

Abstract
Human glucosylcerebrosidase 2 (GBA2) of the CAZy family GH116 is responsible for the breakdown of glycosphingolipids on the cytoplasmic face of the endoplasmic reticulum and Golgi apparatus. Genetic defects in GBA2 result in spastic paraplegia and cerebellar ataxia, while cross-talk between GBA2 and GBA1 glucosylceramidases may affect Gaucher disease. Here, we report the first three-dimensional structure for any GH116 enzyme, Thermoanaerobacterium xylanolyticum TxGH116 β-glucosidase, alone and in complex with diverse ligands. These structures allow identification of the glucoside binding and active site residues, which are shown to be conserved with GBA2. Mutagenic analysis of TxGH116 and structural modeling of GBA2 provide a detailed structural and functional rationale for pathogenic missense mutations of GBA2.
Human glucosylcerebrosidase 2 (GBA2, nonlysosomal glucosylcerebrosidase, E.C. 3.2.1.45, bile acid β-glucosidase E.C. 3.2.1.21) is a member of a family of sequence-related glycoside hydrolases (GH) designated GH116 (see www.cazy.org), with members in animals, plants, fungi, archaea, and eubacteria.1−3 Originally identified as a bile acid β-glucosidase,4 GBA2 was later found to hydrolyze the glycolipids glucosylceramide (GlcCer) and glucosyllysosphingosine, and its defect was found to disrupt glycolipids in sperm and cause infertility in knockout mice.5
GBA2 is a peripheral membrane protein localized to the cytoplasmic face of the endoplasmic reticulum and/or Golgi apparatus membrane.6 GBA2 and the lysosomal family GH30 glucosylcerebrosidase GBA1 (GBA, acid β-glucosidase) are thought to be the primary enzymes responsible for the breakdown of GlcCer.7 GlcCer is the most abundant glycosylceramide, a common precursor for the majority of more complex glycosphingolipids, and an important messenger in its own right.8,9 While there is extensive structural and functional knowledge of GBA1, genetic deficiency of which results in the lysosomal storage disease Gaucher disease,10,11much less is known of GBA2.
Mutations in the human GBA2 gene cause hereditary spastic paraplegia (HSP) and autosomal recessive cerebellar ataxia (ARCA).12−15 Zebrafish with the corresponding gene knockout also show neurological defects.12 Significantly, knockout of GBA2 in mouse models for Gaucher disease and Niemann-Pick disease, in which there is a secondary deficiency in GBA1, partially alleviated symptoms.16,17 Furthermore, overexpression of GBA2 was found to be toxic to melanoma cells.18 Taken together, this suggests that specific inhibitors and activators of GBA2 could be useful for the treatment of Gaucher disease, hereditary ataxias, and possibly melanoma. Although GBA2 inhibitors have been identified,7,19,20 the lack of any structural insight for GBA2, or indeed any other GH116 protein, has limited both the rational development of more potent inhibitors and the molecular understanding of pathogenic mutations.
Glycoside hydrolase family GH116 includes enzymes with diverse specificities, including a β-xylosidase/β-glucosidase3 and an N-acetylglucosaminidase,21 the biochemical analysis of which has been useful in defining the basic catalytic machinery, including the general acid/base and catalytic nucleophile. However, these biochemically characterized members are divergent from mammalian GBA2 and lack conservation of residues that have been identified to be mutated in GBA2-related disorders. Here, we describe the three-dimensional structure of a bacterial homologue of the human GBA2, the first 3-D structure for this important family of glycosidases, in native and ligand complexes. Through functional analysis of this highly tractable model, in which all pathogenic GBA2 mutation sites are strictly conserved, we provide detailed insight into the molecular bases of human GBA2 mutations leading to ataxias and paraplegias.
Results and Discussion
Enzyme Activity and Small Molecule Inhibition
Given the absence of any 3-D model or structure for the human enzyme, we sought to work on a close bacterial homologue of GBA2. Thermoanaerobacterium xylanolyticum GH116 (TxGH116) shares approximately 40% sequence identity over the catalytic domain, and we predicted it would share glucose-specificity and identity in the glucose-binding residues in the active site. Recombinant TxGH116 is indeed active on 4-nitrophenyl (4NP) β-d-glucoside (4NPGlc) and other glucosides (Table 1). Michaelis Menten kinetics, determined at 60 °C (the temperature of the Frying Pan Hot Springs, from which T. xylanolyticum was isolated22), revealed that 4NPGlc is hydrolyzed 17 times as efficiently, in terms of kcat/KM, than the corresponding β-d-galactoside, 4NPGal. TxGH116 has a pH optimum of 5.5 and shows high activity toward both β-1,3- and β-1,4-linked glucooligosaccharides, with similar kcat and KM values for both cellobiose and laminaribiose (Table 2). Since T. xylanolyticum was isolated based on its growth on plant cell wall polysaccharides,22 these oligosaccharides are likely natural substrates for the enzyme. Although we were unable to detect hydrolysis of glucosylceramide, TxGH116 does hydrolyze alkyl glycosides including n-heptyl glucoside and n-octyl glucoside. With knowledge that the enzyme was a β-glucosidase, and given its high sequence similarity to GBA2, we were encouraged to determine the three-dimensional structure of TxGH116, as a model for family GH116.
Table 1. Specific Activity of TxGH116 and Catalytic Residue Mutants with Aryl and Alkyl Glycosides and Oligosaccharides.
| specific
activity (μmol mg–1 min–1)a,b |
|||
|---|---|---|---|
| substrate | TxGH116 wild type | D593A | E441A |
| 4NP β-d-glucoside | 27.3 (100%) | 0.0772 (100%) | 0.000528 |
| 4NP β-d-galactoside | 8.90 (33%) | -c | -c |
| 4NP N-acetyl-β-d-glucosaminide | 0.123 (0.45%) | - | - |
| 4NP α-d-galactoside | < 0.01 (<0.04%) | - | - |
| 4NP α-l-arabinoside | < 0.01 (<0.04%) | - | - |
| 4NP β-d-fucoside | < 0.01 (<0.04%) | - | - |
| 4NP α-d-glucoside | < 0.01 (<0.04%) | - | - |
| 4NP β-d-mannoside | < 0.01 (<0.04%) | - | - |
| 4NP β-d-xyloside | < 0.01 (<0.04%) | - | - |
| 4NP β-cellobioside | 0.302 (1.1%) | - | - |
| gentiobiose | 2.23 (8.2%) | - | - |
| sophorose | 4.10 (15%) | - | - |
| laminaribiose | 11.7 (43%) | 0.000245 (0.32%) | - |
| laminaritriose | 16.1 (59%) | 0.000478 (0.62%) | - |
| laminaritetraose | 15.0 (55%) | 0.000490 (0.62%) | - |
| laminaripentaose | 16.5 (60%) | 0.000524 (0.68%) | - |
| cellobiose | 13.4 (49%) | 0.000345 (0.45%) | - |
| cellotriose | 15.6 (57%) | 0.000538 (0.70%) | - |
| cellotetraose | 14.9 (55%) | 0.000498 (0.65%) | - |
| cellopentaose | 13.8 (51%) | 0.000448 (0.58%) | - |
| cellohexaose | 13.5 (49%) | 0.000422 (0.55%) | - |
| methyl β-d-glucoside | 0.0817 (0.30%) | - | - |
| n-heptyl-β-d-glucoside | 3.74 (14%) | - | - |
| n-octyl β-d-glucoside | 3.58 (13%) | - | - |
The assay contained 1 mM substrate in 50 mM MES buffer, pH 5.5, at 60 °C.
Percent activity relative to glucose or 4NP released from 4NP β-d-glucopyranoside is given in parentheses. The values for disaccharide hydrolysis were determined by dividing the amount of glucose released by 2, since two glucose molecules are released per glycosidic bond hydrolyzed. For oligosaccharides, which may release more than one glucose molecule per substrate molecule due to sequential cleavage, the values quoted are for total glucose released.
The dashes mean that the values were not determined for the mutant enzymes due to low activity.
Table 2. Kinetic Parameters of TxGH116 and Its Mutants for Hydrolysis of 4NP-Glycosides and Oligosaccharides.
| kinetic
parameters (60 °C) |
temperature |
|||||
|---|---|---|---|---|---|---|
| protein | substrate | KM (mM) | kcat (s–1) | kcat/KM (mM–1 s–1) | optimum | Tm |
| WT without N-term taga | 4NP β-d-glucoside | 0.18 ± 0.008 | 49.0 ± 0.8 | 272 | 75 °C | - |
| 4NP β-d-galactoside | 16.3 ± 0.94 | 255 ± 5 | 15.6 | |||
| cellobiose | 0.25 ± 0.016 | 44.4 ± 0.7 | 178 | |||
| laminaribiose | 0.27 ± 0.018 | 41.4 ± 0.7 | 153 | |||
| WT with N-term tagb | 4NP β-d-glucoside | 0.21 ± 0.012 | 37.6 ± 0.6 | 179 | 75 °C | 81.3 °C |
| D508H | 4NP β-d-glucoside | 110 ± 3 | 3.47 ± 0.17 | 0.031 | 65 °C | 66.5 °C |
| D508N | 4NP β-d-glucoside | 41.0 ± 2.5 | 30.2 ± 0.71 | 0.74 | 60 °C | 67.2 °C |
| R544W | 4NP β-d-glucoside | 0.26 ± 0.011 | 48.2 ± 0.53 | 186 | 60 °C | 76.2 °C |
| R786H | 4NP β-d-glucoside | 15.7 ± 0.50 | 193 ± 2.4 | 12.3 | 70 °C | 73.9 °C |
Wild type without the N-terminal tag is used for comparison to R544W and R786H, which were purified in the same way as the crystallized protein.
Wild type with the N-terminal tag is used for comparison to D508H and D508N, which could not be digested with enterokinase without internal cleavage of the protein. Temperature optimum curves are shown in SI Figure S8A. Melting temperatures (Tm) for all mutant proteins are those for the fusion protein without removal of the N-terminal tag. All Tm values were determined based on the change in circular dichroism at 220 nm upon heating. The melting curves are provided in SI Figure S8B.
Three-Dimensional Structures of TxGH116
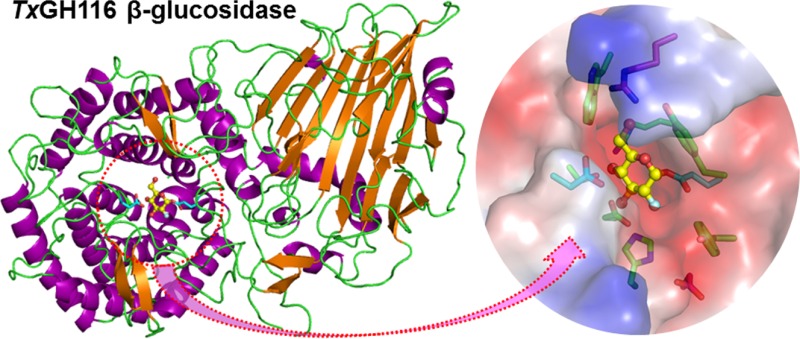
TxGH116 crystallized in space groups P21212 and P61, and its structure was solved and refined at 1.61 Å (Table 3). The absence of structural homologues necessitated a SeMet-derivatized protein for structure solution by single wavelength anomalous dispersion (see Methods). The structure consists of an N-terminal domain, primarily formed by a two-sheet β-sandwich, and a C-terminal (α/α)6 solenoid domain, Figure 1A. The C-terminal domain contains the residues previously proposed as the catalytic nucleophile and general acid/base in the archaeal β-glycosidase from Sulfolobus solfataricus and human GBA2.3,7The putative catalytic nucleophile, E441 (equivalent to human GBA2 E527), lies near the end of a long loop between the first (H5) and second (H6) α-helices of the C-terminal domain, while the putative catalytic acid/base, D593 (equivalent to human GBA2 D677), lies in a long loop between the fifth (H9) and sixth (H10) helices of the solenoid (Figure 1A). This loop also contains the binding site for a structural Ca2+ ion (Supporting Information, Figure S1A).
Table 3. X-ray Data Collection, Phasing, and Refinement Statistics (SAD).
| data seta (PDB ID) |
|||||||
|---|---|---|---|---|---|---|---|
| native-form1 TxGH116 (5FJS) | native-form2 TxGH116 (5BVU) | SeMet TxGH116 | TxGH116 G2F (5BX2) | TxGH116 DNJ (5BX3) | TxGH116 GIM (5BX4) | TxGH116 Glc (5BX5) | |
| data collection | |||||||
| space group | P61 | P21212 | P21212 | P21212 | P21212 | P21212 | P21212 |
| cell dimensions | |||||||
| a, b, c (Å) | a = 187.8, b = 187.8, c = 99.3 | a = 177.7, b = 54.3, c = 83.2 | a = 177.3, b = 54.5, c = 83.2 | a = 177.1, b = 54.7, c = 83.3 | a = 177.3, b = 54.7, c = 83.1 | a = 177.2, b = 54.5, c = 83.2 | a = 177.8, b = 54.6, c = 83.2 |
| α, β, γ (deg) | 90, 90, 120 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| resolution (Å) | 163–2.60 (2.67–2.60) | 50–1.61 (1.64–1.61)a | 50–1.90 (1.93–1.90) | 50–1.61 (1.67–1.61)a | 50–1.96 (2.03–1.96) | 50–1.65 (1.71–1.65) | 50–1.85 (1.88–0.85) |
| Rmerge (%) | 3.7 (79.4) | 5.0 (32.2) | 13.7 (68) | 6 (39) | 7.8 (28.8) | 6.8 (50.4) | 7.8 (45.3) |
| I/σI | 51.2 (3.0) | 34.3 (5.9) | 34.5 (7.9) | 34.5 (6.5) | 28.4 (10.2) | 27.0 (3.9) | 37.7 (8.25) |
| completeness (%) | 100 (99.9) | 99.0 (99.2) | 99.9 (100) | 99.9 (100) | 99.3 (99.3) | 99.9 (99.9) | 95.1 (89.1) |
| redundancy | 9.4 (10.5) | 3.5 (3.5) | 9.1 (9.5) | 7.3 (7.4) | 6.7 (7.1) | 6.6 (6.1) | 6.4 (6.2) |
| refinement | |||||||
| resolution (Å) | 162–2.6 | 89–1.61 | 177–1.61 | 177–1.96 | 89–1.65 | 89–1.85 | |
| no. reflections | 61406 | 97906 | 99862 | 55692 | 91591 | 63333 | |
| Rwork/Rfree (%) | 19/25 | 15.5/17.5 | 15.6/17.6 | 14.8/17.7 | 15.8/18.1 | 14.9/17.8 | |
| no. atoms | |||||||
| protein | 12083 | 6283 | 6292 | 6273 | 6295 | 6268 | |
| carbohydrate | 11 | 11 | 14 | 12 | |||
| hetero | 2 | 101 | 107 | 95 | 107 | 71 | |
| water | 78 | 583 | 628 | 441 | 619 | 474 | |
| B-factors (Å2) | |||||||
| protein | 68 | 19.4 | 14.7 | 17.9 | 15.4 | 20.6 | |
| carbohydrate | - | 9.6 | 11.8 | 10.4 | 17.5 | ||
| hetero | 68 | 37.5 | 32.4 | 39.2 | 33.6 | 38.8 | |
| water | 55 | 31.5 | 27.5 | 29.1 | 28.4 | 30.1 | |
| RMS deviations | |||||||
| bond lengths (Å) | 0.014 | 0.008 | 0.007 | 0.009 | 0.007 | 0.010 | |
| bond angles (deg) | 1.73 | 1.29 | 1.21 | 1.35 | 1.26 | 1.38 | |
| Ramachandran plot | |||||||
| residues in most favorable regions (%) | 93.4 | 88.5 | 89.3 | 89.7 | 88.8 | 89.3 | |
| residues in allowed regions (%) | 5.5 | 11.5 | 10.7 | 10.3 | 11.2 | 10.7 | |
Each data set comprises data from a single crystal. Values in parentheses are for the highest-resolution shell.
Figure 1.

Three-dimensional structure of TxGH116 β-glucosidase. (A) Three views of the three-dimensional structure of TxGH116. The N-terminal domain consists of β-sheets (sheets 1–3) surrounded by α-helices (H1–H4). The C-terminal (α/α)6 solenoid domain contains the active site and consists of 12 α-helices (H5–H16) in six outer helix plus inner helix pairs, with H5 situated between the two domains. A long loop with a two-strand β-sheet (sheet 5) lies between helices H7 and H8, near the active site (E441 and D593). α-Helices are shown in blue and β-strands in yellow. (B) Active site residues of the native structure of TxGH116, showing the distance between the catalytic nucleophile (E441) and acid/base (D593) carbons (red labels). (C) Active site of the TxGH116 covalent intermediate complex with 2-deoxy-2-fluoroglucose, showing the distance between the catalytic residues is decreased in this intermediate. Distances between the geometric means of the carboxyl groups are slightly shorter and are given in the text. Distances are in Å.
GBA2 was previously thought to be an intrinsic membrane protein with residues 689–708 predicted as a transmembrane domain but is easily extracted from cells in buffer without detergents.2,6 The previously predicted hypothetical transmembrane domain corresponds to H10, an internal helix in the catalytic domain of TxGH116 and the derived GBA2 model, which is incompatible with it being a transmembrane helix. Our model therefore supports the peripheral membrane localization of GBA2, where it may bind to an intrinsic membrane protein or polar lipid head groups.
TxGH116 shows no structural similarity to other retaining β-glucosidases, which fall into families including GH1, GH2, GH3, GH5, and GH30 (SI, Figure S2). Comparison to structures in the PDB database with PDBeFold23 found that the TxGH116 structure is most similar to the family GH52 β-xylosidase from Geobacillus thermoglucosidasius (GtGH52, PDB ID: 4C1O, 4C1P),24 which upon global superposition yields an RMSD of 3.2 Å over 560 matched Cα atoms (PDBeFold Z score 8.3; SI, Figure S3A). However, the sequence similarity of GH52 and GH116 is negligible (GH52 enzymes are not detected using PSI-BLAST with the TxGH116 sequence as a search query), GH52 lacks the N-terminal loop that contributes to the entrance to the active site in GH116, and different numbers of strands are found in the major β-sheets of the N-terminal domain (see SI). Nonetheless, the active sites also share the same geometry for the putative acid/base and nucleophile and a few sugar binding residues (SI Figure S3) allowing us to propose that these two divergent families be grouped as a “clan” (GH-O) of related 3-D structures.25
Catalytic Residues and Active Site Structure
GH116 family enzymes act via a double-displacement mechanism in which a covalent glycosyl enzyme intermediate is formed and subsequently hydrolyzed via oxocarbenium-ion like transition states (SI Figure S4). Such a mechanism demands two key residues: a general acid/base and a nucleophile. The putative acid/base, D593, is located approximately 8 Å (carboxylate to carboxylate) away from the nucleophile (E441; Figure 1B,C) in the P21212 structure and 10 Å away in the P61 structure due to a displacement of the loop containing D593 by 2 Å (not shown). This is a longer distance than is observed in most retaining β-glucosidases, which typically have the acid/base within 4.5–6.5 Å of the nucleophile.26−29 Given this ambiguity, we verified that the enzyme is retaining by identifying the initial hydrolysis product as β-d-glucose, which retains the anomeric stereochemistry of the substrate, by monitoring the reaction time course by NMR spectroscopy (SI, Figure S5A). Moreover, we demonstrated that E441 is the catalytic nucleophile and D593 the catalytic acid/base by mutation of these residues to alanine (which resulted in large losses of activity, Table 1), followed by chemical rescue30 using small nucleophiles (see SI, Figures S5B–D).
Several established β-glucosidase inhibitors were examined to identify ligands that could bind in the active site. TxGH116 is inhibited by the covalent inactivator 2,4-dinitrophenyl 2-deoxy-2-fluoro-β-d-glucoside (DNP2FG),31 and by the noncovalent inhibitors isofagomine (IFG), deoxynojirimycin (DNJ), glucoimidazole (GIM), δ-gluconolactone (weak inhibition at 1 mM), and the product d-glucose (Glc). Inhibition by IFG, DNJ, GIM, and Glc is competitive with Ki values of 2.9 ± 0.1 nM, 0.13 ± 0.01 μM, 0.34 ± 0.03 μM, and 4.0 ± 0.3 mM, respectively (SI Figure S6). TxGH116 was not inhibited by conduritol B epoxide (CBE), an inhibitor that has been used to distinguish GBA1 (sensitive) from GBA2 (less sensitive20). In contrast, cyclophellitol, a related epoxide that contains a hydroxymethyl group corresponding to the hydroxymethyl group of glucose, rapidly inactivated TxGH116. The similarity of the inhibition profile of human GBA2 and TxGH116 with sugar-shaped inhibitors shows that TxGH116 may be considered an informative functional model for the glycone-binding (−1) subsite of the human enzyme.
Insight into the active site and catalytic residues was obtained by solving the structures of TxGH116 in complexes with a trapped intermediate derived from DNP2FG, the inhibitors DNJ and GIM, and the product (Glc) (Figure 2). The 2-fluoroglucosyl moiety binds to the catalytic nucleophile E441, consistent with the equivalent residue labeled by DNP2FG in studies with Sulfolobus solfataricus (E335) and human enzymes (E527),3,7 and the mutagenesis and chemical rescue studies described in the SI. The putative acid/base, D593, is located in an unorthodox position above the pyranose ring, rather than in a typical position in the plane of the sugar ring which provides for syn- or anti- lateral protonation of the glycosidic oxygen.32 Its position above the plane of the sugar ring prevents the efficient lateral protonation of GIM that is required for its strong binding, thus explaining the weak inhibition by this molecule.32
Figure 2.
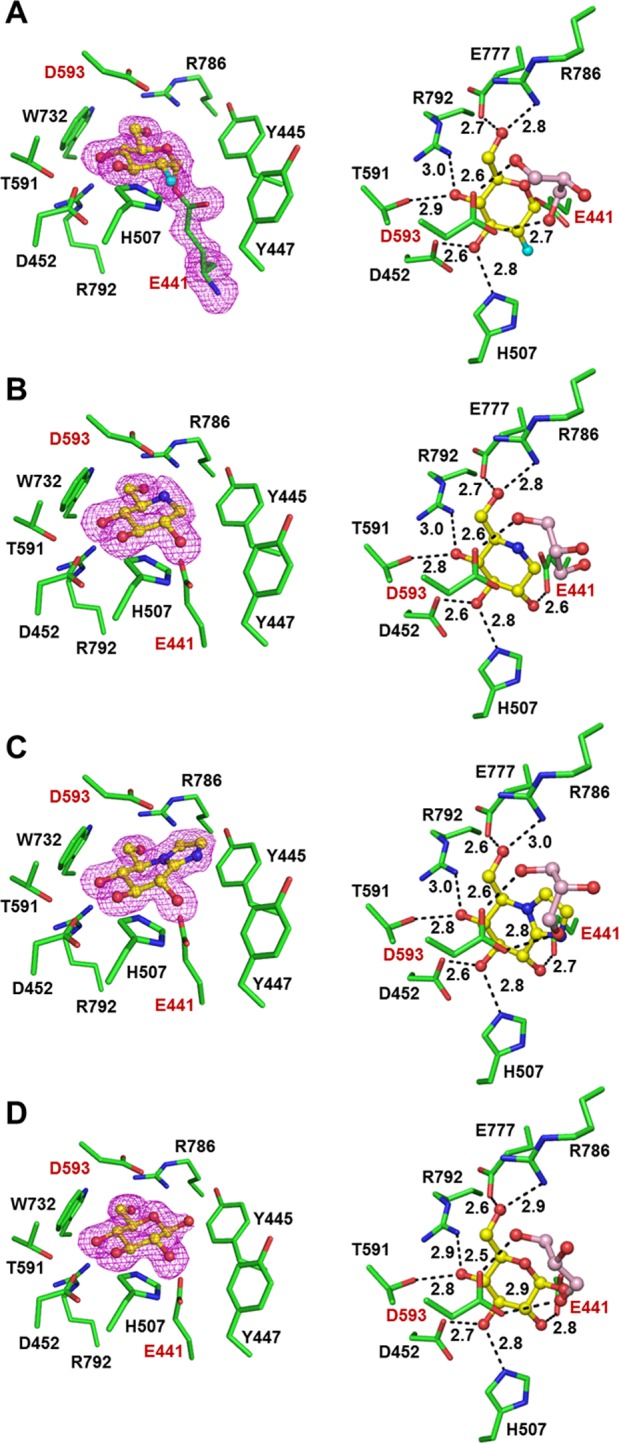
Active site of TxGH116 alone and in complex with inhibitors and glucose. Left panels show the ligand electron density omit maps, contoured at 3σ, in the active sites of the complexes; the right panels show the potential hydrogen bonds between the ligands and surrounding residues, with the distances between the interacting atoms displayed. A glycerol molecule that was omitted from the left panels for clarity is shown in the right panels; catalytic residues are labeled in red. Ligands include (A) 2-deoxy-2-fluoroglucosyl (G2F), (B) deoxynojirimycin (DNJ), (C) glucoimidazole (GIM), and (D) β-d-glucose. Distances are in Å.
The structure of the glycosyl enzyme (Figure 2A) enabled identification of residues interacting with the substrate in the −1 subsite. The amino acid residues capable of forming hydrogen bonding interactions include E441 at OH2 (F2 in G2F); H507 and D452 at OH3; D452, T591, and R792 at OH4; and E777 and R786 at the OH6. In the last case, OH6 appears to mediate a salt bridge between E777 and R786. The interactions at OH6 may explain why cyclophellitol, bearing a hydroxymethyl group equivalent to the substrate C6-hydroxy-methyl group, inhibits TxGH116, but CBE, which lacks this group, does not. All of the glucosyl binding residues in TxGH116 are conserved with human GBA2, whereas only a few of these could be reliably aligned with a conserved residue in the two biochemically characterized archaeal enzymes, SSO1353 and SSO3039 (SI Figure S7).
The complexes with DNJ and GIM and the product Glc reveal similar interactions to those seen in the G2F intermediate (Figure 2B–D). In the complexes with Glc and DNJ, the pyranose/piperidine rings are in 4C1 chairs, while GIM adopts a 4H3 conformation, corresponding to the preferred low energy conformations of these ligands.33
Model of Human GBA2 and Context of Disease-Causing Mutations
Human GBA2 and TxGH116 share 37% sequence identity over the region of GBA2 residues 63 to 893, allowing an informative homology model of GBA2 to be generated from TxGH116. Two longer loops in GBA2, one of which may contribute to the aglycone-binding cleft leading into the active site, as does the corresponding loop in TxGH116 (Figure 3A), could not be modeled with high confidence. Notably, there is excellent correspondence between the active site residues, with all the residues noted as binding the sugar in TxGH116 conserved in human GBA2 (Figure 3C).
Figure 3.

Human GBA2 structural model. (A) Overall structure of the human GBA2 model (purple) superimposed with the TxGH116 structure (green). The loop adjacent to the active site, which is longer in GBA2, is marked with a red arrow, while the longer loop between helices H5 and H6 of the (α/α)6 solenoid domain is marked with a black arrow. (B) Diagram of human GBA2 model showing positions of residues mutated in human disease (space-filling) and interacting residues and ligand (sticks). (C) Superposition of active site residues of human GBA2 and the TxGH116 complex with glucose. All of the sugar binding residues are conserved between TxGH116 and GBA2, as well as the aspartate (D508) that interacts with H507 in TxGH116. The H-bonds between residues involved in human autosomal recessive cerebellar ataxia mutations and the sugar residue are shown as dashed lines. The catalytic nucleophile and acid/base labels are underlined. (D) Superposition of human GBA2 model (purple) on TxGH116 structure (green) in the area of the R630 residue mutated in certain hereditary spastic paraplegia cases. R630 and the nearby carboxylate residues D631 and E555 are conserved with TxGH116 residues. (E) View of GBA2 F419 and the conserved interacting aromatic groups in the overlay of the GBA2 and TxGH116 models. (F) Position of the GBA2 G683R mutation, showing that, although it is near the surface of the protein, G683 is tightly opposed to S741 (both shown in magenta spheres) and the bound calcium cation (green sphere) and is also linked to the catalytic acid/base D677 shown next to the glucosyl residue (cyan carbon sticks).
Known pathogenic human GBA2 mutations include five missense mutations, F419V, D594H, R630W, G683R, and R873H (Figure 3B).12−15,34 The amino acid residues corresponding to the sites of these mutations are conserved between GBA2 and TxGH116 (F347, D508, R544, G599, and R786, respectively), as well as other GH116 enzymes in subfamily 1, which includes eukaryotic and bacterial enzymes (SI Figure S7).21 As shown in Figure 3C, the GBA2 D594 and R873 residues (TxGH116: D508 and R786) are located within the active site, providing an immediate catalytic implication for these mutations. As noted earlier, TxGH116 R786 makes two hydrogen bonds to the glucosyl 6OH group, which are predicted to be conserved in GBA2 R873. The mutation R873H will result in a shorter side chain, which will not form an effective hydrogen bond to O6 of Glc. In TxGH116, D508 hydrogen bonds to H507, which in turn makes a hydrogen bond to OH3, and the D594 residue makes the same interaction with the conserved H593 in the GBA2 model. The mutation D594H is expected to disrupt this interaction. The fact that two human pathogenic mutations involve sugar-binding residues emphasizes the importance of this model.
In contrast to the D584H and R873H, the other three missense mutations are located distal to the active site. The R630W mutation is approximately 15 Å away from the active site, between the layers of helices of the catalytic (α/α)6 solenoid domain. The GBA2 model shows that R630 can form salt bridge interactions with two conserved carboxylates, E555 and D631 (Figure 3D). The mutation R630W will result in the loss of a charged residue, disturbing the charge balance, and introduction of a somewhat larger Trp residue, which together may result in destabilization of the protein. The only pathogenic mutation found in the N-terminal β-sandwich domain is F419V. F419 is positioned at the interface of the two domains, and the aromatic ring appears to form dispersive interactions with the conserved neighboring aromatic residues Y534 in the long loop between the H5 and H6 helices and W603 in the loop between the H7 and H8 helices of the C-terminal (α/α)6 solenoid domain (Figure 3B,E). V419 cannot make such interactions, so the F419V mutation may destabilize the interaction between the two domains. The G683R mutation, located approximately 20 Å from the active site, is in an outside turn of the long loop including the catalytic acid/base (D677) and the sugar-binding residue T675 (Figure 3F). G683 is located very close to the neighboring loop, in which S741 is closest to G683, such that a replacement with arginine would result in a clash. It is also next to the binding site for the calcium cation in TxGH116, suggesting that a positively charged Arg could disrupt this binding.
It was recently shown that expression of GBA genes possessing all known pathogenic mutations in COS-7 or HeLa cells resulted in reduced activity in crude cell extracts relative to wild-type.34 We sought to gain insight into the physical and mechanistic effects of the pathogenic GBA2 D594H, R630W, and R873H mutations by analysis of corresponding mutations in TxGH116. For all three mutations, TxGH116 D508H, R544W, and R786H (corresponding to GBA2 D594H, R630W, and R873H), large decreases in activity were observed at the temperature optimum of the wild type enzyme (Table 2, SI Figure S8). For the TxGH116 R786H mutation (corresponding to GBA2 R873H), a 90-fold increase in the KM value led to a reduction in kcat/KM of more than 20-fold (Table 2), which is consistent with the weaker binding of the sugar expected based on this residue’s interaction with the glucose 6OH. Recapitulation of the GBA2 D594H mutation in TxGH116 D508H resulted in both decreased stability and lowered activity (Table 2). Most notably, the kcat/KM value decreased 5800-fold, owing mostly to a >500-fold increase in the KM value, with a small reduction in the kcat value. The large decrease in kcat/KM and relatively small effect on kcat suggests a large effect on the binding and stabilization of the substrate and transition state of the first committed step of hydrolysis (glycosylation), whereas the rate limiting step is less affected. The wild type kcat values for 4NPGlc (possessing a leaving group with a relatively low pKa value and thus a relatively low energy barrier for departure) and cellobiose (possessing a poor glucosyl leaving group with a high pKa value) are similar (Table 2), suggesting that the deglycosylation step (for which the reaction is the same) is rate-limiting. The small change in kcat in the D508H mutant suggests that this is most likely also true for this mutant.35 To investigate the importance for catalysis of the charge of D508, the isosteric mutation D508N, which should maintain hydrogen bonding to H507, was studied. D508N exhibited a >240-fold reduction in kcat/KM, and thus, the negative charge on D508 appears to be important for catalysis.
The effect of the GBA2 R630W mutation was studied through the TxGH116 R544W mutant. The R544W mutant exhibited a slightly increased KM value and little change in the melting temperature but caused a decrease in the temperature of the optimum catalytic activity by 15 °C. These properties suggest the R544W mutation may destabilize the active site, while having little effect on the overall protein stability. This hypothesis is consistent with the observation that a large amount of GBA2 protein accumulated in COS-7 and HeLa cells that expressed the R630W variant, suggesting it is not an unstable protein despite the low level of activity in the standard GBA assay.34
Conclusions
GBA2 has risen to prominence due to its involvement in congenital neurological disorders leading to cerebellar ataxia and spastic paraplegia, as well as its interplay with GBA1, defects in which cause Gaucher disease and contribute to Parkinsonism.36 Modulators of GBA2 activity may therefore provide therapeutic benefits in the treatment of diseases involving defects in GBA1 or GBA2. The TxGH116 structure described here possesses many unique features, particularly in the N-terminal β-sandwich, which is tightly associated with the catalytic domain and contributes to the substrate-binding cleft, and the unusual orientation of the general acid/base residue. The residues binding the glucose in the −1 subsite are highly conserved between TxGH116 and human GBA2, and the structural model provides a useful framework for understanding the molecular basis for binding of ligands to the glycone-binding site. Significantly, key missense mutations causing defects in GBA2 are conserved between the two enzymes, which have allowed identification of likely biochemical and structural reasons underpinning the genetic defects. The present work demonstrates that the missense mutations are located both proximal to and remote from the active site and result in a loss of enzymatic activity by a range of mechanisms including reduction in substrate binding, reduced temperature for optimal catalysis, and reduced enzyme stability.
Methods
Plasmid Construction and Protein Expression
Two independent constructs presenting E. coli-optimized TxGH116 genes were prepared in pET28a(+) and pET30a(+) expression vectors with polyhistidine tags, as described in the Supporting Information (SI). The pET28a(+) construct generated a TxGH116 protein with the first 20 amino acid residues replaced with an N-terminal His6-tag that was designated TxGH116-N. The pET30a(+) construct encoded the TxGH116 protein lacking the first 18 amino acids following the pET30(+) N-terminal tag including a His6 tag, S-tag, and enterokinase site. This fusion protein, designated TxGH116Δ1–18, also included the C-terminal His6 tag from the plasmid. The fusion proteins were produced in E. coli strain BL21(DE3) and purified by immobilized metal affinity chromatography, as described in the SI Methods. Production of selenomethionine-labeled TxGH116 (SeTxGH116) was accomplished with the pET30a/TxGH116Δ1–18 plasmid in E. coli strain B834(DE3) pLysS cells grown in a SeMet core medium (Nihon Jyunyaku Co.; SI Methods).
Enzymatic Characterization
The TxGH116Δ1–18 protein was used for enzymatic characterization. The optimum pH of TxGH116 for hydrolysis of 4NPGlc was determined to be pH 5.5 by incubating 0.05 μg of enzyme with 1 mM 4NPGlc in 100 mM McIlvaine universal (phosphate-citrate) buffer, pH 2.5–8.0 at 0.5 pH unit intervals, at 60 °C for 30 min. The temperature optimum (75 °C) was determined via the hydrolysis of 1 mM 4NPGlc with 0.05 μg of enzyme in 50 mM sodium acetate buffer, pH 5.5, at temperatures of 10–110 °C, for 10 min (SI Figure S8A). For standard activity assays, 1 mM 4NPGlc was incubated with enzyme at 60 °C for 15 min. Chemical rescue activity of E441A and D593A mutants and wild type TxGH116 were assayed under conditions of 50 mM MES, pH 5.5, and 60 °C with 1 mM 4NPGlc, 0.1 μg of wild type for 15 min, 25 μg of E441 for 60 min, and 5 μg for 15 min of D593A with sodium formate and sodium azide concentrations of 0–4 M.
Inhibition constants were determined by assaying triplicate reactions at five 4NPGlc substrate concentrations bracketing the apparent KM value and five inhibitor concentrations. The inhibitors were preincubated with 0.05 μg of enzyme at 37 °C for 10 min before the addition of 4NPGlc and incubation at 60 °C for 15 min. The Ki values were calculated from the x intercept of the derivative plot of the slopes of Lineweaver–Burk reciprocal plots (1/v vs 1/[4NPGlc]) vs the inhibitor concentration (SI Figure S6). Inhibition was competitive in all cases.
NMR Determination of Retaining Mechanism and Catalytic Rescue Products
The proton and STD NMR experiments were performed on a 500 MHz NMR spectrometer (Bruker AVANCE III HD) with a CPP BBO 500 Cyroprobe and BRUKER TOPSPIN 3.2 software. The 1H NMR spectra were collected at a frequency of 500.366 MHz. The spectra of the 4NPGlc (7.3 mM) hydrolysis reactions were collected before and 10 and 30 min after adding 50 μM TxGH116Δ1–18 enzyme.
Mutagenesis
Site-directed mutagenesis was done by the QuikChange method (Stratagene, Agilent Corp.) to make a catalytic nucleophile (E441A) and acid/base (D593A) mutations and replicate pathogenic human GBA2 mutations (D508H and its variant D508N, R544W, and R786H) in the pET30a/TxGH116Δ1–18 expression vector. Mutagenic primers are listed in SI Table S1. The mutations and entire coding sequence were verified by DNA sequencing (Macrogen).
Protein Crystallization
Crystals were obtained after screening and optimization by hanging drop vapor diffusion at 15 °C in a precipitant of 0.2 M ammonium sulfate, 20% PEG 3000, 0.1 M MES, pH 5.5, for TxGH116Δ1–18, and at 19 °C in 0.2 M ammonium sulfate, 15% w/v PEG 6000, 0.1 M Bis-Tris propane, pH 8, for TxGH116N.
X-ray Diffraction and Structure Solution
Crystals of TxGH116Δ1–18 were soaked briefly in precipitant containing 20% glycerol. For ligand complexes, the soaking solution also contained either 10 mM DNJ, GIM, 2,4-dinitrophenyl 2-deoxy-2-fluoro-β-d-glucopyranoside (DNPG2F), or 100 mM d-glucose. The crystals were flash vitrified in liquid nitrogen and data collected at 100 K. Diffraction data were collected at the SPring-8 synchrotron beamline BL44XL with 0.9000 Å X-ray radiation for native and 0.97865 Å for SeMet TxGH116Δ1–18 crystals on a MX-300HE detector (Rayonix). The data were processed with the HKL2000 suite.37 The initial phases were calculated by the single wavelength anomalous dispersion (SAD) method with PHENIX AUTOSOL.38 The SAD calculation revealed one protein molecule per asymmetric unit in the P21212 space group. The Matthews coefficient was 2.20 Å3/Da, and the solvent content was 44.19%. The SeMet-derivatized protein included 26 SeMet, 25 of which were observed in the anomalous difference Fourier map. X-ray data for TxGH116-N were collected at beamline i24 (wavelength: 0.97 Å) of the Diamond Light Source (Oxfordshire, UK) with a Pilatus3 6 M (100 Hz max. frame rate) detector and processed using MOSFLM and scaled in SCALA39 to show two protein molecules per asymmetric unit in the P61 space group. The solvent content was 56%, and the Matthews coefficient was 2.8 Å3/Da.
The initial phases were transformed to native structure by molecular replacement with the MOLREP program40 in the CCP4 suite.39 The structure was built with the COOT graphic program41 in alternation with refinement with REFMAC5.42 The complex and TxGH116-N structures were solved by MOLREP40 and refined in a similar manner. The final models were analyzed with PROCHECK43 and MolProbity44 and validated on the PDB Web site. Data and structure refinement statistics are shown in Table 3.
Molecular Modeling
To construct a homology model of the human GBA2 protein, the first 64 and last 34 residues of the human protein, which do not have corresponding sequences in TxGH116, were deleted and the remaining sequence aligned to the TxGH116 sequence and used to build five models within the Modeler program.45 After the addition of hydrogen and optimization of hydrogen-bonding side chain rotamers with Molprobity,44 the model with the lowest Molprobity (3.31) and Clash (108.73) scores was used as the human GBA2 model. Comparison of the homology model of human GBA2 with the TxGH116 template structure gave an RMSD of 0.543 Å over 712 Cα atoms that could be aligned.
Acknowledgments
We are grateful to Y. Goto for assistance in circular dichroism and the research staff at Osaka Analytical Center of Rigaku Corp. W. Offen is thanked for assistance and critical reading of the manuscript, and J. Svasti is thanked for advice and support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.6b00192.
Supporting Results and Discussion, Methods, Table S1, Figures S1–S8 (PDF)
Author Contributions
+ Should be considered co-first authors.
Funding was provided by The Thailand Research Fund (Grant BRG5680012), Suranaree University of Technology, the National Research University project grant from the Commission on Higher Education of Thailand to SUT, the International Collaborative Research Program (ICR-14–03) from the Institute for Protein Research, Osaka University. G.J.D., I.B., and L.W. were supported by the European Research Council through ERC-2012-AdG-32294 “Glycopoise.” S.J.W. was supported by the Australian Research Council.
The authors declare no competing financial interest.
Supplementary Material
References
- Lombard V.; Golaconda Ramulu H.; Drula E.; Coutinho P. M.; Henrissat B. (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–495. 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matern H.; Boermans H.; Lottspeich F.; Matern S. (2001) Molecular cloning and expression of human bile acid β-glucosidase. J. Biol. Chem. 276, 37929–37933. [DOI] [PubMed] [Google Scholar]
- Cobucci-Ponzano B.; Aurilia V.; Riccio G.; Henrissat B.; Coutinho P. M.; Strazzulli A.; Padula A.; Corsaro M. M.; Pieretti G.; Pocsfalvi G.; Fiume I.; Cannio R.; Rossi M.; Moracci M. (2010) A new archaeal β-glycosidase from Sulfolobus solfataricus: seeding a novel retaining β-glycan-specific glycoside hydrolase family along with the human non-lysosomal glucosylceramidase GBA2. J. Biol. Chem. 285, 20691–20703. 10.1074/jbc.M109.086470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matern H.; Heinemann H.; Legler G. N.; Matern S. (1997) Purification and characterization of a microsomal bile acid β-glucosidase from human liver. J. Biol. Chem. 272, 11261–11267. 10.1074/jbc.272.17.11261. [DOI] [PubMed] [Google Scholar]
- Yildiz Y.; Matern H.; Thompson B.; Allegood J. C.; Warren R. L.; Ramirez D. M.; Hammer R. E.; Hamra F. K.; Matern S.; Russell D. W. (2006) Mutation of β-glucosidase 2 causes glycolipid storage disease and impaired male fertility. J. Clin. Invest. 116, 2985–2994. 10.1172/JCI29224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Körschen H. G.; Yildiz Y.; Raju D. N.; Schonauer S.; Bonigk W.; Jansen V.; Kremmer E.; Kaupp U. B.; Wachten D. (2013) The non-lysosomal β-glucosidase GBA2 is a non-integral membrane-associated protein at the endoplasmic reticulum (ER) and Golgi. J. Biol. Chem. 288, 3381–3393. 10.1074/jbc.M112.414714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallemeijn W. W.; Witte M. D.; Voorn-Brouwer T. M.; Walvoort M. T.; Li K. Y.; Codee J. D.; van der Marel G. A.; Boot R. G.; Overkleeft H. S.; Aerts J. M. (2014) A sensitive gel-based method combining distinct cyclophellitol-based probes for the identification of acid/base residues in human retaining β-glucosidases. J. Biol. Chem. 289, 35351–35362. 10.1074/jbc.M114.593376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingwood C. A. (2011) Glycosphingolipid functions. Cold Spring Harbor Perspect. Biol. 3, a004788. 10.1101/cshperspect.a004788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendelac A.; Savage P. B.; Teyton L. (2007) The biology of NKT cells. Annu. Rev. Immunol. 25, 297–336. 10.1146/annurev.immunol.25.022106.141711. [DOI] [PubMed] [Google Scholar]
- Jmoudiak M.; Futerman A. H. (2005) Gaucher disease: pathological mechanisms and modern management. Br. J. Haematol. 129, 178–188. 10.1111/j.1365-2141.2004.05351.x. [DOI] [PubMed] [Google Scholar]
- Butters T. D. (2007) Gaucher disease. Curr. Opin. Chem. Biol. 11, 412–418. 10.1016/j.cbpa.2007.05.035. [DOI] [PubMed] [Google Scholar]
- Martin E.; Schule R.; Smets K.; Rastetter A.; Boukhris A.; Loureiro J. L.; Gonzalez M. A.; Mundwiller E.; Deconinck T.; Wessner M.; Jornea L.; Oteyza A. C.; Durr A.; Martin J. J.; Schols L.; Mhiri C.; Lamari F.; Zuchner S.; De Jonghe P.; Kabashi E.; Brice A.; Stevanin G. (2013) Loss of function of glucocerebrosidase GBA2 is responsible for motor neuron defects in hereditary spastic paraplegia. Am. J. Hum. Genet. 92, 238–244. 10.1016/j.ajhg.2012.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer M. B.; Eleuch-Fayache G.; Schottlaender L. V.; Nehdi H.; Gibbs J. R.; Arepalli S. K.; Chong S. B.; Hernandez D. G.; Sailer A.; Liu G.; Mistry P. K.; Cai H.; Shrader G.; Sassi C.; Bouhlal Y.; Houlden H.; Hentati F.; Amouri R.; Singleton A. B. (2013) Mutations in GBA2 cause autosomal-recessive cerebellar ataxia with spasticity. Am. J. Hum. Genet. 92, 245–251. 10.1016/j.ajhg.2012.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Votsi C.; Zamba-Papanicolaou E.; Middleton L. T.; Pantzaris M.; Christodoulou K. (2014) A novel GBA2 gene missense mutation in spastic ataxia. Ann. Hum. Genet. 78, 13–22. 10.1111/ahg.12045. [DOI] [PubMed] [Google Scholar]
- Citterio A.; Arnoldi A.; Panzeri E.; D’Angelo M. G.; Filosto M.; Dilena R.; Arrigoni F.; Castelli M.; Maghini C.; Germiniasi C.; Menni F.; Martinuzzi A.; Bresolin N.; Bassi M. T. (2014) Mutations in CYP2U1, DDHD2 and GBA2 genes are rare causes of complicated forms of hereditary spastic paraparesis. J. Neurol. 261, 373–381. 10.1007/s00415-013-7206-6. [DOI] [PubMed] [Google Scholar]
- Mistry P. K.; Liu J.; Sun L.; Chuang W. L.; Yuen T.; Yang R.; Lu P.; Zhang K.; Li J.; Keutzer J.; Stachnik A.; Mennone A.; Boyer J. L.; Jain D.; Brady R. O.; New M. I.; Zaidi M. (2014) Glucocerebrosidase 2 gene deletion rescues type 1 Gaucher disease. Proc. Natl. Acad. Sci. U. S. A. 111, 4934–4939. 10.1073/pnas.1400768111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques A. R. A.; Aten J.; Ottenhoff R.; van Roomen C. P. A. A.; Herrera Moro D.; Claessen N.; Veloz M. F. V.; Zhou K.; Lin Z.; Mirzaian M.; Boot R. G.; De Zeeuw C. I.; Overkleeft H. S.; Yildiz Y.; Aerts J. M. F. G. (2015) Reducing GBA2 activity ameliorates neuropathology in Niemann-Pick Type C mice. PLoS One 10, e0135889. 10.1371/journal.pone.0135889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorli S. C.; Colie S.; Albinet V.; Dubrac A.; Touriol C.; Guilbaud N.; Bedia C.; Fabrias G.; Casas J.; Segui B.; Levade T.; Andrieu-Abadie N. (2013) The nonlysosomal β-glucosidase GBA2 promotes endoplasmic reticulum stress and impairs tumorigenicity of human melanoma cells. FASEB J. 27, 489–498. 10.1096/fj.12-215152. [DOI] [PubMed] [Google Scholar]
- Overkleeft H. S.; Renkema G. H.; Neele J.; Vianello P.; Hung I. O.; Strijland A.; van der Burg A. M.; Koomen G. J.; Pandit U. K.; Aerts J. M. (1998) Generation of specific deoxynojirimycin-type inhibitors of the non-lysosomal glucosylceramidase. J. Biol. Chem. 273, 26522–26527. 10.1074/jbc.273.41.26522. [DOI] [PubMed] [Google Scholar]
- Ridley C. M.; Thur K. E.; Shanahan J.; Thillaiappan N. B.; Shen A.; Uhl K.; Walden C. M.; Rahim A. A.; Waddington S. N.; Platt F. M.; van der Spoel A. C. (2013) β-Glucosidase 2 (GBA2) activity and imino sugar pharmacology. J. Biol. Chem. 288, 26052–26066. 10.1074/jbc.M113.463562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara M. C.; Cobucci-Ponzano B.; Carpentieri A.; Henrissat B.; Rossi M.; Amoresano A.; Moracci M. (2014) The identification and molecular characterization of the first archaeal bifunctional exo-beta-glucosidase/N-acetyl-beta-glucosaminidase demonstrate that family GH116 is made of three functionally distinct subfamilies. Biochim. Biophys. Acta 1840, 367–377. 10.1016/j.bbagen.2013.09.022. [DOI] [PubMed] [Google Scholar]
- Lee Y.-E.; Jain M. K.; Lee C.; Lowe S. E.; Zeikus J. G. (1993) Taxonomic distinction of saccharolytic thermophilic anaerobes: Description of Thermoanaerobacterium xylanolyticum gen. nov., sp. nov., and Thermoanaerobacterium saccharolyticum gen. nov., sp. nov.; Reclassification of Thermoanaerobium brockii, Clostridium thermosulfurogenes, and Clostridium thermohydrosulfiricum ElO0–69 as Thermoanaerobacter brockii comb. nov., Thermoanaerobacterium thermosulfurigenes comb. nov., and Thermoanaerobacter thermohydrosulfuricus comb. nov.,respectively; and transfer of Clostridium thermohydrosulfuricum 39E to Thermoanaerobacter ethanolicus. Int. J. Syst. Bacteriol. 43, 41–51. 10.1099/00207713-43-1-41. [DOI] [Google Scholar]
- Krissinel E.; Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2256–2268. 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- Espina G.; Eley K.; Pompidor G.; Schneider T. R.; Crennell S. J.; Danson M. J. (2014) A novel beta-xylosidase structure from Geobacillus thermoglucosidasius: the first crystal structure of a glycoside hydrolase family GH52 enzyme reveals unpredicted similarity to other glycoside hydrolase folds. Acta Crystallogr., Sect. D: Biol. Crystallogr. 70, 1366–1374. 10.1107/S1399004714002788. [DOI] [PubMed] [Google Scholar]
- Henrissat B.; Davies G. (1997) Structural and sequence-based classification of glycoside hydrolases. Curr. Opin. Struct. Biol. 7, 637–644. 10.1016/S0959-440X(97)80072-3. [DOI] [PubMed] [Google Scholar]
- Barrett T.; Suresh C. G.; Tolley S. P.; Dodson E. J.; Hughes M. A. (1995) The crystal structure of a cyanogenic beta-glucosidase from white clover, a family 1 glycosyl hydrolase. Structure 3, 951–960. 10.1016/S0969-2126(01)00229-5. [DOI] [PubMed] [Google Scholar]
- Varghese J. N.; Hrmova M.; Fincher G. B. (1999) Three-dimensional structure of a barley β-D-glucan exohydrolase, a family 3 glycosyl hydrolase. Structure 7, 179–190. 10.1016/S0969-2126(99)80024-0. [DOI] [PubMed] [Google Scholar]
- Davies G. J.; Dauter M.; Brzozowski A. M.; Bjornvad M. E.; Andersen K. V.; Schulein M. (1998) Structure of the Bacillus agaradherans family 5 endoglucanase at 1.6 Å and its cellobiose complex at 2.0 Å resolution. Biochemistry 37, 1926–1932. 10.1021/bi972162m. [DOI] [PubMed] [Google Scholar]
- Dvir H.; Harel M.; McCarthy A. A.; Toker L.; Silman I.; Futerman A. H.; Sussman J. L. (2003) X-ray structure of human acid-β-glucosidase, the defective enzyme in Gaucher disease. EMBO Rep. 4, 704–709. 10.1038/sj.embor.embor873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly H. D.; Withers S. G. (1999) Mutagenesis of glycosidases. Annu. Rev. Biochem. 68, 487–522. 10.1146/annurev.biochem.68.1.487. [DOI] [PubMed] [Google Scholar]
- Withers S. G.; Warren R. A. J.; Street I. P.; Rupitz K.; Kempton J. B.; Aebersold R. (1990) Unequivocal demonstration of the involvement of a glutamate residue as a nucleophile in the mechanism of a retaining glycosidase. J. Am. Chem. Soc. 112, 5887–5889. 10.1021/ja00171a043. [DOI] [Google Scholar]
- Heightman T. D.; Vasella A. (1999) Recent insights into inhibition, structure, and mechanism of configuration-retaining glycosidases. Angew. Chem., Int. Ed. 38, 750–770. . [DOI] [PubMed] [Google Scholar]
- Granier T.; Panday N.; Vasella A. (1997) Structure-activity relations for imidazo-pyridine-type inhibitors of β-D-glucosidases. Helv. Chim. Acta 80, 979–987. 10.1002/hlca.19970800329. [DOI] [Google Scholar]
- Sultana S.; Reichbauer J.; Schule R.; Mochel F.; Synofzik M.; van der Spoel A. C. (2015) Lack of enzyme activity in GBA2 mutants associated with hereditary spastic paraplegia/cerebellar ataxia (SPG46). Biochem. Biophys. Res. Commun. 465, 35–40. 10.1016/j.bbrc.2015.07.112. [DOI] [PubMed] [Google Scholar]
- MacLeod A. M.; Lindhorst T.; Withers S. G.; Warren R. A. J. (1994) The acid/base catalyst in the exoglucanase/xylanase from Cellulomonas fimi is glutamic acid 127: evidence from detailed kinetic studies of mutants. Biochemistry 33, 6371–6376. 10.1021/bi00186a042. [DOI] [PubMed] [Google Scholar]
- Bultron G.; Kacena K.; Pearson D.; Boxer M.; Yang R.; Sathe S.; Pastores G.; Mistry P. (2010) The risk of Parkinson’s disease in type 1 Gaucher disease. J. Inherited Metab. Dis. 33, 167–173. 10.1007/s10545-010-9055-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode, In Methods in Enzymology, Macromolecular Crystallography, part A (Carter C. W. J., and Sweet R. M., Eds.), pp 307–326, Academic Press, New York. [DOI] [PubMed] [Google Scholar]
- Terwilliger T. C.; Adams P. D.; Read R. J.; McCoy A. J.; Moriarty N. W.; Grosse-Kunstleve R. W.; Afonine P. V.; Zwart P. H.; Hung L.-W. (2009) Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr., Sect. D: Biol. Crystallogr. 65, 582–601. 10.1107/S0907444909012098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 67, 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A.; Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 22–25. 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubak P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 67, 355–367. 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A.; MacArthur M. W.; Moss D. S.; Thornton J. M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291. 10.1107/S0021889892009944. [DOI] [Google Scholar]
- Chen V. B.; Arendall W. B. 3rd; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N., Webb B., Marti-Renom M. A., Madhusudhan M. S., Eramian D., Shen M. Y., Pieper U., and Sali A. (2006) Comparative protein structure modeling using Modeller, In Current Protocols in Bioinformatics, p 5.6.1, Chapter 5, John Wiley and Sons, Inc., Hoboken, NJ. DOI: 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.