

Abstract
Frequent evolutionary birth and death events have created a large quantity of biologically important, lineage‐specific DNA within mammalian genomes. The birth and death of DNA sequences is so frequent that the total number of these insertions and deletions in the human population remains unknown, although there are differences between these groups, e.g. transposable elements contribute predominantly to sequence insertion. Functional turnover – where the activity of a locus is specific to one lineage, but the underlying DNA remains conserved – can also drive birth and death. However, this does not appear to be a major driver of divergent transcriptional regulation. Both sequence and functional turnover have contributed to the birth and death of thousands of functional promoters in the human and mouse genomes. These findings reveal the pervasive nature of evolutionary birth and death and suggest that lineage‐specific regions may play an important but previously underappreciated role in human biology and disease.
Keywords: enhancers, evolution, indels, lineage‐specific biology, promoters, transcriptional regulation
Abbreviations
- CAGE
cap analysis of gene expression
- ChIP
chromatin immunoprecipitation
- DHS
DNaseI hypersensitive site
- H3K27ac
acetylated histone H3 lysine 27
- H3K4me2
dimethylated histone H3 lysine 4
- H3K4me3
trimethylated histone H3 lysine 4
- lncRNA
long non‐coding RNA
- LTR
long terminal repeat
- miRNA
microRNA
- TF
transcription factor
Introduction
The large and varied diversity observed between individuals and across species is reflected in high levels of genetic diversity. The study of this diversity between mammalian species has been possible on a genome‐wide scale since the publication of the first complete drafts of the human and mouse genomes in 2001 1 and 2002 2, respectively. The subsequent emergence of next‐generation sequencing technologies 3 has led to an explosion of whole‐genome sequencing, such that public databases now host many mammalian genomes, and 39 of these can currently be directed viewed and compared through the Ensembl genome browser (www.ensembl.org) 4. The first personal genome sequence was only published in 2007 5, but has now been joined by a number of studies, including the 1,000 Genomes Project which sequenced over 1,000 individual genomes 6 and those from deCODE Genetics which sequenced over 2,500 individual genomes from the Icelandic population 7.
This wealth of data has stimulated the field of comparative genomics, which investigates both the similarities and differences between genomes. Much early work focussed on identifying shared features between sequenced genomes and restricted itself to the small proportion of the genome which encodes for protein‐coding genes. Many of these genes have been deeply conserved throughout evolution from yeast to human, the lineages of which diverged approximately one billion years ago 8. The number of protein‐coding genes found in vertebrate species is relatively constant and, unexpectedly, does not appear to correlate with our assumptions regarding organismal complexity 9.
Not all protein‐coding genes, however, are evolutionarily ancient. For example, C20orf203 is found only in the human genome, and is absent from closely related primates. This gene is highly expressed in the brain, and is further upregulated in Alzheimer's disease, which suggests a potential role for this lineage‐specific gene in the development of the disease 10. There are now several reports of genes that have been born and died in various species, through a variety of mechanisms (fully reviewed by Kaessmann 11).
Comparative genomics has also been applied to studying the remaining, non‐coding, regions of the genome, which make up almost 99% of the genome 12 and contain a wealth of transcriptional and regulatory elements. MicroRNAs (miRNAs) – short, approximately 22 nt long, non‐coding RNA genes primarily involved in negative regulation of protein‐coding genes 13 – are often deeply conserved across a range of divergent species 14. Long non‐coding RNAs (lncRNAs) are a relatively unstudied class of non‐coding transcripts that are over 200 bp long 15. lncRNAs show modest evolutionary constraint, which has been interpreted as indicating that these sequences have been conserved across species because they encode a biological function 16, 17, 18. Similarly, non‐coding regulatory elements such as enhancers, which positively regulate gene expression at a distance 19, have been computationally predicted in regions which show increased evolutionary conservation across species. This approach has been demonstrated to have a 45% success rate in predicting enhancers using comparative genomics alone 20.
As for protein‐coding genes, there are also corresponding examples of non‐coding RNA genes that have been emerged during evolution, such as the mouse‐specific lncRNA Poldi. This lncRNA is restricted to the post‐meiotic cells of the testis, and promotes sperm motility and testis development 21. Similarly, the miRNA miR‐941 – which was born in the human lineage between one and six million years ago from the expansion of an evolutionarily unstable tandem repeat sequence – was recently discovered to be important for neurotransmitter signalling in the brain 22.
There are a handful of known examples of enhancer birth and death in the human genome. For example, the non‐coding element HACNS1 has evolved rapidly in humans and only the human sequence, but not the orthologous primate sequence, is able to function as a limb enhancer in mouse reporter experiments 23. Alternatively, an enhancer for the AR gene, which is conserved across most mammalian species has been deleted from the human genome. The activity of this enhancer is correlated with the formation of whiskers and penile spines in non‐human mammals, and it has been speculated that this loss in humans may be linked to increased monogamous reproductive strategies relative to other primates 24.
On a genome‐wide scale, non‐coding elements are less conserved between mammalian species than protein‐coding genes 25, 26. This has led many to speculate that organismal diversity is not in fact driven by changes to the protein‐coding gene set, but by divergence in the regulatory mechanisms responsible for controlling their expression 27. Due to their increased volatility, much recent work has therefore focussed on the birth and death of such non‐coding, regulatory elements within the human genome.
In this essay, I will first discuss sequence turnover, where sequence is either inserted (born) or deleted (dies) along a lineage. I will then describe analyses of functional elements that have been identified through experimental profiling (Box 1) and were subsequently shown to completely turn over between lineages despite conservation of the underlying DNA sequence (functional turnover). Where such profiling has been done in multiple species, it is possible to further define functional turnovers as gains and losses down individual lineages and throughout this essay I will also refer to these as birth and death events, respectively. Finally, I will examine those studies that have considered both the birth and death of sequence and function in the same experimental system. I will show that the birth and death of entire regulatory elements are frequent occurrences within the human genome, and will suggest that future research is likely to focus on both the transcriptional regulatory and phenotypic consequences of these events to normal and perhaps also pathogenic human diversity.
Box 1. Functional genomics technologies discussed in this review.
Chromatin immunoprecipitation followed by sequencing (ChIP‐seq): Identifies the locations of histone modifications or the sites of a DNA‐binding protein, such as a transcription factor, by high‐throughput sequencing of DNA pulled down with an antibody specific to the modification or protein of interest.
DNase 1 hypersensitivity sites sequencing (DNase‐seq): Discovers all classes of active regulatory elements by digesting accessible chromatin which is not packaged into nucleosomes, followed by high‐throughput DNA sequencing.
Cap analysis of gene expression (CAGE): Clones the 5′ ends of transcribed mRNA molecules and then subjects them to high‐throughput sequencing to precisely identify the sites of transcription initiation at promoters and other transcribed elements, such as enhancers.
Sequence turnover is common in the human genome
The insertion or deletion of sequence along one of the lineages that separates two species (collectively known as ‘indels’) results in gaps in the sequence alignments which describe the relationship between orthologous sequences within the two genomes being compared. Insertions can be discriminated from deletions by comparing the sequences of three or more genomes simultaneously. The principle of parsimony makes the assumption that the most likely evolutionary history for a set of related sequences is the scenario that can be explained by the minimal number of mutations. In this way, a sequence is defined as having been deleted if it is present in the outgroup species, while an inserted sequence will be absent from this species (Fig. 1). This type of analysis also allows one to identify the lineage along which the insertion or deletion has taken place. Most alignment tools and analysis programs, however, treat these gaps as missing data.
Figure 1.
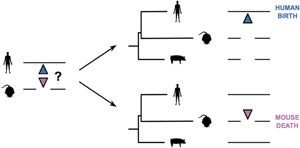
By comparison to a third outgroup species, the lineage along which a mutation took place can be identified and an alignment gap (indel) can be classified as an insertion or deletion. In this example, a gap in the orthologous sequence in both the mouse and pig genomes reveals that novel sequence has been inserted in the human genome – as shown by the blue triangle – and that there has been a birth of sequence on the human lineage. Conversely, if there is no gap in the orthologous pig sequence, then a sequence death (pink triangle) is inferred to have taken place on the mouse lineage.
Within the human population, there are likely to be many millions of polymorphic indels which are found in some, but not all, individuals. A study of 79 diverse human genomes reported almost two million small indels 28. However, the limited overlap between this and other studies suggests that this is an under‐estimation of the total number of indels segregating within the human population, and that there are many indels yet to be discovered 28. Longer regions of sequence that has either been inserted or deleted within an individual genome – known as structural variants and generally defined as longer than 1 kb – are less common, and only approximately 20,000 have so far been detected by the 1,000 Genomes Project 6. Within individuals, these variants of different lengths have been found to be associated with differences in gene expression 29, 30.
Polymorphic indels disrupt the coding sequence of over 6% of annotated human genes, but 72% of genes contain an intronic indel 31. The reduced frequency of evolutionarily conserved, functional material within intronic sequences 32 implies that these coding sequence‐disrupting indels are likely to confer a substantial genetic load. Many deletions are shared across populations, and have been present since humans migrated out of Africa 33. The lower average frequency of deletions relative to insertions in the population suggests that sequence loss is more damaging than the birth of new sequence 34.
The distribution of indels throughout the genome has been used to quantify the amount of functional, but lineage‐specific, sequence within the genome. This model assumes that indels occur randomly within the genome and that unexpectedly large distances between indels therefore contain sequence which is preserved by natural selection 35, presumably because this sequence conveys a biological function as yet unknown. By comparing the quantity of sequence defined to be functional using this metric from a range of different pairs of species alignments, the authors determined that this quantity rapidly decreases as the evolutionary distance between the species being compared increased 36. This implies that most functional material is conserved only within a narrow range of related species, and that there must be a rapid turnover of functional sequence and a large quantity of lineage‐specific sequence within mammalian genomes. This rate of sequence turnover is not constant between genomes, and appears to be higher along the mouse lineage, where sequence is preferentially deleted at a particularly high rate 37, 38. The vast majority of this evolutionary volatile sequence is found outside protein‐coding gene borders, and it has been predicted that 110–143 Mb (50%) of functional non‐coding DNA sequence within the human genome has turned over in the last 130 million years 39.
What mechanisms drive sequence birth and death in the genome?
There are a number of molecular mutations that insert or delete sequence in the genome. Transposable elements, which are capable of jumping around the genome, make up approximately half of the human genome 40 and are divided into two major classes. Retrotransposons duplicate via an RNA intermediate (Fig. 2A) before the new copy is re‐integrated into the genome at a distant site. Retrotransposition does not typically include the copying or movement of intronic and surrounding regulatory DNA. The other class of DNA transposons use a cut‐and‐paste mechanism (Fig. 2B) in which the entire DNA sequence is excised and then re‐integrated into the genome. Repetitive elements, and particularly retrotransposons, are enriched at both species‐specific enhancers and gene promoters 41. Promoters, which are the site of RNA polymerase II complex assembly and transcription initiation 42, are enriched for repetitive elements only at sequences that have been inserted, rather than deleted, in both the human and mouse genomes 43. One class of transposons, known as long terminal repeats (LTRs), is particularly common at tissue‐restricted promoters, which is consistent with the previously reported role for LTRs in driving such a limited expression profile 44, 45. Despite the association between simple repetitive elements and sequence deletion 46, no such relationship was found between repetitive sequences and promoters which have been deleted along either the human or mouse lineages 43. Instead, simple repeats were found to be enriched at newly inserted promoters that are broadly expressed, but it remains unknown which types of this family of repeats are responsible or the manner in which they drive widespread expression across tissues.
Figure 2.
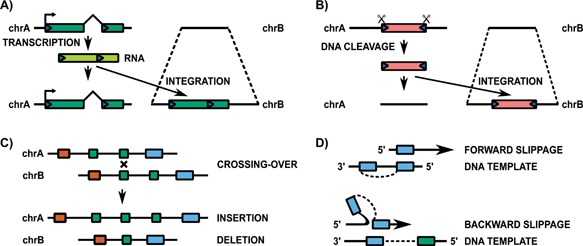
DNA sequences can be inserted and deleted by various mechanisms. Retrotransposons replicate via an RNA intermediate (A) while other transposable elements use a cut‐and‐paste mechanism to integrate the DNA sequence at a distant site (B). Unequal crossing‐over during cell division (C) and replication slippage (D) can also result in the birth and death of DNA sequences.
Sequence can also be inserted or deleted from the genome through the activity of normal cellular processes, such as recombination and replication. Unequal crossing‐over takes place when non‐homologous regions are paired during cell division. This can result in one chromosome gaining sequence and the other losing the same sequence (Fig. 2C), but this exchange of sequence need not be reciprocal as shown here. Indeed, a comparative analysis of the human, chimp and macaque genomes has suggested that recombination is more associated with the gain of sequence than sequence loss in the human genome 47. DNA replication can also create indels through replication slippage (Fig. 2D), if the DNA polymerase skips over a region (known as ‘forward slippage’) to remove sequence or skips back to replicate a region twice (known as ‘backward slippage’), resulting in the insertion of a second copy of the sequence. These replication errors are most frequent at regions containing nearby tandem duplications and, although it has been suggested that they are responsible for most births of recently arisen short insertions in the human lineage 48, replicative errors are actually more likely to be associated with the loss of sequence across the entire genome 47.
Despite reports of sequence gain and loss within the human genome, their accurate discovery remains a difficult task, requiring the development of specialised computational pipelines 49. The two commonly used genome‐wide alignments have been built with different methods – the Ensembl EPO pipeline 50 builds alignments and reconstructs candidate ancestral genome sequences across multiple species simultaneously while the UCSC BLASTZ alignments 51 are generated from small, local sequence alignments of two species which are then extended into larger blocks of related DNA sequences. Multi‐species alignments are then build separately using these pairwise alignments. The difference in these approaches results in substantial differences in the amount of aligning sequence, e.g. UCSC aligns 1.0 Gb (33%) of the human genome to mouse while Ensembl aligns only 820 Mb (26%), and similar discrepancies in the amount of sequence which is estimated to have been gained or lost within the human genome. Furthermore, progressive alignment algorithms incur a greater penalty when creating insertions rather than deletions 52, which hinders robust discrimination of these separate classes of mutations. Further improvements in our ability to identify regions which are inserted and deleted within whole‐genome alignments and the human population are likely to take account of the different mechanisms, and sometimes complicated, evolutionary histories which generate these events. Our increased knowledge of driving forces between these events should improve our ability to predict and accurately detect when an insertion or deletion has truly taken place, rather than as now solely defining them as positions within the genome where alignment pipelines fail to identify orthologous sequences.
Tissue‐restricted regulatory elements show frequent functional turnover
Genome sequencing projects have been followed by a second wave of functional genomics studies, as exemplified by the work of the ENCODE consortium 53. Functional genomics combines experimental techniques with advances in DNA sequencing to investigate the functional role of genes and other regulatory DNA sequences throughout the genome (see Box 1).
Large‐scale functional turnover of both transcription factor (TF) binding and promoter locations have been reported. A comparison of four liver‐specific TFs (FOXA2, HNF1A, HNF4A and HNF6) in human and mouse revealed that 41–89% of their binding locations within aligning sequence were found in only one of these two species 54, implying a substantial rate of functional turnover. This suggests that there are many births and deaths of these binding sites along the two lineages, but this could not be confirmed from the data published in this study (Fig. 1). This high rate of TF binding turnover in the liver takes place across much of the animal clade (Fig. 3A) 26 and can even be detected between individual rodent lineages, suggesting that these turnover events are evolutionarily very rapid 55.
Figure 3.
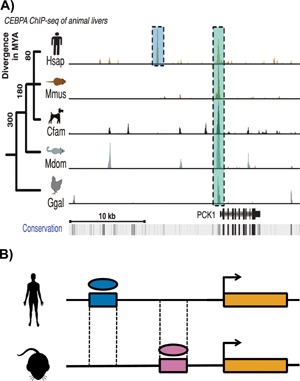
A: TF binding at the PCK1 locus in the livers of five vertebrates (human, mouse, dog, monodelphis, chicken). A conserved binding site is highlighted by the green box, and a human‐inserted site by the blue box. Other binding sites show more complicated evolutionary histories. Reproduced with permission from Schmidt et al. 26. B: Compensatory turnover of TF binding sites. The locus retains TF binding and a similar transcriptional response in both species, but each TF binding site has turned over.
Complete functional turnover of genetic elements, such as enhancers and promoters, is also frequent between mammalian species. While turnover is less prevalent in cis‐regulatory modules that contain multiple TFs bound to the same locus 55, only 279 (less than 1%) of enhancers active in the liver alone are conserved across 10 placental genomes 41. Promoters defined epigenetically by the presence of trimethylated histone H3 lysine 4 (H3K4me3) in the same system seem to be less susceptible than enhancers to this type of functional turnover 41, but this may not reflect the true turnover rate as promoters defined by their transcriptional output using CAGE turn over more frequently 56 (see also below ‘Both sequence and functional turnover contribute to the birth and death of functional promoters’ for a more detailed discussion where promoter turnovers were polarised into births and deaths). There may be further differences between promoter and TF‐binding site functional turnover because these events at promoters are often accompanied by changes to the underlying sequence 43 while binding site locations within rodents can turn over without changes to the underlying DNA sequence 55. It remains unclear what mechanisms, such as the lack of cooperative binding partners or a compaction of the local chromatin state, are responsible for driving these TF turnover events.
The function of DNA‐binding factors or the length of their DNA‐binding motif may also be related to their evolutionary volatility. For example, the binding locations of the insulator protein CTCF – which also has an unusually long binding motif – are much more conserved between mammalian species than most transcription factors, and therefore less likely to be gained or lost between mammalian species 57. Fifteen per cent (5,178/33,966) of alignable binding sites in human are also present in each of macaque, mouse, rat and dog 58. Unlike TFs which often possess tissue‐specific roles in regulating gene expression, CTCF binding sites are largely consistent across tissues 59. CTCF is important for regulating the three‐dimensional structure of the genome, e.g. by insulating transcriptionally active from inactive regions 60 and it also demarcates the borders of DNA sequences which are anchored to the nuclear periphery 61. Like individual CTCF binding sites, this structural role for CTCF appears to be conserved as is the higher‐order genome structure which it regulates 62.
Both the mouse ENCODE 63 and FANTOM5 64 collaborative projects have carried out comparative functional genomics analyses across a range of tissues, and confirmed this rapid functional turnover throughout the human and mouse genomes. Tissue‐restricted elements are more susceptible to turning over, perhaps due to the increased functional constraints on pleiotropic elements that are active across tissues, and the immune system and testis appear to be the tissues throughout the body with the greatest rates of turnover 43. While it is likely that the many of the changes observed within immune cells are driven by positive natural selection to avoid host pathogenicity 65, it is currently unclear to what extent sexual selection and the locally elevated mutation rate at active sites in the germ cells 43, 66, 67, 68 contribute to the functional element turnover within the testis.
The transcriptional and phenotypic consequences of functional turnover remain unclear
There is evidence that the turnover of an individual binding site can be compensated by the birth of a binding site at a second site within a gene locus (Fig. 3B). For example, approximately 25% of species‐specific TF binding site functional losses in the liver were mirrored by the gain of a separate, species‐specific, binding site within 10 kb 26. Similarly, while 53% of genes targeted in an OCT4 knockdown in embryonic stem cells in both human and mouse contained nearby OCT4‐NANOG binding, only 15% of these binding sites were found at the same position in both species 57. These observations suggest that, while the rapid evolutionary turnover of TF binding sites may be driven by a high mutation rate at these sites 68, this may be matched by a strong selective pressure to prevent a subsequent divergence in the transcriptional output regulated by these factors 69.
Furthermore, the trans environment within the cell is more conserved than the individual elements themselves, as TF‐to‐TF interactions 70 and TF network topologies 71 are similar between human and mouse. These results are consistent with the independent observation that human chromosome 21, when inserted into mouse hepatocytes, behaves in largely the same manner as the human chromosome when in human cells. This again suggests that species‐specific binding is due to changes at binding sites themselves but that the same trans‐acting TFs can still drive DNA binding in both species 72.
There is little evidence that the majority of these lineage‐specific elements, although defined by their functional activity, are directly involved in transcriptional regulation. It is known that not all TF binding sites have a direct effect on gene expression 73, 74. Binding sites that are identified in the liver and are conserved in multiple mammalian species show greater functional enrichments (e.g. disease ontology annotations), and are found near genes with a higher expression than species‐specific binding sites 75, suggesting that those sites that have been gained or lost down individual lineages are less likely to be functionally important in positively driving gene expression. Furthermore, gene expression and nearby TF binding divergence in the liver do not appear to be generally correlated within closely related mouse species 69. However, these conclusions are contradicted by the observation that mouse‐ and human‐specific binding of the glucocorticoid receptor in macrophages were associated with species‐specific upregulation of neighbouring genes upon glucocorticoid stimulation 76. Further studies of this type are required to determine whether the liver or the macrophage is the more representative system.
Population genetics studies also support the argument that few regulatory elements that have been born along the human lineage possess a biological function. Although enriched for disease‐ and trait‐associated variants, the nucleotide diversity for human‐specific DHSs is relatively high and comparable to that of fourfold‐degenerate sites in exonic sequence, which further suggests that DHSs possess a relatively limited proportion of functional sequence 77. These elements do, however, experience at least some purifying selection, as indicated by their reduced diversity relative to sequences defined biochemically to be inactive 78.
The ultimate test of functionality of lineage‐specific regions is to disrupt them to determine their biological role. Whether, as predicted computationally 36, these elements are frequently responsible for a phenotype has yet to be tested in a systematic manner.
Functional births and deaths of regulatory elements may be associated with expression changes at nearby genes
With data from only two species, these studies are largely limited to describing turnover events, and matched data from a third species is required to discriminate functional births from deaths along individual lineages (Fig. 1). Shibata et al. 79 measured DNaseI hypersensitive sites (DHSs) in human, chimp and macaque fibroblasts and identified hundreds of gains and losses along both the human and chimp lineages. DHSs that were born along each lineage were associated with up‐regulation of nearby target genes, while DHSs which died were associated with the concomitant down‐regulation of nearby genes. However, most differential gene expression could not be explained by the simple gain or loss of DHS sites. Both gained and lost DHSs were more likely to be experiencing positive selection specifically along the lineage in which they had been gained and lost, respectively. Active enhancers and promoters were similarly identified in embryonic limbs from human, macaque and mouse by profiling the location of acetylated histone H3 lysine 27 (H3K27ac) 80. Promoters identified using this data have gained activity along the human lineage more rapidly than enhancers (13 vs. 11%), but the vast majority of both classes of elements are gained through the co‐option (exaptation) of existing sequence rather than insertion of novel sequence 80. A similar study in the same species mapped two epigenetic marks (dimethylated histone H3 lysine 4, H3K4me2) and H3K27ac during human, macaque and mouse corticogenesis to confirm a high rate of human‐specific promoter and enhancer birth 81. These human‐specific elements were frequently found at, or near to, genes important for cortical development, suggesting that they may play important roles in regulating human‐specific aspects of this important biological process. A collection of histone modifications and protein‐binding sites have also been profiled in matched human, mouse and pig pluripotent stem cells, where divergence in the intensity of these binding factors at gene promoters is correlated with gene expression divergence 82. However, these authors did not explicitly examine functional birth and death of these elements between the lineages studied.
These three‐species experiments also differ from those mentioned above in their methodology for detecting lineage‐specific elements. Those studies described above which focused on liver‐specific transcription factors identified binding regions in each species independently and then defined lineage‐specific regions as those in orthologous regions for which no binding peak had been discovered in other species. The description of an individual region as being lineage‐specific is dependent on the genome‐wide alignments used to identify orthology as these show clear discrepancies in the amount of sequence which can be aligned between species (see ‘What mechanisms drive sequence birth and death in the genome?’ above). The degree of overlap required to identify orthologous regions also affects the detection of functional turnover events. Some studies consider a single 1 bp overlap between regions as sufficient to define them as being conserved while others have required at least a 50% overlap in reciprocal comparisons between species 41, which will reduce the number of lineage‐specific regions that can be identified from the same data. In contrast, these studies describing functional genomics data from other tissues 79, 80, 81, combine these alignments with statistical methods, such as edgeR 83, to detect lineage‐specific regions as those orthologous regions which also show differential levels of histone modifications or chromatin accessibility between species. This approach does not depend on calling peaks in all species and will therefore account for regions with evidence for binding that just misses the threshold for calling a peak as significant within one of the related species. The use of a statistical framework also makes it possible to quantitatively measure the confidence in a single region being truly lineage‐specific and how these regions differ from those identified in the same system which show binding in all species, albeit at significantly different levels. Despite being dependent on replicated functional genomics datasets to make these statistical assessments, these more complex approaches, using more than simple genomic overlaps, will likely be considered the more robust approach to detect functional turnover in future.
Both sequence and functional turnover contribute to the birth and death of functional promoters
While both mechanisms of birth and death in the genome – sequence and functional turnover – are clearly important contributors to lineage‐specific genomics, it is only recently that they have been explicitly investigated simultaneously in the same experimental system.
The FANTOM5 project, which identified promoter locations across a range of matched human and mouse cell lines and tissues, described the half‐life of promoters when aligned to increasingly divergent species 64. Evolutionary history varied with both expression profile and promoter class, where broadly expressed protein‐coding promoters and tissue‐restricted ncRNA promoters were more deeply conserved. These patterns have been similarly observed within aligning exonic sequence in both protein‐coding genes in human 84 and lncRNAs in Drosophila 18.
The sequences of a large number of promoters have been born or died along the human lineage (conservatively 2,472 and 2,818, respectively), since its divergence with mouse 43. As seen for regulatory elements within the ENCODE datasets, the gain and loss of promoters is enriched within immune cells and the testes and brain‐biased promoters were less likely to show either type of sequence turnover. Genes that experienced at least one of these turnover events were enriched for evidence of positive selection acting on their coding sequence, suggesting promoter turnover may be related to adaptive evolution throughout the encoded protein, and not just at the turnover site 79. However, within the human population, both inserted and deleted promoters showed no evidence of either positive or purifying selection, suggesting that, as for the species‐specific TF binding sites described above, many of these may not be phenotypically relevant.
Many promoters whose sequence has been conserved between human and mouse have experienced functional turnover (22 and 13% of aligned promoters in human and mouse, respectively), as they show no detectable evidence of transcription in the opposing species. These species‐specific promoters are specifically associated with decreased evolutionary constraint at the promoter elements 43. Similar levels of evolutionary constraint were seen at promoters with matched, divergent or reduced expression between species, suggesting that differences in transcriptional output were not driven by sequence changes at the promoter or at cis‐regulatory elements found at a constant distance from the promoter. This contrasts with the inverse correlation seen between expression and substitution rate divergence at promoters activated in lipopolysaccharide‐stimulated macrophages 85. Whether these differences are specific to the macrophage timecourse profiled here, or are a general feature of stress‐response genes remains unclear.
While sequence gain and loss are clearly important factors in promoter evolution along the mouse and human lineages, the lack of well‐matched data across more species remains the limiting factor for resolving the large number of functional turnovers into births and deaths at aligned sequence. As shown in Fig. 4, beyond the liver, the number of mammalian genomes that has been sequenced outnumbers the number of tissues that have been comprehensively profiled experimentally in multiple species. Even when datasets from multiple tissues are available from consortia such as ENCODE and FANTOM, matched samples are usually only available for human and mouse, hence precluding this discrimination of functional gain from loss at aligning sequences.
Figure 4.
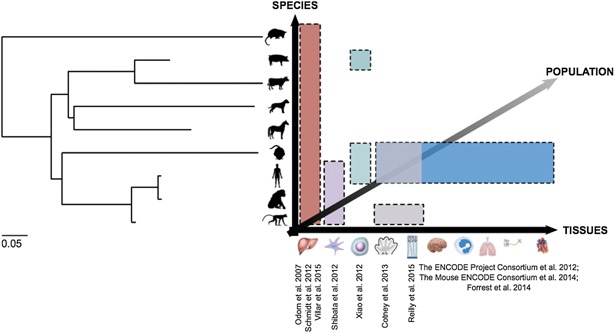
Current state of published, functional genomics data from various mammalian species which are related as shown in the phylogenetic tree. Branch lengths indicate the genome‐wide estimate of the neutral substitution rate at fourfold degenerate sites. ChIP‐seq datasets describing the location of several TFs 53, CTCF 57, H3K27ac and H3K4me3 40 in the liver are available for up to 20 mammalian species, nine of which are shown here. Further studies of three species simultaneously have examined DHSs in fibroblasts 72; various histone modifications and TFs in pluripotent stem cells 75; H3K27ac in the developing limb 73; and H3K4me2 and H3K27ac during corticogenesis 74. The ENCODE, mouse ENCODE and FANTOM consortiums have published large collections of datasets from human and mouse tissues and cell lines. Comparative functional genomics studies within populations of the same species are likely to be a focus of future research.
Conclusions and prospects
The birth and death of both sequence and function is a common occurrence within the human genome, and represents an important contributor to genetic diversity. These turnover events have been observed at both distal regulatory elements and functional promoters and confirm that, while a useful predictor, evolutionary conservation is not required to identify functioning, lineage‐specific elements in the human genome.
The availability of large amounts of functional genomics data in both human and mouse have already allowed evolutionary turnover to be investigated across a number of tissues. Extending the available datasets to more distantly related species will permit functional turnover events to be resolved into births and deaths along individual lineages (Fig. 4). Investigating the dynamics of births and deaths within the human population should reveal any phenotypic consequences, and whether they are associated with disease, such as autoimmune disorders, which could aid in the development of personalised strategies to treat these.
Investigations into the combined effects of sequence and functional turnover in the birth and death of genetic elements have only recently been attempted. Further work will likely focus on the direct relationship between these, for example one might ask if functional deaths result in biologically unimportant sequence that is then a target for sequence deletion and the complete removal of the element from the genome.
Despite this work, the biological relevance of these evolutionarily volatile elements still remains unclear. Do they drive the diversification of gene expression profiles, or do they simply represent the neutral churn of redundant genetic elements in the genome? By carefully matching current datasets to increasing amounts of functional genomics data from multiple species 86, we now have an exciting opportunity to reveal the role of evolutionary birth and death in shaping the mammalian genome and its regulatory apparatus.
The author has declared no conflicts of interest.
Acknowledgements
This work was supported by the UK Medical Research Council. I thank Martin Taylor, Vera Kaiser and Colin Semple for constructive discussions and comments.
Figures 1 and 4 were updated on June 23, 2016 to correct a formatting error. The scientific content was not affected.
References
- 1.International Human Genome Sequencing C, Adekoya E, Ait‐Zahra M, Allen N, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921. [DOI] [PubMed] [Google Scholar]
- 2.Mouse Genome Sequencing C, Waterston RH, Lindblad‐Toh K, Birney E, et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–62. [DOI] [PubMed] [Google Scholar]
- 3. Mardis ER. 2013. Next‐generation sequencing platforms. Ann Rev Anal Chem 6: 287–303. [DOI] [PubMed] [Google Scholar]
- 4. Flicek P, Amode MR, Barrell D, Beal K, et al. 2014. Ensembl 2014. Nucleic Acids Res 42: D749–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Levy S, Sutton G, Ng PC, Feuk L, et al. 2007. The diploid genome sequence of an individual human. PLoS Biol 5: e254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McVean GA, Abecasis GR, Auton A, Brooks LD, et al. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491: 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gudbjartsson DF, Helgason H, Gudjonsson SA, Zink F, et al. 2015. Large‐scale whole‐genome sequencing of the Icelandic population. Nat Genet 47: 435–44. [DOI] [PubMed] [Google Scholar]
- 8. Douzery EJ, Snell EA, Bapteste E, Delsuc F, et al. 2004. The timing of eukaryotic evolution: does a relaxed molecular clock reconcile proteins and fossils? Proc Natl Acad Sci U S A 101: 15386–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hahn MW, Wray GA. 2002. The g‐value paradox. Evol Dev 4: 73–5. [DOI] [PubMed] [Google Scholar]
- 10. Li CY, Zhang Y, Wang Z, Zhang Y, et al. 2010. A human‐specific de novo protein‐coding gene associated with human brain functions. PLoS Comput Biol 6: e1000734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kaessmann H. 2010. Origins, evolution, and phenotypic impact of new genes. Genome Res 20: 1313–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Church DM, Goodstadt L, Hillier LW, Zody MC, et al. 2009. Lineage‐specific biology revealed by a finished genome assembly of the mouse. PLoS Biol 7: e1000112‐e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wheeler BM, Heimberg AM, Moy VN, Sperling EA, et al. 2009. The deep evolution of metazoan microRNAs. Evol Dev 11: 50–68. [DOI] [PubMed] [Google Scholar]
- 15. Ponting CP, Oliver PL, Reik W. 2009. Evolution and functions of long noncoding RNAs. Cell 136: 629–41. [DOI] [PubMed] [Google Scholar]
- 16. Ponjavic J, Ponting CP, Lunter G. 2007. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res 17: 556–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marques AC, Ponting CP. 2009. Catalogues of mammalian long noncoding RNAs: modest conservation and incompleteness. Genome Biol 10: R124‐R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Young RS, Marques AC, Tibbit C, Haerty W, et al. 2012. Identification and properties of 1,119 candidate lincRNA loci in the Drosophila melanogaster genome. Genome Biol Evol 4: 427–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bulger M, Groudine M. 2010. Enhancers: the abundance and function of regulatory sequences beyond promoters. Dev Biol 339: 250–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, et al. 2006. In vivo enhancer analysis of human conserved non‐coding sequences. Nature 444: 499–502. [DOI] [PubMed] [Google Scholar]
- 21. Heinen TJ, Staubach F, Haming D, Tautz D. 2009. Emergence of a new gene from an intergenic region. Curr Biol 19: 1527–31. [DOI] [PubMed] [Google Scholar]
- 22. Hu HY, He L, Fominykh K, Yan Z, et al. 2012. Evolution of the human‐specific microRNA miR‐941. Nat Commun 3: 1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Prabhakar S, Visel A, Akiyama JA, Shoukry M, et al. 2008. Human‐specific gain of function in a developmental enhancer. Science 321: 1346–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McLean CY, Reno PL, Pollen AA, Bassan AI, et al. 2011. Human‐specific loss of regulatory DNA and the evolution of human‐specific traits. Nature 471: 216–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kutter C, Watt S, Stefflova K, Wilson MD, et al. 2012. Rapid turnover of long noncoding RNAs and the evolution of gene expression. PLoS Genet 8: e1002841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schmidt D, Wilson MD, Ballester B, Schwalie PC, et al. 2010. Five‐vertebrate ChIP‐seq reveals the evolutionary dynamics of transcription factor binding. Science 328: 1036–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wittkopp PJ, Kalay G. 2012. Cis‐regulatory elements: molecular mechanisms and evolutionary processes underlying divergence. Nat Rev Genet 13: 59–69. [DOI] [PubMed] [Google Scholar]
- 28. Mills RE, Pittard WS, Mullaney JM, Farooq U, et al. 2011. Natural genetic variation caused by small insertions and deletions in the human genome. Genome Res 21: 830–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stranger BE, Forrest MS, Dunning M, Ingle CE, et al. 2007. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 315: 848–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gymrek M, Willems T, Guilmatre A, Zeng H, et al. 2016. Abundant contribution of short tandem repeats to gene expression variation in humans. Nat Genet 48: 22–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen FC, Chen CJ, Li WH, Chuang TJ. 2007. Human‐specific insertions and deletions inferred from mammalian genome sequences. Genome Res 17: 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, et al. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15: 1034–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McCarroll SA, Hadnott TN, Perry GH, Sabeti PC, et al. 2006. Common deletion polymorphisms in the human genome. Nat Genet 38: 86–92. [DOI] [PubMed] [Google Scholar]
- 34. Sjodin P, Bataillon T, Schierup MH. 2010. Insertion and deletion processes in recent human history. PLoS ONE 5: e8650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lunter G, Ponting CP, Hein J. 2006. Genome‐wide identification of human functional DNA using a neutral indel model. PLoS Comput Biol 2: e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meader S, Ponting CP, Lunter G. 2010. Massive turnover of functional sequence in human and other mammalian genomes. Genome Res 20: 1335–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Laurie S, Toll‐Riera M, Rado‐Trilla N, Alba MM. 2012. Sequence shortening in the rodent ancestor. Genome Res 22: 478–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Taylor MS, Ponting CP, Copley RR. 2004. Occurrence and consequences of coding sequence insertions and deletions in Mammalian genomes. Genome Res 14: 555–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rands CM, Meader S, Ponting CP, Lunter G. 2014. 8.2% of the human genome is constrained: variation in rates of turnover across functional element classes in the human lineage. PLoS Genet 10: e1004525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kuhn RM, Karolchik D, Zweig AS, Trumbower H, et al. 2007. The UCSC genome browser database: update 2007. Nucleic Acids Res 35: D668–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Villar D, Berthelot C, Aldridge S, Rayner TF, et al. 2015. Enhancer evolution across 20 mammalian species. Cell 160: 554–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sandelin A, Carninci P, Lenhard B, Ponjavic J, et al. 2007. Mammalian RNA polymerase II core promoters: insights from genome‐wide studies. Nat Rev Genet 8: 424–36. [DOI] [PubMed] [Google Scholar]
- 43. Young RS, Hayashizaki Y, Andersson R, Sandelin A, et al. 2015. The frequent evolutionary birth and death of functional promoters in mouse and human. Genome Res 25: 1546–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Faulkner GJ, Kimura Y, Daub CO, Wani S, et al. 2009. The regulated retrotransposon transcriptome of mammalian cells. Nat Genet 41: 563–71. [DOI] [PubMed] [Google Scholar]
- 45. Fort A, Hashimoto K, Yamada D, Salimullah M, et al. 2014. Deep transcriptome profiling of mammalian stem cells supports a regulatory role for retrotransposons in pluripotency maintenance. Nat Genet 46: 558–66. [DOI] [PubMed] [Google Scholar]
- 46. Usdin K. 2008. The biological effects of simple tandem repeats: lessons from the repeat expansion diseases. Genome Res 18: 1011–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kvikstad EM, Tyekucheva S, Chiaromonte F, Makova KD. 2007. A macaque's‐eye view of human insertions and deletions: differences in mechanisms. PLoS Comput Biol 3: 1772–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Messer PW, Arndt PF. 2007. The majority of recent short DNA insertions in the human genome are tandem duplications. Mol Biol Evol 24: 1190–7. [DOI] [PubMed] [Google Scholar]
- 49. Mills RE, Luttig CT, Larkins CE, Beauchamp A, et al. 2006. An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res 16: 1182–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Paten B, Herrero J, Beal K, Fitzgerald S, et al. 2008. Enredo and Pecan: genome‐wide mammalian consistency‐based multiple alignment with paralogs. Genome Res 18: 1814–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schwartz S, Kent WJ, Smit A, Zhang Z, et al. 2003. Human‐mouse alignments with BLASTZ. Genome Res 13: 103–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Loytynoja A, Goldman N. 2008. Phylogeny‐aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science 320: 1632–5. [DOI] [PubMed] [Google Scholar]
- 53. Consortium EP. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Odom DT, Dowell RD, Jacobsen ES, Gordon W, et al. 2007. Tissue‐specific transcriptional regulation has diverged significantly between human and mouse. Nat Genet 39: 730–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Stefflova K, Thybert D, Wilson MD, Streeter I, et al. 2013. Cooperativity and rapid evolution of cobound transcription factors in closely related mammals. Cell 154: 530–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Frith MC, Ponjavic J, Fredman D, Kai C, et al. 2006. Evolutionary turnover of mammalian transcription start sites. Genome Res 16: 713–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kunarso G, Chia NY, Jeyakani J, Hwang C, et al. 2010. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat Genet 42: 631–4. [DOI] [PubMed] [Google Scholar]
- 58. Schmidt D, Schwalie PC, Wilson MD, Ballester B, et al. 2012. Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Cell 148: 335–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shen Y, Yue F, McCleary DF, Ye Z, et al. 2012. A map of the cis‐regulatory sequences in the mouse genome. Nature 488: 116–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Herold M, Bartkuhn M, Renkawitz R. 2012. CTCF: insights into insulator function during development. Development 139: 1045–57. [DOI] [PubMed] [Google Scholar]
- 61. Guelen L, Pagie L, Brasset E, Meuleman W, et al. 2008. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453: 948–51. [DOI] [PubMed] [Google Scholar]
- 62. Chambers EV, Bickmore WA, Semple CA. 2013. Divergence of mammalian higher order chromatin structure is associated with developmental loci. PLoS Comput Biol 9: e1003017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yue F, Cheng Y, Breschi A, Vierstra J, et al. 2014. A comparative encyclopedia of DNA elements in the mouse genome. Nature 515: 355–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Forrest AR, Kawaji H, Rehli M, Baillie JK, et al. 2014. A promoter‐level mammalian expression atlas. Nature 507: 462–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vallender EJ, Lahn BT. 2004. Positive selection on the human genome. Hum Mol Genet 13: R245–54. [DOI] [PubMed] [Google Scholar]
- 66. Haygood R, Fedrigo O, Hanson B, Yokoyama KD, et al. 2007. Promoter regions of many neural‐ and nutrition‐related genes have experienced positive selection during human evolution. Nat Genet 39: 1140–4. [DOI] [PubMed] [Google Scholar]
- 67. Taylor MS, Massingham T, Hayashizaki Y, Carninci P, et al. 2008. Rapidly evolving human promoter regions. Nat Genet 40: 1262–3; author reply 3–4. [DOI] [PubMed] [Google Scholar]
- 68. Reijns MA, Kemp H, Ding J, de Proce SM, et al. 2015. Lagging‐strand replication shapes the mutational landscape of the genome. Nature 518: 502–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wong ES, Thybert D, Schmitt BM, Stefflova K, et al. 2015. Decoupling of evolutionary changes in transcription factor binding and gene expression in mammals. Genome Res 25: 167–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Neph S, Vierstra J, Stergachis AB, Reynolds AP, et al. 2012. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature 489: 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Stergachis AB, Neph S, Sandstrom R, Haugen E, et al. 2014. Conservation of trans‐acting circuitry during mammalian regulatory evolution. Nature 515: 365–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wilson MD, Barbosa‐Morais NL, Schmidt D, Conboy CM, et al. 2008. Species‐specific transcription in mice carrying human chromosome 21. Science 322: 434–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Biggin MD. 2011. Animal transcription networks as highly connected, quantitative continua. Dev Cell 21: 611–26. [DOI] [PubMed] [Google Scholar]
- 74. Bradley RK, Li XY, Trapnell C, Davidson S, et al. 2010. Binding site turnover produces pervasive quantitative changes in transcription factor binding between closely related Drosophila species. PLoS Biol 8: e1000343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ballester B, Medina‐Rivera A, Schmidt D, Gonzalez‐Porta M, et al. 2014. Multi‐species, multi‐transcription factor binding highlights conserved control of tissue‐specific biological pathways. eLife 3: e02626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jubb AW, Young RS, Hume DA, Bickmore WA. 2016. Enhancer turnover is associated with a divergent transcriptional response to glucocorticoid in mouse and human macrophages. J Immunol 196: 813–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Vierstra J, Rynes E, Sandstrom R, Zhang M, et al. 2014. Mouse regulatory DNA landscapes reveal global principles of cis‐regulatory evolution. Science 346: 1007–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ward LD, Kellis M. 2012. Evidence of abundant purifying selection in humans for recently acquired regulatory functions. Science 337: 1675–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Shibata Y, Sheffield NC, Fedrigo O, Babbitt CC, et al. 2012. Extensive evolutionary changes in regulatory element activity during human origins are associated with altered gene expression and positive selection. PLoS Genet 8: e1002789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cotney J, Leng J, Yin J, Reilly SK, et al. 2013. The evolution of lineage‐specific regulatory activities in the human embryonic limb. Cell 154: 185–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Reilly SK, Yin J, Ayoub AE, Emera D, et al. 2015. Evolutionary genomics. Evolutionary changes in promoter and enhancer activity during human corticogenesis. Science 347: 1155–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Xiao S, Xie D, Cao X, Yu P, et al. 2012. Comparative epigenomic annotation of regulatory DNA. Cell 149: 1381–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Winter EE, Goodstadt L, Ponting CP. 2004. Elevated rates of protein secretion, evolution, and disease among tissue‐specific genes. Genome Res 14: 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Schroder K, Irvine KM, Taylor MS, Bokil NJ, et al. 2012. Conservation and divergence in toll‐like receptor 4‐regulated gene expression in primary human versus mouse macrophages. Proc Natl Acad Sci U S A 109: E944–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Andersson L, Archibald AL, Bottema CD, Brauning R, et al. 2015. Coordinated international action to accelerate genome‐to‐phenome with FAANG, the Functional Annotation of Animal Genomes project. Genome Biol 16: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]