

Abstract
Activation of KISS1 receptor (KISS1R or GPR54) by its ligands (kisspeptins) regulates a diverse function both in normal physiology and pathophysiology. In cancer, KISS1-induced KISS1R signaling is known to inhibit tumor angiogenesis and metastasis. However, roles of KISS1 and KISS1R in earlier stages of tumor progression and metastasis in vivo are still unknown. In this study, we demonstrate a critical role for Kiss1r in early stages of tumor progression using mouse tumor models. PyMT/Kiss1r mice with different Kiss1r genotypes were obtained by crossing MMTV-PyMT transgenic mouse with Kiss1r heterozygous mouse (Kiss1r+/−). Kiss1r heterozygosity attenuated breast tumor initiation, growth, latency, multiplicity and metastasis in MMTV-PyMT/Kiss1r+/− mouse models. To confirm the effects of Kiss1r in tumor progression and limit any effect of endogenous hormones, we isolated primary tumor cells from PyMT/Kiss1r+/+ or PyMT/Kiss1r+/− mice and performed in vitro and in vivo tumorigenesis assays. Kiss1r heterozygosity inhibited PyMT-induced in vitro tumorigeneity and in vivo tumor growth in NOD.SCID/NCr mice. To understand the underlying mechanism, we showed that activation of KISS1R by kisspeptin-10 led to RhoA activation and RhoA-dependent gene expression through Gαq-p63RhoGEF signaling pathway. Furthermore, anchorage-independent growth was tightly linked to the dosage-dependent regulation of RhoA by KISS1R. When MCF10A cells overexpressing H-RasV12 were subjected to in vitro tumorigenesis assays, knockdown of KISS1R or inactivation of RhoA in MCF10A cells reduced Ras-induced anchorage-independent growth, similar to our data obtained from PyMT-Kiss1r+/− mouse models. Altogether, we conclude that Kiss1r haploinsufficiency delays breast tumor initiation, progression and metastasis through its downstream Gαq-p63RhoGEF-RhoA signaling pathway.
Keywords: Kiss1, Kiss1r, GPR54, GEFT/p63RhoGEF, RhoA, tumor progression, metastasis
Introduction
KISS1 receptor (KISS1R, also named GPR54) coupled to its endogenous ligands, kisspeptins (KISS1 gene product), has been revealed to suppress cancer metastasis and to play a pivotal role for the onset of puberty (1–7). Recently, we and others found that kisspeptins regulated cell proliferation, migration, and invasion in different cell line models via KISS1 receptor/GPR54 (8–10). In pubertal development, Kiss1 was revealed as a phenocopy of Kiss1r, since in knockout mouse models where Kiss1 or Kiss1r was deleted, similar phenotypes were observed in both Kiss1 and Kiss1r knockout mice (1, 3, 11). In cancer, various experimental and clinical studies have shown that kisspeptins could suppress cancer metastasis (12). We recently found that KISS1 gene located at chromosome 1q32 was regulated by genes at chromosome 6q21-32, which was often lost in metastatic breast cancer and melanoma (9, 13). Notably, genomic studies have revealed a gain of chromosome 1q32 in primary breast cancer and loss of chromosome 6q21-32 in aggressive breast cancer (14–21). Those genomic studies with recent findings suggest that KISS1 and KISS1R expression may be increased at the early stage of tumor development. Earlier studies focused on KISS1 function in cancer metastasis on the basis of KISS1 loss in metastatic cancer, and revealed a role of kisspeptin-activated KISS1R signaling (hereafter, KISS1/KISS1R signaling) for metastasis suppression. However, a role of endogenous KISS1/KISS1R signaling in early stages of cancer progression is still unclear, while it has been shown that the expression of KISS1 and KISS1R was higher in non-aggressive cancer than in normal and/or metastatic cancer (9, 13, 22–24).
MMTV (mouse mammary tumor virus)-PyMT (polyoma virus middle T antigen) mouse model is widely used to investigate a relationship between human and mouse breast cancer development and metastasis (25, 26). The MMTV-PyMT mouse model is time-saving for investigating tumor progression, since PyMT-induced hyperplasia is usually detected as early as the onset of puberty at 3 weeks and aggressive carcinoma with lung metastasis is found at 11 weeks (25, 26). An absence of mammary gland development in Kiss1- or Kiss1r-deficient female mice was closely linked to the absence of central Kiss1/Kiss1r signaling for the onset of puberty (1, 3, 11). However, the heterozygous mice for Kiss1 or Kiss1r did not cause any defects in pubertal development, including postnatal mammary gland development (1, 3, 11), suggesting that Kiss1r heterozygous condition in MMTV-PyMT mouse might be a good model to understand breast-restricted Kiss1/Kiss1r signaling in the early stage of breast cancer development.
In this study, we found that Kiss1r heterozygosity delayed PyMT-induced breast cancer development and metastasis. Notably, Kiss1r heterozygosity (Kiss1r+/−) attenuated breast tumor initiation, tumor growth, latency, multiplicity and metastasis induced in MMTV-PyMT/Kiss1r mouse models. Kiss1 or Kiss1r silencing in pubertal breast epithelia confirmed that Kiss1/Kiss1r signaling in breast epithelial cells was sufficient for breast hyperplasia. To limit any effect of endogenous hormones, we isolated mouse primary breast cancer cells from MMTV-PyMT/Kiss1r+/+ and MMTV-PyMT/Kiss1r+/− mice and examined the tumorigenesis in vitro and in vivo. We found that Kiss1r heterozygosity (Kiss1r+/−) attenuated breast tumor growth when tumor cells were orthotopically injected into NOD.SCID/NCr mice. To understand the molecular mechanism of Kiss1r regulation of tumorigenesis, we further determined that dosage-dependent regulation of RhoA activity by KISS1R was critical for Ras-induced tumorigeneity. In summary, our study suggests that autocrine Kiss1/Kiss1r signaling sufficiently regulates breast tumor initiation and progression by activating Gαq-p63RhoGEF-RhoA signaling pathway.
Materials and methods
Animal Studies
Transgenic FVB/N mice expressing the polyoma middle T antigen under the control of MMTV long terminal repeat promoter (MMTV-PyMT) were received from Dr. Jeffrey M. Rosen and Dr. Jianming Xu at Baylor College of Medicine (Houston, TX). All matings were performed with male mice heterozygous for the PyMT transgene and female C57BL/6 Kiss1r+/− mice received from Dr. Eric L. Gustafson at Schering-Plough Research Institute (Kenilworth, NJ). Genotyping for the PyMT transgene was performed according to the protocol of Jackson Laboratories (Bar Harbor, ME). All mice analyzed in this study were virgin females. Animal protocols used for this study were approved by the IACUC, Texas A&M Health Science Center.
In vivo studies
Mice were observed three times a week for mammary tumors by eye examination and finger palpation. Tumor length (L) and width (W) were measured once a week by a caliper and tumor volume was estimated by the formula, (W2 × L) / 2. Mice were euthanized at different stages of mammary tumorigenesis, and their mammary glands and tumors were collected for morphologic and biochemical analyses. For the whole-mount staining, the fourth inguinal mammary fat pads were excised, fixed with ethanol, and then stained with carmine alum. For histological study, tissues were formalin fixed for 24 hrs, and embedded in paraffin. Tissues were cut at 5μm in width, stained with hematoxylin and eosin, and evaluated by three different observers. Analyses and descriptions were followed in accordance with the guidelines in the NIH mammary gland pathology. Anti-Kiss1 and anti-Kiss1r antibodies from Santa Cruz Biotechnology (Santa Cruz, CA) were used for the immunohistochemistry. HistoStain® Plus Broad Spectrum (DAB) (Invitrogen, Camarillo, CA) and Vectorstain ABC kit with Vector® NovaRED™ Substrate kit (Vector Laboratories, Burlingame, CA) were used. Lentiviral plasmids were obtained from Open Biosystems (Huntsville, AL). At two weeks of age, 4th mammary glands were injected with lentiviruses through the nipple. At 5 week, mice were sacrificed, and mammary glands were stained and counted for hyperplastic nodules or lyzed for Western blot. Primary tumors cells were isolated from breast tumors in 11 week-old PyMT/Kiss1r+/+ and PyMT/Kiss1r+/− female mice. For the orthotopic injection of primary tumor cells, 1×106 cells were mixed with 0.1% matrigel and then injected into the left 4th mammary fat pad of NOD.SCID/NCr mouse (NCI-Frederick).
In vitro studies
Primary tumor cells were cultured in DMEM/F12 medium supplemented with 5% horse serum, 1% streptomycin/penicillin, 20ng/ml EGF, 0.5mg/ml hydrocortisone, 100ng/ml cholera toxin, 10μg/ml insulin at first day, and then in DMEM medium. To examine cell proliferation, cells were manually counted every day until 12 days. In order to examine cell migration, Boyden chamber assays were used. For colony formation assays, cells were cultured in 0.35% soft agar for 2 weeks and then colonies were stained with crystal violet. MCF10A cells were cultured in DMEM/F12 medium supplemented with 5% horse serum, 1% streptomycin/penicillin, 20ng/ml EGF, 0.5mg/ml hydrocortisone, 100ng/ml cholera toxin, 10μg/ml insulin. Mammary tumors from PyMT/Kiss1r+/+ and PyMT/Kiss1r+/− mice at the indicated time points were isolated and RNAs were extracted. For quantification, real-time PCR analyses were performed using SYBR green reagents in Prism 7300 a real-time PCR machine (Applied Biosystems, Foster City, CA), and ΔΔCt values were calculated. Gapdh as a reference was used for normalization. Anti-KiSS1, KiSS1R, RhoA, Cdc42, Rac1, Ras, and Actin antibodies were used for the Western blot (Santa Cruz Biotechnology, Santa Cruz, CA). For the Rho GTPases activity was performed using pull-down assays with GST-PAK PBD or GST-Rhotekin RBD, respectively. To detect active Rho GTPases, the appropriate antibody was used. For the luciferase assay, luciferase assay kit was used according to the manufacture’s protocol (Promega, Madison, MI). Cells were transfected with pSRF-luc or pSRE-luc for 6hrs to 24hrs and then subjected to the luciferase activity.
Statistics
Statistics were done using MedCalc (Medcalc Software, Mariakerke, Belgium). All statistics are two-sided. One-way analysis of variance and student’s t test were appropriately done. Tumor-free survival curves were performed by Kaplan-Meier method and compared with log-rank test. All data were expressed as means with 95% confidential intervals. A P value < .05 was considered as statistical significance.
Results
Kiss1r heterozygosity delays mouse breast tumor development and lung metastasis
To investigate the roles of Kiss1 receptor (Kiss1r) in breast tumor development and metastasis in vivo, we generated the MMTV-PyMT/Kiss1r mouse models. As described in the materials and methods section, we obtained PyMT/Kiss1r wild type (PyMT/Kiss1r+/+), heterozygous (PyMT/Kiss1r+/−), and Kiss1r null (PyMT/Kiss1r−/−) mice for our analysis of Kiss1r in tumor progression and metastasis. For breast tumor development, we examined palpable tumors in different groups of mouse models, including PyMT/Kiss1r+/+ (n = 30), PyMT/Kiss1r+/− (n = 50), and PyMT/Kiss1r−/− mice (n = 20) until 16 weeks. As shown in the Kaplan-Meier plot, PyMT/Kiss1r+/− mice in comparison with PyMT/Kiss1r+/+ mice showed a delay of breast tumor development as measured by the percentage of tumor-free mice (Fig. 1A). In PyMT/Kiss1r+/− mice, 50% of mice developed tumors in 11 weeks compared to 7 weeks in PyMT.Kiss1r+/+ mice (P < .0001), whereas PyMT/Kiss1r−/− mice generated no palpable tumor (Fig. 1A). Thus, our data indicated that Kiss1r heterozygosity affected tumor latency and Kiss1r is essential for breast tumor development.
Figure 1. Kiss1r heterozygosity affects mammary tumor latency, multiplicity, growth and lung metastasis.
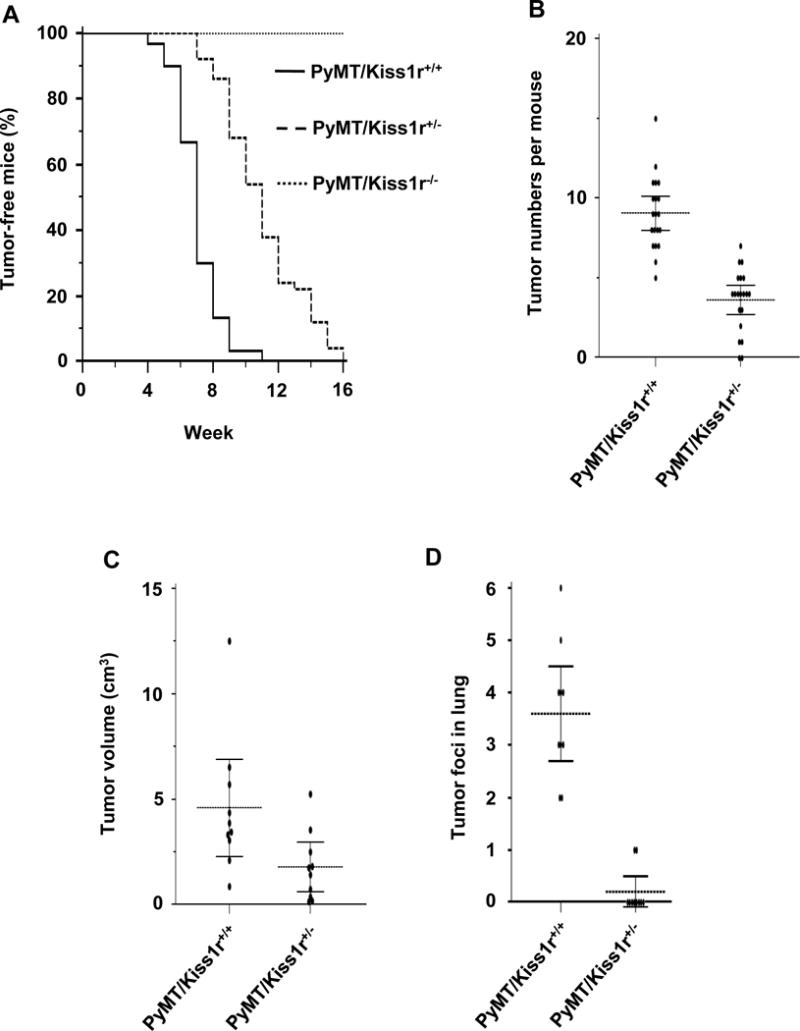
(A) Kaplan-Meier plot for palpable tumors until 16 weeks. (B) Breast tumor numbers in different mammary fat pads between PyMT-Kiss1r wild type (Kiss1r+/+) and PyMT-Kiss1r+/− (Kiss1r+/−). (C) The volume of primary tumors in 11 week-old mice. (D) Metastatic tumor foci number in lung tissues at 11 weeks of age in wild-type (Kiss1r+/+) and heterozygosity mice (Kiss1r+/−).
Meanwhile, pubertal phenotypes such as vaginal opening at the precise time schedule showed no differences between PyMT/Kiss1r+/+ and PyMT/Kiss1r+/− mice, which was consistent with previous reports that Kiss1r (as well as Kiss1) heterozygous condition did not significantly alter the level of sexual hormones and does not affect pubertal development (1, 11). Lack of tumor development in PyMT/Kiss1r−/− mice seemed to suggest that hypothalamic Kiss1/Kiss1r signaling regulate postnatal mammary development, which was consistent with previous findings that Kiss1r knockout mice did not have postnatal mammary gland development and that PyMT-induced tumors arose at the time of pubertal mammary gland development (1, 11, 26). Altogether, Kiss1r heterozygosity resulted in the haploinsufficiency for PyMT-induced mammary tumor development, which was not due to the pubertal defect of mammary gland development.
We next counted the number of mammary tumors in different mammary fat pads of each mouse at 11 weeks. Kiss1r heterozygosity (Kiss1r+/−) reduced the number of tumors per mouse (Fig. 1B; n = 20 per group, P < .00001). Thus, Kiss1r heterozygosity further affected mammary tumor multiplicity. We then measured tumor burden at 15 weeks to analyze whether Kiss1r heterozygosity further affected tumor growth. Tumor volume in PyMT/Kiss1r+/− mice was smaller than that in PyMT/Kiss1r+/+ mice (Fig. 1C; n = 10 per group, P = .029). Therefore, our data indicate that Kiss1r haploinsufficiency attenuates breast tumor latency and multiplicity, and further affects tumor growth.
PyMT-induced breast tumors primarily metastasize to lung, which is normally detected at 11 weeks of age (25, 26). To investigate whether Kiss1r heterozygosity affected breast tumor metastatic growth at lung, we prepared lung tissue sections and counted metastatic tumor foci in PyMT/Kiss1r+/+ and PyMT/Kiss1r+/− mice. At 11 weeks of age, the number of tumor foci found in PyMT/Kiss1r+/+ lung was significantly higher than that in PyMT/Kiss1r+/− lung (Fig. 1D; n = 10 per group, P < .0001). Thus, our data indicate that Kiss1r haploinsufficiency further affected breast tumor lung metastasis.
Kiss1r heterozygosity delays the incidence of mouse breast hyperplasia, tumor formation, and malignancy
As Kiss1r heterozygosity affected tumor latency, multiplicity, and growth, we next examined whether those results were due to a delay of tumor incidence. To analyze mammary tumor incidence, we prepared whole-mounting analysis on the fourth inguinal mammary fat pads at the age of 5 to 9 weeks since whole mounting preparations were preferred to address premalignant lesions of mammary glands, termed mammary intraepithelial neoplasia (MIN) or hyperplastic atypia that results in mammary tumors. When hyperplastic nodules in the 4th inguinal mammary fat pads of PyMT/Kiss1r+/+ (n = 20), PyMT/Kiss1r+/− (n = 21) and PyMT/Kiss1r−/− (n = 11) mice at ages from 5 to 9 weeks were counted, respectively, we found that the total number of hyperplastic nodules in the 4th inguinal mammary fat pad of PyMT/Kiss1r+/− mice was much less than that in the PyMT/Kiss1r+/+ mice (Fig. 2A and 2B; P < .0001). In addition, PyMT/Kiss1r−/− did not develop hyperplastic mammary gland as a result of the defective postnatal mammary gland development (Fig. 2B; P < .0001). At 5 weeks, mammary ductal hyperplasia in PyMT/Kiss1r+/− mice was much less severe than that in PyMT/Kiss1r+/+ mice (Fig. 2A, inboxes in second panels). At 7 to 9 weeks, tumor sizes was smaller in PyMT/Kiss1r+/− mice than that in PyMT/Kiss1r+/+ mice (Figure 2A, arrows in third and forth panels). Therefore, our data indicate that Kiss1r heterozygosity affects tumor incidence and tumor formation.
Figure 2. Kiss1r heterozygosity attenuates the incidence of mammary hyperplasia, solid tumor formation and tumor malignance.
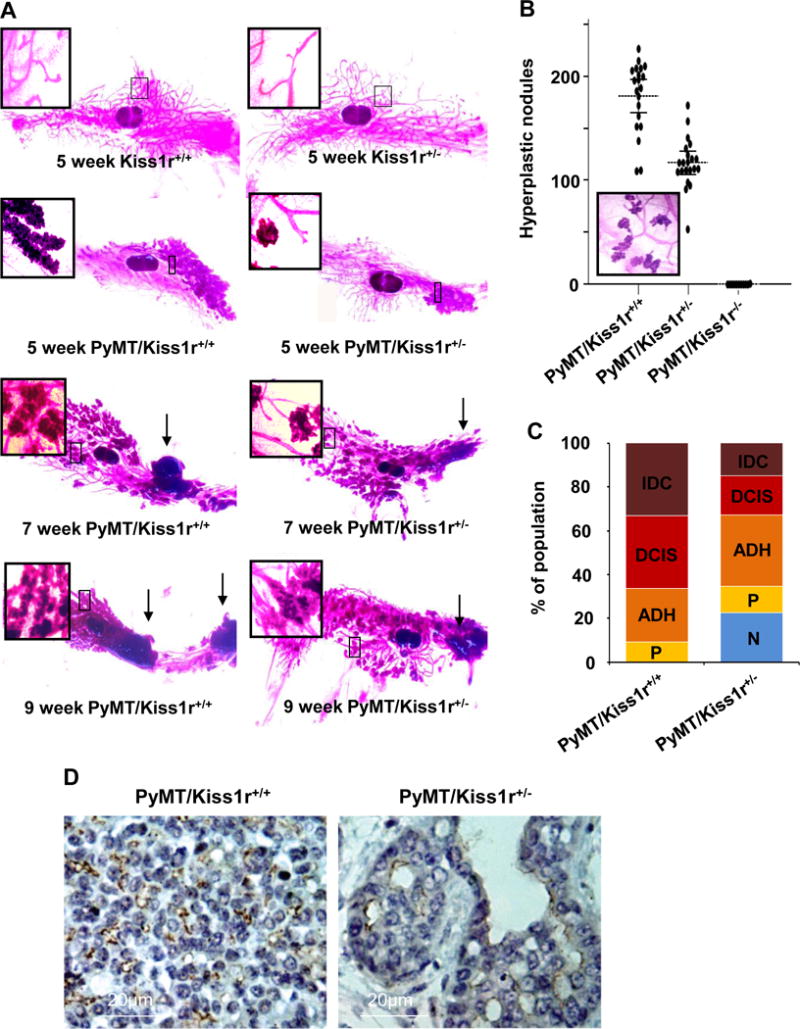
(A) Primary tumor development in 4th inguinal mammary fat pad from 5 to 9 weeks of age. Arrows indicate primary tumors. (B) Hyperplastic nodules (the inbox) at 5 weeks of age. Inbox represents the hyperplastic nodule image. (C) Tumor malignancy in PyMT/Kiss1r+/+ (wild type) and PyMT/Kiss1r+/− (heterozygous mice). (D) Immunohistochemistry for Kiss1r in tumor tissues.
To investigate whether Kiss1r heterozygosity further affects tumor malignancy, we analyzed tumor cohorts in 11 week-old PyMT/Kiss1r+/+ and PyMT/Kiss1r+/− mice (n = 7 per group). Tumor cohorts were more complex and advanced in the histology samples of PyMT/Kiss1r+/+ mice than those in PyMT/Kiss1r+/− mice (Fig. 2C). Normal mammary gland (N; terminal ductal lobular unit: TDLU) was not found in PyMT/Kiss1r+/+ mice, but approximately 22% of N was detected in PyMT/Kiss1r+/− mice (Fig. 2C; P < .0001). There was no significant difference in distribution of papilloma (P) and atypical ductal hyperplasia (ADH) between PyMT/Kiss1r+/− tumor cohort and PyMT/Kiss1r+/+ tumor cohort. However, Kiss1r heterozygosity markedly delayed mammary tumor progression with a decrease of ductal carcinoma in situ (DCIS) by approximately 20% (Fig. 2C; P = 0.0068). Moreover, the amount of invasive ductal carcinoma (IDC) in PyMT/Kiss1r+/− mice was significantly lower than those in PyMT/Kiss1r+/+ mice, amounting to an approximate 14% in 11 week-old PyMT/Kiss1r+/− mice compared to an approximate 37% in 11 week-old PyMT/Kiss1r+/+ mice (Fig. 2C; P = 0.0213). Thus, our data indicate that Kiss1r heterozygosity delays tumor malignancy and progression.
We next examined an expression of Kiss1r in tumor tissues using the immunohistochemistry. Compared to PyMT/Kiss1r+/− tumor, PyMT/Kiss1r+/+ tumor expressed higher Kiss1r (Fig. 2D). Although Kiss1r was detected in fibroblast and endothelium, both Kiss1 and Kiss1r were found in luminal epithelial tumor cells (Fig. 2D), suggesting that Kiss1/Kiss1r signaling in an autocrine manner.
Autocrine Kiss1/Kiss1r signaling is sufficient for mouse breast tumor initiation
To investigate whether Kiss1 and Kiss1r locally function for hyperplasia, lentiviruses for shRNAs specific for Kiss1 and Kiss1r were injected into 4th nipples at 2 weeks of age prior to pubertal mammary development. Mice were sacrificed at 5 weeks of age when hyperplasia was developed. Lentiviral knockdown strategies repressed the expression levels of either Kiss1 or Kiss1r in breast epithelia (Fig. 3A and B). Compared to the silencing with control shRNA (scr), both Kiss1 and Kiss1r silencing in breast epithelia caused a significant decrease of hyperplastic nodule number (Fig. 3B and 3C; n = 5 per group, Kiss1 shRNA vs. scr shRNA, P = 0.0062; Kiss1r shRNA vs. scr shRNA, P = 0.0097), supporting that local Kiss1 and Kiss1 receptor (Kiss1r) are involved in breast tissue hyperplasia and tumor initiation. Data from knockdown of Kiss1 or Kiss1r were consistent with our data from Kiss1r heterozygosity, which indicates that autocrine Kiss1/Kiss1r signaling in breast epithelial cells play a critical role in breast tumor development.
Figure 3. Autocrine Kiss1/Kiss1r signaling is sufficient for breast tissue hyperplasia.
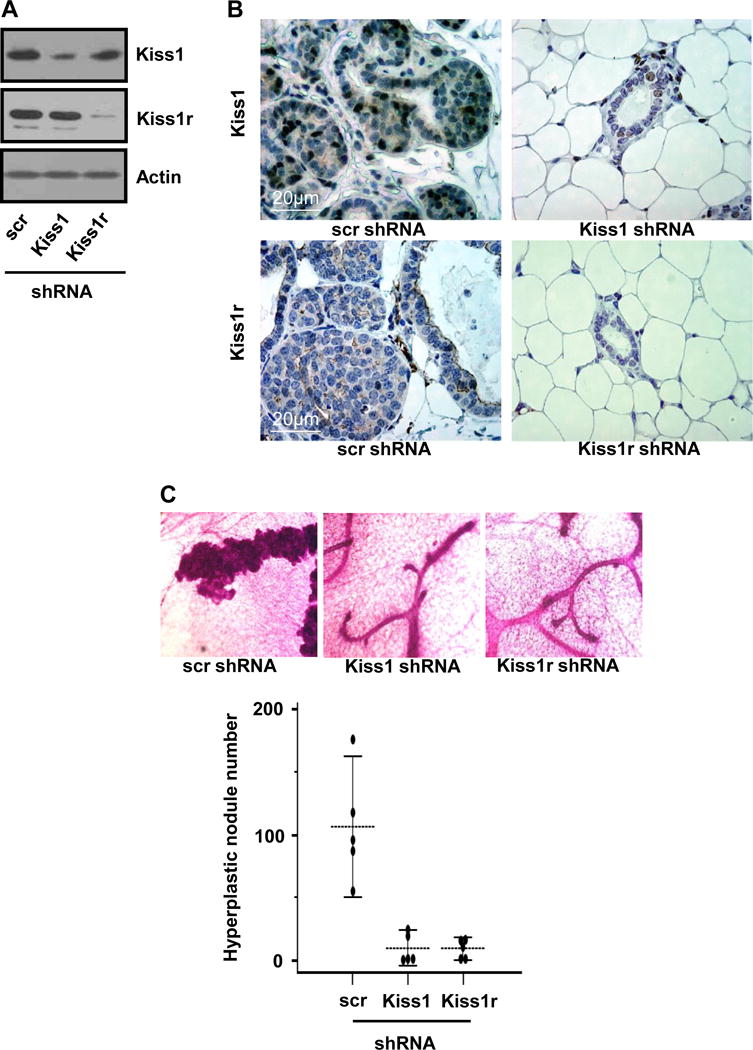
(A) Expression of Kiss1 and Kiss1r in mammary glands injected with lentiviral shRNA for scr (control), Kiss1 or Kiss1r, showing down-regulation of Kiss1 and Kiss1r in mammary glands. (B) Immunohistochemistry staining for breast tissues infected with lentivial shRNA for scr, Kiss1 or Kiss1r. (C) Hyperplastic nodule numbers in mammary glands infected with different shRNAs for control, Kiss1 and Kiss1r, respectively. The representative images were shown in the top of the figure.
Kiss1r haploinsufficiency affects tumorigenesis in isolated primary tumor cells and orthotopic mouse tumor models
To investigate the function of Kiss1/Kiss1r signaling in breast tumor cells, we isolated breast tumor cells from tumor burdens in 11 week-old PyMT/Kiss1r+/+ and PyMT/Kiss1r+/− mice and examined the expression of Kiss1 and Kiss1r, respectively. As shown in figure 4A, PyMT/Kiss1r+/− primary tumor cells showed a significant reduction of Kiss1r expression compared with that in PyMT/Kiss1r+/+ primary tumor cells. On the other hand, no significant alteration of Kiss1 expression was detected (Fig. 4A). To examine whether Kiss1r heterozygosity affects cell growth, primary tumor cells isolated from PyMT/Kiss1r+/+ and PyMT/Kiss1r+/− mice were cultured until 12 days and the number of cells was counted every day to calculate the doubling time. Doubling time for PyMT/Kiss1r+/+ and PyMT/Kiss1r+/− primary tumor cells was 29.33 hr and 31.76 hr, respectively (Fig. 4A; n = 3 per group, P = 0.0007), suggesting that Kiss1r heterozygosity negatively regulates mammary tumor cell proliferation.
Figure 4. Kiss1/Kiss1r signaling regulates tumorigeneity.
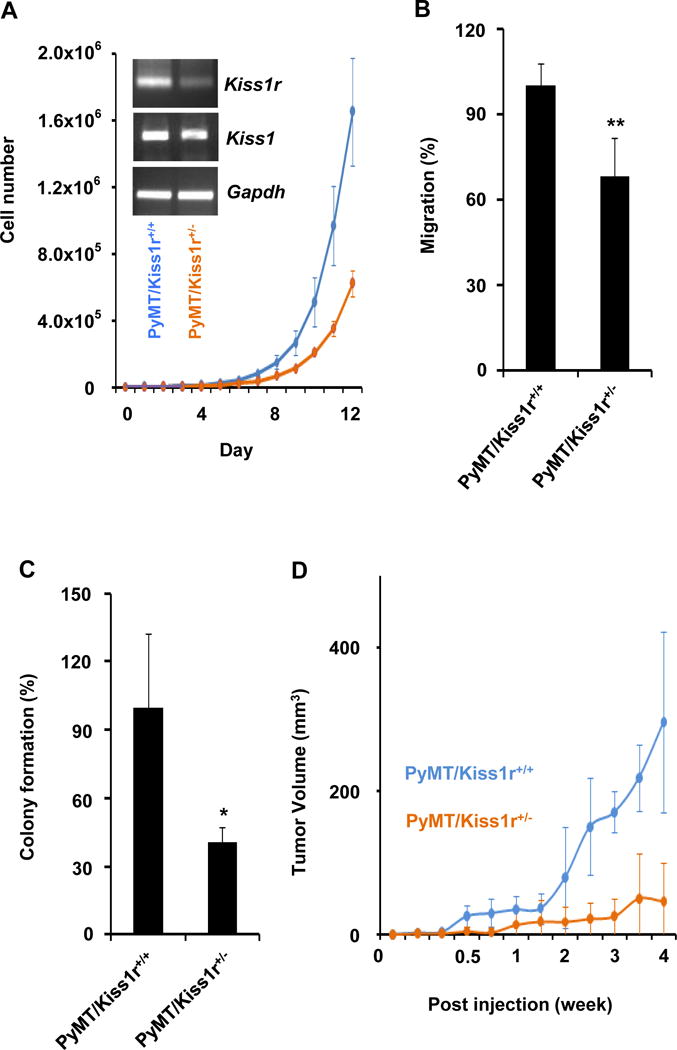
(A–C) Kiss1r heterozygosity suppressed primary tumor cell growth (A), cell migration and invasion (B), and anchorage-independent colony formation (C). (A- insert) Kiss1r and Kiss1 mRNA level in primary tumor cells isolated from the PyMT/Kiss1r+/+ and PyMT/Kiss1r+/− mice. Gapdh used as the internal control of PCR reaction. (D) Kiss1r heterozygosity significantly inhibits orthotopic tumor growth in xenograft mouse tumor model using primary tumor cells isolated from the PyMT/Kiss1r+/+ and PyMT/Kiss1r+/− mouse models, respectively.
Since Kiss1/Kiss1r signaling was implicated in cell motility and invasion, we next investigated cell migration of PyMT/Kiss1r+/+ and PyMT/Kiss1r+/− primary tumor cells. Cells were cultured on Boyden chamber precoated with Matrigel and then the number of cells passed through the Matrigel was counted after 24hr. The invasion capacity of PyMT/Kiss1r+/+ cells was approximately 31% higher than that of PyMT/Kiss1r+/− cells (Fig. 4B; n = 4 per group, P = .0099), indicating that Kiss1r heterozygosity affects tumor cell motility and invasion.
Next, we performed the anchorage-independent colony formation assays to examine whether Kiss1r heterozygosity in mammary tumor cells affects tumorigeneity. Primary tumor cells isolated from PyMT/Kiss1r+/+ and PyMT/Kiss1r+/− mice were cultured in soft agar for 20 days and then the number of colonies were counted. Compared to PyMT/Kiss1r+/+ cells, the number of colonies formed by PyMT/Kiss1r+/− cells was reduced by approximately 59% (Fig. 4C; P = .00375). Thus, our data suggest that Kiss1r heterozygosity causes haploinsufficiency and negatively regulates breast tumorigeneity in vitro.
To further test that Kiss1r heterozygosity in breast epithelial tumor cells affects tumor growth in vivo, we performed orthotopic injection assays in NOD.SCID/NCr mice using primary cultured tumor cell lines isolated from PyMT/Kiss1r+/+ and PyMT/Kiss1r+/− mice, respectively. Tumor volume obtained from PyMT/Kiss1r+/− tumor cells in NOD.SCID/NCr mice was significantly smaller than those obtained from PyMT/Kiss1r+/+ tumor cells (Fig. 4D; n = 13 per group, P < 0.0001), indicating that Kiss1r haploinsufficiency in breast cancer cells significantly inhibited tumor growth in vivo.
Kiss1r sufficiently regulates tumorigeneity via RhoA
To understand the molecular mechanisms of Kiss1r signaling in the regulation of tumorigeneity, we next examined the activation of Rho GTPases in breast tumor cells since Rho GTPases are reported to regulate tumor progression and metastasis (27, 28). In PyMT/Kiss1r+/+ primary tumor cells, knockdown of Kiss1r expression significantly decreased RhoA activity in a dose-dependent manner but did not affect the activity of either Cdc42 or Rac1 (Fig. 5A). RhoA activity in PyMT/Kiss1r+/− tumor cells was also decreased when compared to that in PyMT/Kiss1r+/+ tumor cells (Fig. 5A), suggesting that Kiss1r signaling in breast tumor cells regulate the activation of RhoA GTPase. To link the finding to our previous observation that autocrine Kiss1/Kiss1r signaling affected tumorigeneity, we next performed the anchorage-independent growth assays. Dominant negative RhoA (RhoA DN; RhoAN17) decreased the anchorage-independent growth of PyMT/Kiss1r+/+ cells by approximately 47% (n = 6, RhoA DN vs. empty, P = .0001), but wild type and constitutively active RhoA (RhoA DA; RhoAV14) increased the anchorage-independent colony growth by approximately 29% and 35%, respectively (n= 6; RhoA WT vs. empty, P = .0146; RhoA DA vs. empty: RhoA DA vs. empty, P = .0045). Similar data were also observed in PyMT/Kiss1r+/− tumor cells although the colony number was reduced compared to wild type tumor cells (Fig. 5B; n = 6; empty/PyMT/Kiss1r+/− vs. empty/PyMT/Kiss1r+/+, P = .0004; RhoA DN/PyMT/Kiss1r+/− vs. empty/PyMT/Kiss1r+/−, P = .0003; RhoA WT/PyMT/Kiss1r+/− vs. empty/PyMT/Kiss1r+/−, P = .0003; RhoA DA/PyMT/Kiss1r+/− vs. empty/PyMT/Kiss1r+/−, P = .0001). Altogether, our data indicate that RhoA is a key protein downstream of Kiss1/Kiss1r signaling for breast tumorigeneity and tumor progression.
Figure 5. Kiss1/Kiss1r regulates tumorigenesis by activating RhoA.
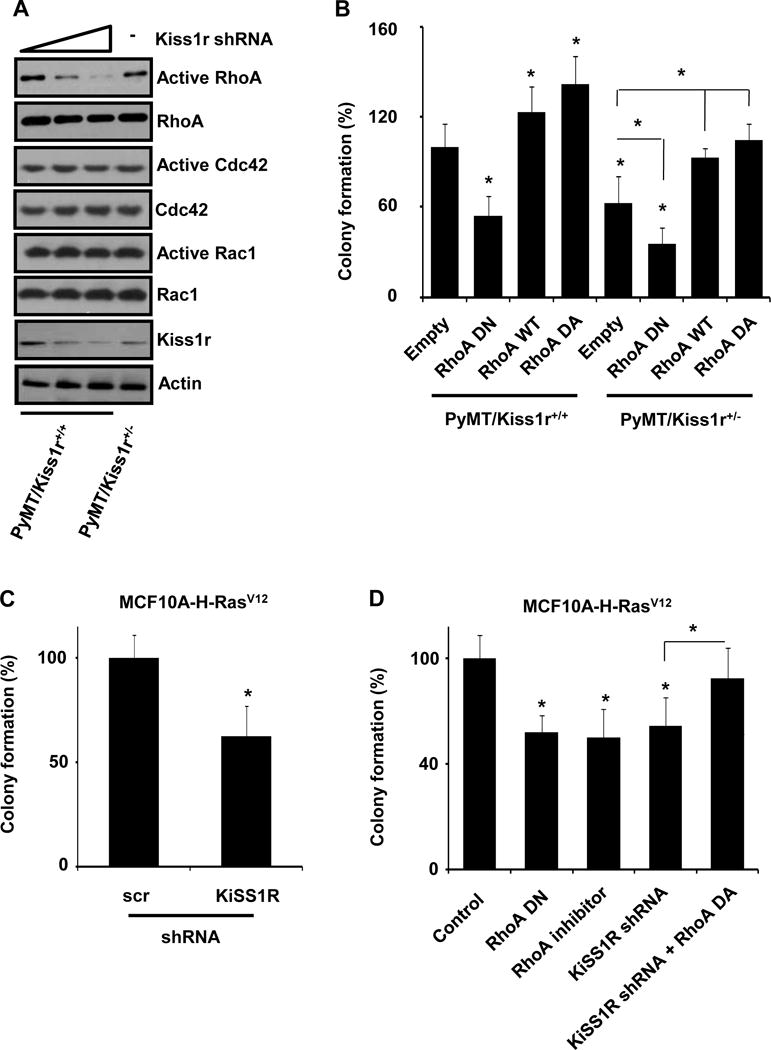
(A) Activation of Rho family of GTPases by suppressing Kiss1r expression. Kiss1r activates RhoA in a dose-dependent manner while Rac1 and Cdc42 are not affected by the downregulation of Kiss1r. (B) RhoA and its mutants regulate anchorage-independent colony formation in tumor cells isolated from PyMT/Kiss1r+/+ and PyMT/Kiss1r+/− mouse models. (C) Knockdown of KISS1R in Ras-transformed MCF10A cell inhibited anchorage-independent cell growth and colony formation. (D) Ras-induced anchorage-independent colony formation of MCF10A cells depends on RhoA signaling pathway using different RhoA plasmids or treated with RhoA inhibitor, Y-27632 (10μmol/L).
To confirm the roles of human KISS1R for tumorigeneity in human breast epithelium, we transformed MCF10A human normal breast epithelial cells by overexpressing the constitutively active H-Ras (H-RasV12) to induce tumorigenesis, as PyMT-induced tumorigenesis was known to require oncogenic activation of Ras (29, 30). As shown in figure 5C, overexpression of the active H-RasV12 transformed the MCF10A breast epithelial cells and induced anchorage-independent colony growth on soft agar. Knockdown of KISS1R using specific shRNA for human KISS1R reduced Ras-induced anchorage-independent colony formation by approximately 37% (Fig. 5C; n = 7, KISS1R shRNA vs. scr shRNA, P = .0002), suggesting that human KISS1R plays a key role in Ras-induced MCF10A cell tumorigenesis. We further examined whether KISS1R-mediated RhoA activation was involved in Ras-induced tumorigenesis using the anchorage-independent colony growth assays. Inactive mutant of RhoA (RhoA DN) decreased Ras-induced colony formation of MCF10A (Fig. 5D; n = 7, RhoA DN vs. control, P < .0001). In addition, RhoA inhibitor (Y27632) blocked Ras-induced tumorigenesis in MCF10A cells that express RhoA, indicating that Ras-induced tumorigenesis requires RhoA activation (Fig. 5D; n = 7, RhoA inhibitor vs. control, P = .0001). On the other hand, dominant active form of RhoA (RhoA DA) recovered the inhibitory effect of KISS1R deficiency (knockdown) in Ras-induced tumorigenesis (n = 7, RhoA DA + KiSS1R shRNA vs. KiSS1R shRNA, P = .0097), suggesting that RhoA is the key downstream target of KISS1R in Ras-induced MCF10A cell tumorigenesis. Together, our data indicate that KISS1R-mediated RhoA activation is important for Ras-induced tumorigenesis, similar to our results found in the PyMT/Kiss1r mouse tumor models.
Kisspeptin activates RhoA-mediated transcription through KISS1R-Gαq-p63RhoGEF
To delineate KISS1/KISS1R signaling to RhoA, we performed RhoA activity assays when cells were serum-starved for 24hr and then incubated for 6hr in the absence or presence of kisspeptin-10 (Kp10, 100nM). Kisspeptin-10 (100nM) increased GTP-bound RhoA in HEK293 cells overexpressing KISS1R (Fig. 6A). Since serum response factor (SRF) transcriptional activity is widely used for the readout of RhoA GTPase activation (31–33), we performed the luciferase assays for SRF-dependent promoter activity as the readout of RhoA activation. Kisspeptin-10 increased SRF-luciferase activity in cells transfected with KISS1R (Kisspeptin-10 in KiSS1R cells vs. none in KiSS1R cells, P = 0.0309) but not with empty vector (Fig. 6B). Consistently, kisspeptin-10 also activated serum response element (SRE)-luciferase activity via KISS1R (data not shown). Therefore, our data indicate that KISS1/KISS1R signaling directly activates RhoA.
Figure 6. KiSS1/KiSS1R regulates RhoA-dependent gene expression through Gaq-p63RhoGEF-RhoA signaling.
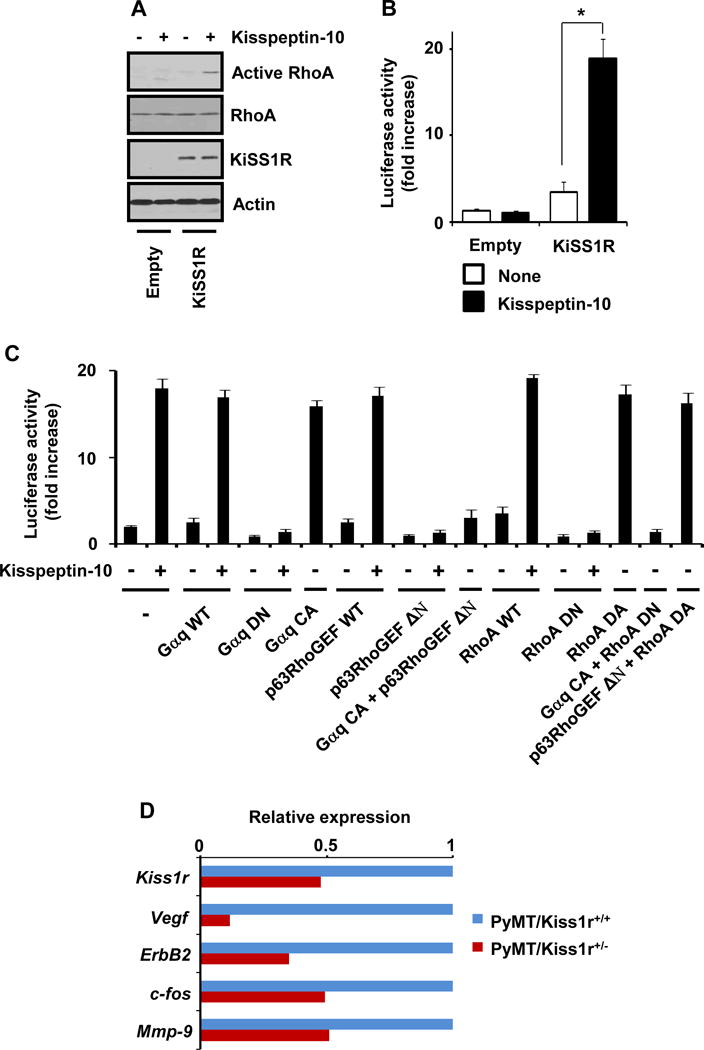
(A) Kisspeptin-10 activation of RhoA is dependent on the expression of KiSS1R. (B) Activation of SRF-dependent transcriptional activity by Kisspeptin-KiSS1R. (C) Activation of SRF-dependent transcriptional activity by Gαq, p63RhoGEF and RhoA signaling pathway as indicated conditions. (D) Regulation of gene expression levels in Kiss1r heterozygosity mouse models.
We next investigated whether KISS1/KISS1R signaling increases SRF transcriptional activity through RhoA activation. Dominant active form of RhoA (RhoA DA) highly increased SRF transcriptional activity in HEK293 cells stably expressing KISS1R (RhoA DA vs. empty, P = .0027). RhoA wild type (RhoA WT) has little effect on the basal SRF transcriptional activity (RhoA WT plus kisspeptin-10 vs. RhoA WT, P = .0002) (Fig. 6C, 13th, 14th, and 17th bars). A dominant negative mutant of RhoA (RhoA DN) suppressed kisspeptin-10 activation of SRF transcriptional activity (Fig. 6C, 15th and 16th bars). Thus, our data indicate that KISS1/KISS1R-mediated SRF activation requires RhoA activation.
To understand how KISS1 receptor activates RhoA, we examined whether the activation of RhoA was mediated by Gαq, as it was revealed that KISS1R selectively coupled to Gαq upon kisspeptin stimulus (1, 4, 8, 34). In HEK293 cells stably transfected with KISS1R, kisspeptin-10 stimulation or constitutively active mutant of Gαq (Gαq CA) induced SRF transcriptional activity (Gαq CA vs. empty, P = .0015; Gαq WT plus kisspeptin-10 vs. Gαq WT, P = .0003), while Gαq wild type (Gαq WT) alone did not affect SRF transcriptional activity (Fig. 6C, 3st, 4nd, and 7th bars). A dominant negative mutant of Gαq (Gαq DN) attenuated kisspeptin-10-induced SRF transcriptional activation (Fig. 6C, 5rd and 6th bars). Thus, our data suggest that the activation of RhoA by kisspeptin-10 is mediated by KiSS1R-Gαq signaling pathway.
It has been reported that Gαq activates RhoA via p63RhoGEF (also called GEFT) (35–37), we further examined the Gαq coupling to p63RhoGEF. In KISS1R-overexpressing cells, p63RhoGEF wild type (p63RhoGEF WT) did not affect basal or kisspeptin-10-induced SRE transcriptional activity (p63RhoGEF WT plus kisspeptin-10 vs. p63RhoGEF WT, P =.0003), but catalytic domain-deleted mutant of p63RhoGEF (p63RhoGEF ΔN) inhibited kisspeptin-10 activation of SRF transcriptional activity (Fig. 6C, 8th to 11th bars). Furthermore, p63RhoGEF ΔN suppressed SRF transcriptional activation by Gαq CA (Fig. 6C, 12th bar; Gαq CA vs. p63RhoGEF ΔN plus Gαq CA, P = .0001), indicating that p63RhoGEF is downstream of Gαq in the activation of KISS1R. We further examined whether KISS1/KISS1R signaling activated RhoA-dependent SRF transcriptional activity through Gαq-p63RhoGEF. RhoA DN inhibited Gαq CA-induced SRF transcriptional activation (Gαq CA vs. RhoA DN plus Gαq CA, P = .0001), and p63RhoGEF ΔN did not inhibit RhoA DA-increased SRF transcriptional activity (Fig. 6C, 18th and 19th bars). Therefore, our data indicate that KiSS1/KiSS1R signaling activates Gαq-p63RhoGEF-RhoA signaling pathway.
Finally, we examined whether Kiss1r heterozygosity regulated RhoA-dependent gene expression in tumor progression using real-time PCR assays and MatInspector software (38). Among genes of which promoter region contained SRE, we found that the expression levels of Vegf, ErbB2, c-fos, and Mmp-9 in PyMT/Kiss1r+/− tumor compared to that in PyMT/Kiss1r+/+ tumor was significantly reduced (Fig. 6D), indicating that Kiss1r heterozygosity selectively affected RhoA-dependent expression level of key genes involved in cancer progression.
Discussion
Kiss1/Kiss1r signaling has been implicated in cancer metastasis, which was first revealed in xenograft tumor growth and experimental metastasis models (4, 6). However, its functional requirement in the early stages of cancer development was unclear. In this study, we demonstrated that mouse Kiss1r heterozygosity delayed breast cancer initiation, progression and metastasis in MMTV-PyMT mouse model system. In our PyMT/Kiss1r mouse models, Kiss1r heterozygosity (PyMT/Kiss1r+/−) diminished hyperplasia, resulting in the delay of tumor formation, progression, and lung metastasis. Furthermore, we confirmed that knockdown of Kiss1 and Kiss1r locally inhibited mammary gland hyperplasia and that the dosage-dependent regulation of breast tumor initiation by Kiss1r was connected to RhoA activity. Accordingly, we demonstrate that Kiss1/Kiss1r signaling in autocrine manner regulates early steps of tumor development through RhoA activation by coupling to Gαq-p63RhoGEF.
In xenograft mouse tumor model systems, KISS1 overexpression blocked cancer metastases without affecting tumorigenesis (4, 6, 7). However, xenograft mouse tumor model has limitations to understand KISS1/KISS1R signaling in early stages of tumor progression since it is not able to recapitulate some aspects of tumorigenesis (25). For example, tumor cells used in xenograft assays already have tumor characteristics, indicating that tumorigenic process is not recapitulated in xenograft assay. Furthermore, the environment where tumor cells are injected is quite different from original environment where tumor is arising, since xenograft assay is usually performed in dorsal skin. Moreover, data from human cancer patients have consistently suggested that the roles of KISS1/KISS1R signaling might not be simple in metastasis because both KISS1 and KISS1R were comparably expressed in normal and/or benign tissues (12). While KISS1/KISS1R signaling inhibited metastasis in xenograft tumor and experimental metastasis mouse model systems, our MMTV-PyMT mouse model system showed that silencing of Kiss1 or Kiss1r as well as Kiss1r heterozygosity in breast epithelial cells attenuated PyMT-induced breast tumor initiation and development. Our ovarectomy study showed that mammary Kiss1r did not affect mammary gland development (data not shown). Thus, in our present study, Kiss1/Kiss1r signaling was selective for PyMT-induced mammary tumor initiation. Meanwhile, our data showed that KISS1/KISS1R signaling seems to be required for tumor cell growth and motility, as knockdown of KISS1 or KISS1R as well as Kiss1r heterozygosity reduced cell growth and motility. Therefore, KISS1/KISS1R signaling may form the fine-tune network in the cells and during tumor progression.
Our study further identified that KiSS1/KiSS1R regulated breast tumorigenesis via the activation of RhoA by Gαq-p63RhoGEF signaling pathway. Somatic mutation of Gαq (GNAQ) at codon 209 was frequently found in uveal melanoma and blue naevi (39, 40); thereby constitutively active mutation of Gαq seems to be involved in melanoma progression. Recent studies delineated that p63RhoGEF linked the activation of Gαq and RhoA (36, 37). Our lab also revealed that p63RhoGEF overexpression induced NIH3T3 transformation (35). RhoA upstream of Cdc42 and Rac1 in regulating dynamics of cell growth and motility is known to affect preneoplastic transformation in primary mammary epithelial cells (41). Consistently, RasV12 required ROCK and Cdc42 for apical growth in Madin-Darby canine kidney (MDCK) epithelial cells, indicating that RhoA-ROCK pathway is critical for tumorigenic cell growth (42). Thus, KiSS1/KiSS1R signaling to RhoA is likely to control the growth of tumorigenic cells. In addition, while RhoA promotes pulmonary metastasis of MDA-MB-231 metastatic human breast cancer cell line where endogenous KISS1/KISS1R signaling is absent (43), kisspeptin-10 treatment inhibits TNFalpha-induced RhoA activation in the same cell line (28). Thus, KiSS1/KiSS1R signaling to RhoA during breast cancer progression is probably stage-specific. However, KiSS1/KiSS1R signaling to RhoA is likely more complex in vivo since kisspeptin-10 inhibited cell migration and invasion in Caki-1 and ACHN renal cancer cells or caused apoptosis of Jurkat human leukemic cells by activating RhoA (27, 44). It is possible that the effect of KiSS1/KiSS1R signaling is also cell type-specific. As tumor microenvironment is complex and changed following cancer progression (45), a different combination of signaling inputs would result in diverse consequences (46).
Genetic alteration in the MMTV-PyMT model may change a predisposition of different breast cell lineages, which can affect particular stages of cancer progression (47–49). In our study, the autocrine Kiss1/Kiss1r signaling showed dosage-dependent regulation of mouse breast tumor initiation and progression. When we examined the expression patterns of various genes in PyMT/Kiss1r+/+ and PyMT/Kiss1r+/− tumors, Kiss1r heterozygosity affected the expression levels of various genes for tumor initiation and development, such as Sca-1 (data not shown). Thus, KiSS1/KiSS1R signaling is likely to alter the expression pattern of endogenous gene sets for tumor initiating-cell propagation. Meanwhile, gene expression studies for Kiss1 and Kiss1r during mouse cancer progression (data not shown) demonstrated that Kiss1/Kiss1r signaling might be upregulated in benign tumor and then downregulated in malignant tumor. Thus, endogenous KiSS1/KiSS1R signaling may sufficiently regulate tumorigenesis, and be sustained or enhanced during cancer progression, and then disappeared by mechanisms that we previously reported (9, 13). Thus, proper schedules for the usage of exogenous kisspeptin should consider the endogenous KiSS1/KiSS1R signaling in tumor progression since recent studies have shown that exogenous KiSS1/KiSSR1 signaling could suppress tumor angiogenesis as well as distant metastasis (7, 50). However, if KiSS1/KiSS1R signaling exhibits a tonic effect on tumor progression including tumorigenesis and metastasis, exceeding KiSS1/KiSS1R signaling by administrating kisspeptins may target any steps of cancer progression including metastasis through the net effect as mentioned above.
Altogether, we conclude that Kiss1r heterozygosity attenuates breast cancer initiation, progression and metastasis and that autocrine Kiss1/Kiss1r signaling via RhoA is sufficient for breast tumor development and progression.
Acknowledgments
We appreciate Dr. Eric L. Gustafson and Ms. Galya Vassileva at Schering-Plough Research Institute (Kenilworth, NJ) for their great help to provide the Gpr54+/− mouse (male and female) line. We thank Drs. Jianming Xu and Jeffrey M. Rosen at the Baylor College of Medicine (Houston, TX) for providing MMTV-PyMT mouse, and Dr. Steven H. Safe at the Texas A&M University (College Station, TX) and Dr. Bharat B. Aggarwal at UT-MDA (Houston, TX) for experimental materials. This work was supported by grants from National Cancer Institute (NIH) (ML: 1R01CA106479 and 5R01CA134731), and in part by a Predoctoral Award (W81XWH-05-1-0353 to SG C) from the Department of Defense Breast Cancer Program.
References
- 1.Seminara SB, Messager S, Chatzidaki EE, et al. The GPR54 gene as a regulator of puberty. N Engl J Med. 2003;349:1614–27. doi: 10.1056/NEJMoa035322. [DOI] [PubMed] [Google Scholar]
- 2.de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci U S A. 2003;100:10972–6. doi: 10.1073/pnas.1834399100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Funes S, Hedrick JA, Vassileva G, et al. The KiSS-1 receptor GPR54 is essential for the development of the murine reproductive system. Biochem Biophys Res Commun. 2003;312:1357–63. doi: 10.1016/j.bbrc.2003.11.066. [DOI] [PubMed] [Google Scholar]
- 4.Ohtaki T, Shintani Y, Honda S, et al. Metastasis suppressor gene KiSS-1 encodes peptide ligand of a G-protein-coupled receptor. Nature. 2001;411:613–7. doi: 10.1038/35079135. [DOI] [PubMed] [Google Scholar]
- 5.Lee JH, Miele ME, Hicks DJ, et al. KiSS-1, a novel human malignant melanoma metastasis-suppressor gene. J Natl Cancer Inst. 1996;88:1731–7. doi: 10.1093/jnci/88.23.1731. [DOI] [PubMed] [Google Scholar]
- 6.Lee JH, Welch DR. Suppression of metastasis in human breast carcinoma MDA-MB-435 cells after transfection with the metastasis suppressor gene, KiSS-1. Cancer Res. 1997;57:2384–7. [PubMed] [Google Scholar]
- 7.Nash KT, Phadke PA, Navenot JM, et al. Requirement of KISS1 secretion for multiple organ metastasis suppression and maintenance of tumor dormancy. J Natl Cancer Inst. 2007;99:309–21. doi: 10.1093/jnci/djk053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stafford LJ, Xia C, Ma W, Cai Y, Liu M. Identification and characterization of mouse metastasis-suppressor KiSS1 and its G-protein-coupled receptor. Cancer Res. 2002;62:5399–404. [PubMed] [Google Scholar]
- 9.Mitchell DC, Stafford LJ, Li D, Bar-Eli M, Liu M. Transcriptional regulation of KiSS-1 gene expression in metastatic melanoma by specificity protein-1 and its coactivator DRIP-130. Oncogene. 2007;26:1739–47. doi: 10.1038/sj.onc.1209963. [DOI] [PubMed] [Google Scholar]
- 10.Mead EJ, Maguire JJ, Kuc RE, Davenport AP. Kisspeptins: a multifunctional peptide system with a role in reproduction, cancer and the cardiovascular system. Br J Pharmacol. 2007;151:1143–53. doi: 10.1038/sj.bjp.0707295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lapatto R, Pallais JC, Zhang D, et al. Kiss1−/− mice exhibit more variable hypogonadism than Gpr54−/− mice. Endocrinology. 2007;148:4927–36. doi: 10.1210/en.2007-0078. [DOI] [PubMed] [Google Scholar]
- 12.Nash KT, Welch DR. The KISS1 metastasis suppressor: mechanistic insights and clinical utility. Front Biosci. 2006;11:647–59. doi: 10.2741/1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mitchell DC, Abdelrahim M, Weng J, et al. Regulation of KiSS-1 metastasis suppressor gene expression in breast cancer cells by direct interaction of transcription factors activator protein-2alpha and specificity protein-1. J Biol Chem. 2006;281:51–8. doi: 10.1074/jbc.M506245200. [DOI] [PubMed] [Google Scholar]
- 14.Tsuji K, Kawauchi S, Saito S, et al. Breast cancer cell lines carry cell line-specific genomic alterations that are distinct from aberrations in breast cancer tissues: comparison of the CGH profiles between cancer cell lines and primary cancer tissues. BMC Cancer. 2010;10:15. doi: 10.1186/1471-2407-10-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orsetti B, Nugoli M, Cervera N, et al. Genetic profiling of chromosome 1 in breast cancer: mapping of regions of gains and losses and identification of candidate genes on 1q. Br J Cancer. 2006;95:1439–47. doi: 10.1038/sj.bjc.6603433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Argos M, Kibriya MG, Jasmine F, et al. Genomewide scan for loss of heterozygosity and chromosomal amplification in breast carcinoma using single-nucleotide polymorphism arrays. Cancer Genet Cytogenet. 2008;182:69–74. doi: 10.1016/j.cancergencyto.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ried T, Just KE, Holtgreve-Grez H, et al. Comparative genomic hybridization of formalin-fixed, paraffin-embedded breast tumors reveals different patterns of chromosomal gains and losses in fibroadenomas and diploid and aneuploid carcinomas. Cancer Res. 1995;55:5415–23. [PubMed] [Google Scholar]
- 18.Forozan F, Mahlamaki EH, Monni O, et al. Comparative genomic hybridization analysis of 38 breast cancer cell lines: a basis for interpreting complementary DNA microarray data. Cancer Res. 2000;60:4519–25. [PubMed] [Google Scholar]
- 19.Negrini M, Sabbioni S, Possati L, et al. Suppression of tumorigenicity of breast cancer cells by microcell-mediated chromosome transfer: studies on chromosomes 6 and 11. Cancer Res. 1994;54:1331–6. [PubMed] [Google Scholar]
- 20.Bilanges B, Varrault A, Basyuk E, et al. Loss of expression of the candidate tumor suppressor gene ZAC in breast cancer cell lines and primary tumors. Oncogene. 1999;18:3979–88. doi: 10.1038/sj.onc.1202933. [DOI] [PubMed] [Google Scholar]
- 21.Cingoz S, Altungoz O, Canda T, Saydam S, Aksakoglu G, Sakizli M. DNA copy number changes detected by comparative genomic hybridization and their association with clinicopathologic parameters in breast tumors. Cancer Genet Cytogenet. 2003;145:108–14. doi: 10.1016/s0165-4608(03)00094-3. [DOI] [PubMed] [Google Scholar]
- 22.Prentice LM, Klausen C, Kalloger S, et al. Kisspeptin and GPR54 immunoreactivity in a cohort of 518 patients defines favourable prognosis and clear cell subtype in ovarian carcinoma. BMC Med. 2007;5:33. doi: 10.1186/1741-7015-5-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mitra A, Fillmore RA, Metge BJ, et al. Large isoform of MRJ (DNAJB6) reduces malignant activity of breast cancer. Breast Cancer Res. 2008;10:R22. doi: 10.1186/bcr1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stark AM, Tongers K, Maass N, Mehdorn HM, Held-Feindt J. Reduced metastasis-suppressor gene mRNA-expression in breast cancer brain metastases. J Cancer Res Clin Oncol. 2005;131:191–8. doi: 10.1007/s00432-004-0629-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vargo-Gogola T, Rosen JM. Modelling breast cancer: one size does not fit all. Nat Rev Cancer. 2007;7:659–72. doi: 10.1038/nrc2193. [DOI] [PubMed] [Google Scholar]
- 26.Lin EY, Jones JG, Li P, et al. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am J Pathol. 2003;163:2113–26. doi: 10.1016/S0002-9440(10)63568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Navenot JM, Fujii N, Peiper SC. Activation of Rho and Rho-associated kinase by GPR54 and KiSS1 metastasis suppressor gene product induces changes of cell morphology and contributes to apoptosis. Mol Pharmacol. 2009;75:1300–6. doi: 10.1124/mol.109.055095. [DOI] [PubMed] [Google Scholar]
- 28.Cho SG, Li D, Stafford LJ, et al. KiSS1 suppresses TNFalpha-induced breast cancer cell invasion via an inhibition of RhoA-mediated NF-kappaB activation. J Cell Biochem. 2009;107:1139–49. doi: 10.1002/jcb.22216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pylayeva Y, Gillen KM, Gerald W, Beggs HE, Reichardt LF, Giancotti FG. Ras- and PI3K-dependent breast tumorigenesis in mice and humans requires focal adhesion kinase signaling. J Clin Invest. 2009;119:252–66. doi: 10.1172/JCI37160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodriguez-Viciana P, Collins C, Fried M. Polyoma and SV40 proteins differentially regulate PP2A to activate distinct cellular signaling pathways involved in growth control. Proc Natl Acad Sci U S A. 2006;103:19290–5. doi: 10.1073/pnas.0609343103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ueyama T, Sakoda T, Kawashima S, et al. Activated RhoA stimulates c-fos gene expression in myocardial cells. Circ Res. 1997;81:672–8. doi: 10.1161/01.res.81.5.672. [DOI] [PubMed] [Google Scholar]
- 32.Hill CS, Wynne J, Treisman R. The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell. 1995;81:1159–70. doi: 10.1016/s0092-8674(05)80020-0. [DOI] [PubMed] [Google Scholar]
- 33.Fromm C, Coso OA, Montaner S, Xu N, Gutkind JS. The small GTP-binding protein Rho links G protein-coupled receptors and Galpha12 to the serum response element and to cellular transformation. Proc Natl Acad Sci U S A. 1997;94:10098–103. doi: 10.1073/pnas.94.19.10098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wacker JL, Feller DB, Tang XB, et al. Disease-causing mutation in GPR54 reveals the importance of the second intracellular loop for class A G-protein-coupled receptor function. J Biol Chem. 2008;283:31068–78. doi: 10.1074/jbc.M805251200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo X, Stafford LJ, Bryan B, et al. A Rac/Cdc42-specific exchange factor, GEFT, induces cell proliferation, transformation, and migration. J Biol Chem. 2003;278:13207–15. doi: 10.1074/jbc.M208896200. [DOI] [PubMed] [Google Scholar]
- 36.Lutz S, Shankaranarayanan A, Coco C, et al. Structure of Galphaq-p63RhoGEF-RhoA complex reveals a pathway for the activation of RhoA by GPCRs. Science. 2007;318:1923–7. doi: 10.1126/science.1147554. [DOI] [PubMed] [Google Scholar]
- 37.Lutz S, Freichel-Blomquist A, Yang Y, et al. The guanine nucleotide exchange factor p63RhoGEF, a specific link between Gq/11-coupled receptor signaling and RhoA. J Biol Chem. 2005;280:11134–9. doi: 10.1074/jbc.M411322200. [DOI] [PubMed] [Google Scholar]
- 38.Quandt K, Frech K, Karas H, Wingender E, Werner T. MatInd and MatInspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res. 1995;23:4878–84. doi: 10.1093/nar/23.23.4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Raamsdonk CD, Bezrookove V, Green G, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457:599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lamba S, Felicioni L, Buttitta F, et al. Mutational profile of GNAQQ209 in human tumors. PLoS One. 2009;4:e6833. doi: 10.1371/journal.pone.0006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao X, Lu L, Pokhriyal N, et al. Overexpression of RhoA induces preneoplastic transformation of primary mammary epithelial cells. Cancer Res. 2009;69:483–91. doi: 10.1158/0008-5472.CAN-08-2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hogan C, Dupre-Crochet S, Norman M, et al. Characterization of the interface between normal and transformed epithelial cells. Nat Cell Biol. 2009;11:460–7. doi: 10.1038/ncb1853. [DOI] [PubMed] [Google Scholar]
- 43.Chan CH, Lee SW, Li CF, et al. Deciphering the transcriptional complex critical for RhoA gene expression and cancer metastasis. Nat Cell Biol. 2010;12:457–67. doi: 10.1038/ncb2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shoji S, Tang XY, Umemura S, et al. Metastin inhibits migration and invasion of renal cell carcinoma with overexpression of metastin receptor. Eur Urol. 2009;55:441–9. doi: 10.1016/j.eururo.2008.02.048. [DOI] [PubMed] [Google Scholar]
- 45.Butcher DT, Alliston T, Weaver VM. A tense situation: forcing tumour progression. Nat Rev Cancer. 2009;9:108–22. doi: 10.1038/nrc2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bhalla US. Understanding complex signaling networks through models and metaphors. Prog Biophys Mol Biol. 2003;81:45–65. doi: 10.1016/s0079-6107(02)00046-9. [DOI] [PubMed] [Google Scholar]
- 47.Theodorou V, Kimm MA, Boer M, et al. MMTV insertional mutagenesis identifies genes, gene families and pathways involved in mammary cancer. Nat Genet. 2007;39:759–69. doi: 10.1038/ng2034. [DOI] [PubMed] [Google Scholar]
- 48.Varticovski L, Hollingshead MG, Robles AI, et al. Accelerated preclinical testing using transplanted tumors from genetically engineered mouse breast cancer models. Clin Cancer Res. 2007;13:2168–77. doi: 10.1158/1078-0432.CCR-06-0918. [DOI] [PubMed] [Google Scholar]
- 49.Qiu TH, Chandramouli GV, Hunter KW, Alkharouf NW, Green JE, Liu ET. Global expression profiling identifies signatures of tumor virulence in MMTV-PyMT-transgenic mice: correlation to human disease. Cancer Res. 2004;64:5973–81. doi: 10.1158/0008-5472.CAN-04-0242. [DOI] [PubMed] [Google Scholar]
- 50.Cho SG, Yi Z, Pang X, et al. Kisspeptin-10, a KISS1-derived decapeptide, inhibits tumor angiogenesis by suppressing Sp1-mediated VEGF expression and FAK/Rho GTPase activation. Cancer Res. 2009;69:7062–70. doi: 10.1158/0008-5472.CAN-09-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]