein E in Manduca sexta larvae generates particles that bind to the low density lipoprotein receptor . Proc Natl Acad Sci USA 1991. ; 88 : 8530 – 3 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillozet AL, Smiley JF, Mash DC, Mesulam MM . Butyrylcholinesterase in the life cycle of amyloid plaques . Ann Neurol 1997. ; 42 : 909 – 18 . [DOI] [PubMed] [Google Scholar]
- Hone E, Martins IJ, Jeoung M, Ji TH, Gandy SE, Martins RN . Alzheimer's disease amyloid-beta peptide modulates apolipoprotein E isoform specific receptor binding . J Alzheimers Dis 2005. ; 7 : 303 – 14 . [DOI] [PubMed] [Google Scholar]
- Inestrosa NC, Alvarez A, Calderon F . Acetylcholinesterase is a senile plaque component that promotes assembly of amyloid beta-peptide into Alzheimer's filaments . Mol Psychiatry 1996a. ; 1 : 359 – 61 . [PubMed] [Google Scholar]
- Inestrosa NC, Alvarez A, Perez CA, Moreno RD, Vicente M, Linker C, et al. . Acetylcholinesterase accelerates assembly of amyloid-beta-peptides into Alzheimer's fibrils: Possible role of the peripheral site of the enzyme . Neuron 1996b. ; 16 : 881 – 91 . [DOI] [PubMed] [Google Scholar]
- Kar S, Issa AM, Seto D, Auld DS, Collier B, Quirion R . Amyloid beta-peptide inhibits high-affinity choline uptake and acetylcholine release in rat hippocampal slices . J Neurochem 1998. ; 70 : 2179 – 87 . [DOI] [PubMed] [Google Scholar]
- Karami A, Eyjólfsdóttir H, Vijayaraghavan S, Lind G, Almqvist P, Kadir A, et al. . Changes in CSF cholinergic biomarkers in response to cell therapy with NGF in patients with Alzheimer's disease . Alzheimers Dement 2015. . Advance Access published on February 9, 2015, doi: 10.1016/j.jalz.2014.11.008. [DOI] [PubMed] [Google Scholar]
- Kozakov D, Beglov D, Bohnuud T, Mottarella SE, Xia B, Hall DR, et al. . How good is automated protein docking? Proteins 2013. ; 81 : 2159 – 66 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozakov D, Brenke R, Comeau SR, Vajda S . PIPER: an FFT-based protein docking program with pairwise potentials . Proteins 2006. ; 65 : 392 – 406 . [DOI] [PubMed] [Google Scholar]
- Lane R, Feldman HH, Meyer J, He Y, Ferris SH, Nordberg A, et al. . Synergistic effect of apolipoprotein E ϵ4 and butyrylcholinesterase K-variant on progression from mild cognitive impairment to Alzheimer's disease . Pharmacogenet Genomics 2008. ; 18 : 289 – 98 . [DOI] [PubMed] [Google Scholar]
- Lehmann DJ, Johnston C, Smith AD . Synergy between the genes for butyrylcholinesterase K variant and apolipoprotein E4 in late-onset confirmed Alzheimer's disease . Hum Mol Genet 1997. ; 6 : 1933 – 6 . [DOI] [PubMed] [Google Scholar]
- Lehmann DJ, Nagy Z, Litchfield S, Borja MC, Smith AD . Association of butyrylcholinesterase K variant with cholinesterase-positive neuritic plaques in the temporal cortex in late-onset Alzheimer's disease . Hum Genet 2000. ; 106 : 447 – 52 . [DOI] [PubMed] [Google Scholar]
- Malmsten L, Vijayaraghavan S, Hovatta O, Marutle A, Darreh-Shori T . Fibrillar beta-amyloid 1-42 alters cytokine secretion, cholinergic signalling and neuronal differentiation . J Cell Mol Med 2014. ; 18 : 1874 – 88 . doi: 101111/jcmm12343 2014 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam M, Carson K, Price B, Geula C . Cholinesterases in the amyloid angiopathy of Alzheimer's disease . Ann Neurol 1992. ; 31 : 565 – 9 . [DOI] [PubMed] [Google Scholar]
- Mesulam MM, Geula C . Butyrylcholinesterase reactivity differentiates the amyloid plaques of aging from those of dementia . Ann Neurol 1994. ; 36 : 722 – 7 . [DOI] [PubMed] [Google Scholar]
- Nachon F, Carletti E, Ronco C, Trovaslet M, Nicolet Y, Jean L, et al. . Crystal structures of human cholinesterases in complex with huprine W and tacrine: elements of specificity for anti-Alzheimer's drugs targeting acetyl- and butyryl-cholinesterase . Biochem J 2013. ; 453 : 393 – 9 . [DOI] [PubMed] [Google Scholar]
- Naiki H, Gejyo F, Nakakuki K . Concentration-dependent inhibitory effects of apolipoprotein E on Alzheimer's beta-amyloid fibril formation in vitro . Biochemistry 1997. ; 36 : 6243 – 50 . [DOI] [PubMed] [Google Scholar]
- Norvin D, Kim G, Baker-Nigh A, Geula C . Accumulation and age-related elevation of amyloid-beta within basal forebrain cholinergic neurons in the rhesus monkey . Neuroscience 2015. ; 298 : 102 – 11 . [DOI] [PubMed] [Google Scholar]
- Ono K, Condron MM, Teplow DB . Structure-neurotoxicity relationships of amyloid beta-protein oligomers . Proc Natl Acad Sci USA 2009. ; 106 : 14745 – 50 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish WR, Rosas-Ballina M, Gallowitsch-Puerta M, Ochani M, Ochani K, Yang LH, et al. . Modulation of TNF release by choline requires alpha7 subunit nicotinic acetylcholine receptor-mediated signaling . Mol Med 2008. ; 14 : 567 – 74 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry E, McKeith I, Ballard C . Butyrylcholinesterase and progression of cognitive deficits in dementia with Lewy bodies . Neurology 2003. ; 60 : 1852 – 3 . [DOI] [PubMed] [Google Scholar]
- Podoly E, Bruck T, Diamant S, Melamed-Book N, Weiss A, Huang Y, et al. . Human recombinant butyrylcholinesterase purified from the milk of transgenic goats interacts with beta-amyloid fibrils and suppresses their formation in vitro . Neuro-degenerative diseases 2008. ; 5 : 232 – 6 . [DOI] [PubMed] [Google Scholar]
- Ramanan VK, Risacher SL, Nho K, Kim S, Swaminathan S, Shen L, et al. . APOE and BCHE as modulators of cerebral amyloid deposition: a florbetapir PET genome-wide association study . Mol Psychiatry 2014. ; 19 : 351 – 7 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees T, Hammond PI, Soreq H, Younkin S, Brimijoin S . Acetylcholinesterase promotes beta-amyloid plaques in cerebral cortex . Neurobiol Aging 2003. ; 24 : 777 – 87 . [DOI] [PubMed] [Google Scholar]
- Rees TM, Berson A, Sklan EH, Younkin L, Younkin S, Brimijoin S, et al. . Memory deficits correlating with acetylcholinesterase splice shift and amyloid burden in doubly transgenic mice . Curr Alzheimers Res 2005. ; 2 : 291 – 300 . [DOI] [PubMed] [Google Scholar]
- Reid GA, Darvesh S . Butyrylcholinesterase-knockout reduces brain deposition of fibrillar beta-amyloid in an Alzheimer mouse model . Neuroscience 2015. ; 298 : 424 – 35 . [DOI] [PubMed] [Google Scholar]
- Saez-Valero J, de Ceballos ML, Small DH, de Felipe C . Changes in molecular isoform distribution of acetylcholinesterase in rat cortex and cerebrospinal fluid after intracerebroventricular administration of amyloid beta-peptide . Neurosci Lett 2002. ; 325 : 199 – 202 . [DOI] [PubMed] [Google Scholar]
- Soreq H . Checks and balances on cholinergic signaling in brain and body function . Trends Neurosci 2015. ; 38 : 448 – 58 . [DOI] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. . Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease . Proc Natl Acad Sci USA 1993a. ; 90 : 1977 – 81 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, et al. . Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease . Proc Natl Acad Sci USA 1993b. ; 90 : 8098 – 102 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney D, Martins R, LeVine H, 3rd, Smith JD, Gandy S . Similar promotion of Abeta1-42 fibrillogenesis by native apolipoprotein E epsilon3 and epsilon4 isoforms . J Neuroinflammation 2004. ; 1 : 15 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai LM, Mehra S, Shete V, Estus S, Rebeck GW, Bu G, et al. . Soluble apoE/Abeta complex: mechanism and therapeutic target for APOE4-induced AD risk . Mol Neurodegener 2014. ; 9 : 2 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DR, Capetillo-Zarate E, Schultz C, Rub U, Saido TC, Yamaguchi H, et al. . Apolipoprotein E co-localizes with newly formed amyloid beta-protein (Abeta) deposits lacking immunoreactivity against N-terminal epitopes of Abeta in a genotype-dependent manner . Acta Neuropathol 2005. ; 110 : 459 – 71 . [DOI] [PubMed] [Google Scholar]
- Vijayaraghavan S, Karami A, Aeinehband S, Behbahani H, Grandien A, Nilsson B, et al. . Regulated extracellular choline acetyltransferase activity- the plausible missing link of the distant action of acetylcholine in the cholinergic anti-inflammatory pathway . PloS One 2013. ; 8 : e65936 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayaraghavan S, Maetzler W, Reimold M, Lithner CU, Liepelt-Scarfone I, Berg D, et al. . High apolipoprotein E in cerebrospinal fluid of patients with Lewy body disorders is associated with dementia . Alzheimers Dement 2014. ; 10 : 530 – 40 . e1 . [DOI] [PubMed] [Google Scholar]
- Wang R, Lu Y, Wang S . Comparative evaluation of 11 scoring functions for molecular docking . J Med Chem 2003. ; 46 : 2287 – 303 . [DOI] [PubMed] [Google Scholar]
- Wright CI, Geula C, Mesulam MM . Neurological cholinesterases in the normal brain and in Alzheimer's disease: relationship to plaques, tangles, and patterns of selective vulnerability . Ann Neurol 1993. ; 34 : 373 – 84 . [DOI] [PubMed] [Google Scholar]
- Ziabreva I, Ballard CG, Aarsland D, Larsen JP, McKeith IG, Perry RH, et al. . Lewy body disease: thalamic cholinergic activity related to dementia and parkinsonism . Neurobiol Aging 2006. ; 27 : 433 – 8 . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.