

Abstract
This feature article describes the development of synthetic zinc(II)-dipicolylamine (ZnDPA) receptors as selective targeting agents for anionic membranes in cell culture and living subjects. There is a strong connection between anionic cell surface charge and disease, and ZnDPA probes have been employed extensively for molecular imaging and targeted therapeutics. Fluorescence and nuclear imaging applications include detection of diseases such as cancer, neurodegeneration, arthritis, and microbial infection, and also quantification of cell death caused by therapy. Therapeutic applications include selective targeting of cytotoxic agents and drug delivery systems, photodynamic inactivation, and modulation of the immune system. The article concludes with a summary of expected future directions.
Introduction
The primary premise of this feature article is summarized in Scheme 1. That is, anionic membrane surface charge is a biomarker of disease. In mammalian cells, anionic surface charge indicates cell death and dysfunction, signaling vital physiological processes such as the dead cell clearance without immune activation and blood clot formation. Many cancer cells expose high levels of an anionic phospholipid that can promote cancer proliferation by suppressing the local immune response. Similarly, the anionic surface of many microbial pathogens allows them to enter host cells and inhibit the innate immune response for prolonged survival. The strong connection between anionic surface charge and disease has prompted different research programs to create molecular imaging agents and targeted therapeutics for anionic membranes. A major goal of this article is to describe how we approached the problem as a group whose core expertise is small molecule supramolecular chemistry. In general, our research has progressed through an iterative cycle of test tube experiments, cell studies, and pre-clinical evaluation in living subjects. The article is structured in the following order. First, we explain why anionic membrane surface change is considered a biomarker of disease. Then, we describe our research efforts over the last 14 years to develop synthetic zinc(II)-dipicolylamine (ZnDPA) receptors as selective targeting agents for molecular imaging of cell death and bacterial infection.‡ We also summarize our research to date on several different therapeutic applications using ZnDPA receptors, and we conclude with some comments on likely future directions.
Scheme 1.
Anionic membranes are biomarkers of disease.
The anionic membrane: a biomarker of disease and microbial pathogens
Cells are surrounded by a plasma membrane that separates the internal cell contents from the external environment and maintains crucial electrochemical energy gradients.1,2 In addition, the plasma membrane plays important roles in transmembrane signal transduction and cell-cell communication. The structure of the plasma membrane is a dynamic bilayer composed of different polar lipids and proteins. Many of these amphiphilic compounds have appended carbohydrate chains that protrude from the membrane exterior surface and form the cell glycocalyx.3 About 70% of the polar lipids in a typical mammalian cell plasma membrane are glycerophospholipids, with cholesterol, sphingomyelin, and glycosphingolipids making up the other 30%.1 The transmembrane distribution of these polar lipids is not symmetrical, with the outer monolayer enriched in zwitterionic phosphatidylcholine (PC)¶ and sphingomyelin, and the inner monolayer containing most of the phosphatidylserine (PS), the most abundant anionic phospholipid in the plasma membrane (present in the range of 2–10%).4 This transbilayer distribution is maintained in healthy cells by an ATP-dependent translocase that catalyzes the transport of aminophospholipids from the external to the internal membrane surface. 5 As a result, the electrostatic charge on the membrane exterior surface of a healthy mammalian cell is close to neutral. But during the process of programmed cell death or apoptosis, the translocase activity is attenuated leading to PS exposure and increased negative charge on the cell exterior. The exposed PS can trigger innate immune responses such as recognition and engulfment of dead and dying cells by phagocytes.6 This process is responsible for the daily clearance of senescent and damaged cells in the human body. PS exposure also plays an essential role in hemostasis by acting as a key signal in the coagulation cascade.7 Upon activation via injury to the endothelium, blood platelets expose PS on their outer surface which triggers the attachment of several coagulation factors that execute fibrin synthesis and clot formation.8
Excessive cell death beyond the normal level associated with homeostasis is a hallmark of many acute and chronic ailments.9 For example, PS exposure occurs during chronic conditions such as cardiovascular disease, or atherosclerosis, which is an inflammatory state characterized by the thickening of artery walls. Atherosclerosis arises from a sustained presence of dead cells in arterial vessels due to an imbalance between apoptosis and phagocytosis. As arterial walls condense, less oxygenated blood can pass into the heart muscle resulting in myocardial ischemia, which causes hypoxia-induced apoptosis in cardiomyocytes. Glaucoma, one of the leading causes of irreversible blindness, is characterized by progressive nerve degeneration and retinal ganglion cell apoptosis.10-12 Red blood cell exposure of PS is indicative of the stress and dysfunction which typically manifests during an invasion by pathogens such as plasmodium parasites or as a result of blood diseases like sickle cell anemia.13, 14 Natural PS exposure in red blood cells is identified as a risk factor for stroke and chronic renal failure.15
With certain diseases, such as cancer, the therapeutic goal is to cause cell death and increased amounts of PS on the cancer cell surface is considered a hallmark of therapeutic success. Even without cytotoxic treatment, exposed PS is a characteristic cell surface feature of many tumors and their surrounding microenvironment. Numerous cancer cells have been reported to expose PS including leukemic cells,16 neuroblastoma cells,17 malignant melanoma,18 human gastric carcinoma cells,19 and others.20 PS exposure is particularly apparent for undifferentiated tumorigenic cells which can express about five times greater PS at their cell surface than their differentiated, non-tumorigenic equivalents.18 PS is also found in the tumor microenvironment, in the form of tumor-derived microvesicles and on the surface of tumor vasculature, which may promote blood coagulation and fibrin formation to generate a scaffold for tumor growth.21-23 Additionally, high PS exposure may promote cancer proliferation by suppressing the local immune response. The interaction between PS-expressing cells and immune cells triggers immunosuppressive pathways that prevent both local and systemic immune activation and cell clearance. There is evidence that cell surface PS suppresses tumor inflammation by inducing secretion of immunosuppressants such as transforming growth factor-β, which inhibits T cell and cytokine production.24 While these pathways are used by apoptotic cells to inhibit potential immune sequelae against ‘self’, tumors hijack them to enable proliferation.
Similar to cancer cells, obligate intracellular pathogens exploit host cell functions to assure successful entry into host cells to derive nutrients and manipulate cellular machinery for replication. One common strategy is ‘apoptotic mimicry’ where pathogens mimic apoptotic debris by exposing high fractions of PS on their surface (enveloped PS acquisition) or cloaking themselves in cell-derived PS-containing vesicles (non-enveloped PS acquisition) to facilitate binding, entry and immune evasion. This was first discovered in viruses, which often have a protein capsid surrounded by an outer bilayer membrane.25 Upon fusion with host cells, viruses induce cytopathic effects to delay host cell PS exposure most likely to prevent clearance and enhance virion replication. Subsequent budding from PS-rich organelles, such as the Golgi apparatus, endoplasmic reticulum and lipid rafts, allows PS exposure on newborn virions.26-28 Clusters of viruses are packed and released in PS-exposing lipid vesicles, elevating their infectivity. PS-mediated virion uptake also results in the induction of anti-inflammatory cytokine production and inhibition of inflammatory cytokine secretion. Thus, in addition to promoting uptake and binding, apoptotic mimicry by viruses potentiates infection by depleting the host innate immune responses and evading immune recognition. Apoptotic mimicry is utilized by many viral families including vaccinia,29 hepatitis B,30 HIV,31 and dengue.32
Parasitic microorganisms, most notably trypanosomatids, have devised similar methods for entering host tissue.33 Trypanosomatid parasites exist as either promastigotes (extracellular form within vector insects) or amastigotes (intracellular form responsible for disseminating disease through host cells) and execute two infectious life cycles based on apoptotic mimicry: ‘Trojan horse infection’ and ‘bystander infection’. Trojan horse infection is employed by parasites that naturally expose PS such as Leishmania donovani and Tryapanosma brucei, the causative parasites for visceral leishmaniasis and Chagas disease, respectively.34-38 Upon infection of the host via the bite of a carrier insect, PS-exposing promastigotes are engulfed by host neutrophils which are subsequently phagocytosed by larger macrophages enabling intracellular parasite replication. Smuggling promastigotes within Trojan horse neutrophils not only delivers viable parasites into macrophages but also delays the immune response until the first line neutrophilic response is resolved. Bystander infection is utilized by parasites that do not naturally expose PS, such as Leishmania amazonensis and Trypanosoma brucei, but instead possess the enzymatic machinery to generate mammalian apoptotic features upon parasite death, most importantly the externalization of PS.39 For viable infection to occur, a sub-population of parasites naturally die within the insect carrier, which enables neutrophil engulfment of both dead and live bystander parasites upon infection.40 Alternatively, parasites can migrate with the remnants of PS-exposing apoptotic cells which are also engulfed by neutrophils. Persistent infection and intracellular replication modulate PS exposure on the amastigote form to increase parasite infectivity.35 Another apoptosis-mimicking parasite is Toxoplasma gondii, the causative agent of toxoplasmosis. The infective inoculum of T. gondii is comprised of two different populations, PS-exposing and non-exposing parasites, that cooperate to establish infection.41 T. gondii is often characterized as the most successful parasite on earth. Approximately one-third of the human population is infected with T. gondii with most infections being asymptomatic.41, 42
Anionic surface charge is also a ubiquitous feature of the bacterial cell envelope. Bacteria take advantage of phagocytosis in host cells to promote cell-to-cell spread, which was recently discovered in Listeria monocytogenes. L. monocytogenes emerge from infected macrophages packaged in PS-coated vesicles, which interact with healthy macrophages to facilitate uptake.43 Interestingly, bacterial efferocytosis appears to be an effective mechanism for the host to control Mycobacterium species infection, illustrating the fine balance between host cell defense and bacterial infectivity mechanisms.43 Furthermore, most bacterial strains naturally harbor anionic phospholipids such as phosphatidylglycerol (PG) and cardiolipin (CL) within their membrane bilayers.44 For example, the lipid membrane of Gram-positive Streptococcus pyogenes contains significant fractions of PG (~20 %) and CL (~5 %).45 The Staphylococcus aureus membrane contains > 60% of PG and CL.44, 46 Gram-negative Escherichia coli contain 9 and 19% PG and CL in their outer membrane and 18 and 40% PG and CL in their inner membrane, respectively.47-49 In addition to phospholipids, both Gram-positive and Gram-negative bacteria cell walls have other phosphate containing compounds. The teichoic acids of Gram-positive bacteria contain repeating sugar polymers which are phosphate-linked and can extend 42-50 sugar units in length.50 Gram-negative lipopolysaccharide (LPS) harbors lipid A- and O-antigen which contain anionic phosphates.51,52 Bacterial spores, survival structures unique to Bacilli and Clostridia bacteria, contain high quantities of amphiphilic phosphates and carboxylates in their exosporium.53 Networks of negatively charged lipids and polymers help maintain a protective cell envelope and ensure high biofilm integrity.54
The realization that anionic charge on the exterior of the cell plasma membrane is a biomarker of disease (or successful disease treatment) has prompted a search for selective targeting agents that can enable molecular imaging and drug delivery applications.55-58 Imaging applications include detection of disease, staging of disease progression, and monitoring the efficacy of therapy, whereas, therapeutic applications include selective targeting of cytotoxic agents and activation of the immune system.9,59,60 One targeting approach is to use PS-targeting biomolecules such as antibodies, proteins, or peptides. Examples include the antibody bavituximab61, the proteins annexin V62 and lactadherin63, and the C2A domain of the protein synaptotagmin64. The large size of these biological agents enables high selectivity and target affinities, but it also means they are hard to cleanly label with a reporter group. In addition, they may have stability and storage challenges or undesired pharmacokinetic properties such as slow diffusion in restricted sites or slow rates of clearance from the blood pool. Thus, there is a need for small synthetic molecules that can selectively target anionic membranes with easily tunable pharmacokinetic performance. In some cases, simple cationic molecules or nanoparticles have been examined and found to exhibit promising targeting ability.65, 66 In other cases, synthetic peptides have been uncovered using rational design or screening paradigms and converted into targeted molecular probes.67,68 We have contributed to this broad research effort by developing a series of synthetic ZnDPA coordination complexes as low molecular weight targeting agents for anionic membranes, and the next section summarizes the discovery pathway that produced these compounds for imaging and therapy.
Anion recognition using zinc-dipicolylamine (ZnDPA) receptors
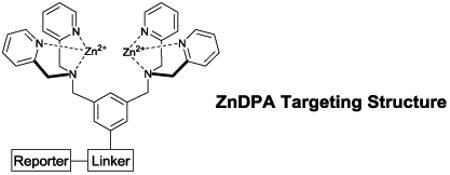
Designing molecular hosts for anion recognition in water is a difficult challenge that must overcome numerous obstacles. First, water forms strong hydrogen bonds with anions and any host/guest complexation process must overcome a large energetic penalty to dehydrate the anion and achieve high-affinity association.69 Second, anions exist in a wide range of molecular geometries (e.g. spherical, linear, trigonal, etc.) and selective binding requires a receptor to have a complementary shape. Additionally, many anions are weak acids or bases and effective anion recognition often only occurs within a specific pH window. Anion recognition in complex biomedical environments is especially challenging due to the many competing non-covalent interactions. To date, the most successful synthetic anion receptors are metal coordination complexes that can reversibly bind target anions with dissociation constants in the millimolar to nanomolar range. The basis of these coordination complexes is an organic scaffold that maintains two or more metal centers at an appropriate distance to allow chelation of a bridging guest anion. In general, there are two anion association mechanisms to consider (Scheme 2). In one limiting case, the scaffold binds the metal cations so strongly that the metal/scaffold complex is essentially a single molecular unit with two Lewis acidic sites whose separation is controlled by the length and rigidity of the scaffold (Scheme 2A). The second association mechanism is when the scaffold has an inherently weak affinity for one or both of the metal cations. However, the presence of a suitable bridging anion induces a three-component assembly process that brings together the scaffold, metal cations, and bridging anion (Scheme 2B). As discussed below, the dipicolylamine (DPA) scaffold can be tuned by structural modification to allow both types of association processes.
Scheme 2.
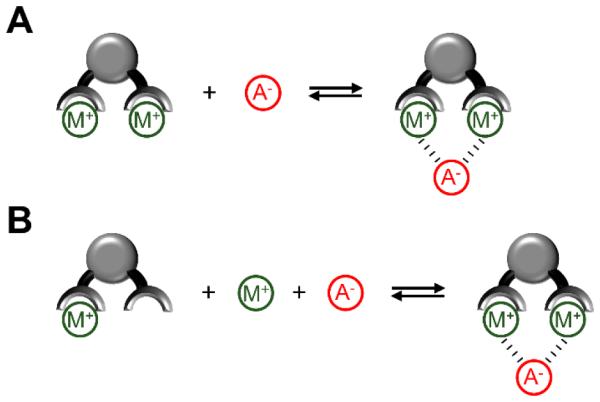
Modes of anion recognition using divalent metal ion complexes. (A) Receptor with strongly bound metal ions acts as a single molecular unit for anion binding. (B) Three-component assembly process in which both metals bind strongly to the receptor scaffold only in the presence of a suitable complementary anion.
The DPA scaffold was reported by Biniecki and Kabzinskato in 1964 to have selective affinity for transition metals such as Zn2+ (association constant of ~107 M−1) over alkali and alkaline metals such as Na+, K+, Mg2+ and Ca2+.70 The change in DPA molecular orbital energies upon coordination to Zn2+ has been exploited extensively over the last two decades as way to design Zn2+ sensors.71-73 The tridentate DPA coordination leaves an open site on the Zn2+ and this Lewis acidic feature has been used to create ZnDPA receptors for anion binding in water (Scheme 2).74 Fluorescent ZnDPA chemosensors were pioneered by Hamachi and co-workers, who reported in 2002 that an anthracene derivative with two appended ZnDPA units (PSVue-380, Scheme 3)75 was a fluorescent sensor of dianionic phosphate derivatives, especially peptides with phosphotyrosine residues. Furthermore, there was a large enhancement of the fluorescence signal upon phosphate association. Mechanistic studies showed that phosphate association with one of the ZnDPA units promotes Zn2+ coordination to the second DPA and loss of a photoinduced electron transfer (PET) quenching effect.76 Various ZnDPA complexes have been designed over the intervening years for aqueous sensing of other phosphate containing molecules with association constants ranging from 105 to 107 M−1.75-80 This work has been described in review articles,81,82 and it remains an active research topic.83-85 In addition, other metal coordination complexes, such as zinc cyclen complexes, have been developed for effective phosphate recognition, and the interested reader is directed to review articles that describe these valuable systems.82 A technical advantage with synthetic zinc coordination complexes is the relative ease in which they can be converted into oligomers with multiple phosphate binding sites. As discussed below, this strategy creates multivalent receptors that can recognize anionic surfaces with altered association thermodynamics and kinetic properties due to cooperativity effects such as enhanced local concentration and binding additivity.82
Scheme 3.
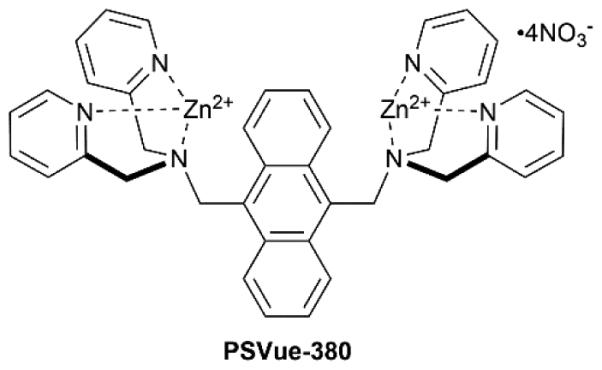
In 2003 we were looking for a synthetic receptor to target anionic membrane surfaces, and we decided to investigate PSVue-380.86 The fluorescence intensity of PSVue-380 was found to increase by 10-fold when titrated with anionic vesicles that contained PS, PG, or phosphatidic acid, but was unchanged when treated with vesicles comprised solely of the zwitterionic PC.87 Association of the sensor with the membrane surface occurs by a ‘three component’ self-assembly process where the DPA scaffold containing one Zn2+ ion and a free Zn2+ ion are attracted to the negatively charged membrane surface and form a bridged complex with the anionic head group of a phospholipid (Scheme 4). The successful targeting of model vesicle systems prompted us to evaluate whether PSVue-380 could operate in more complicated biological environments.
Scheme 4.
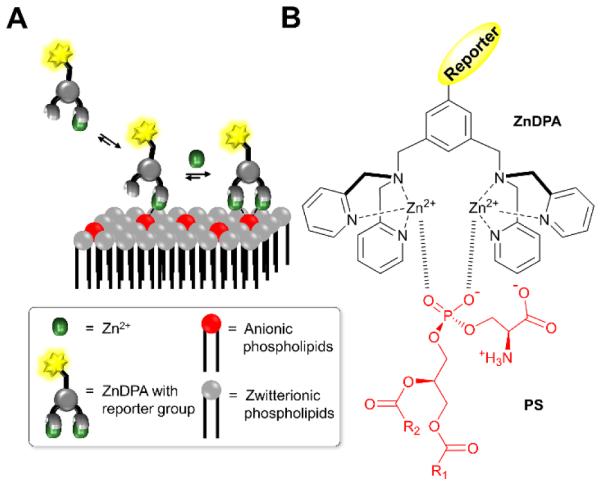
Anionic membrane recognition using ZnDPA receptors. (A) Three-component assembly process for membrane recognition of anionic phospholipids. (B) Structural representation of complexation between anionic PS and ZnDPA receptor.
Molecular imaging of dead and dying cells
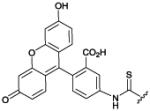
We initially explored fluorescent ZnDPA probes for detecting cell death in cultured cell samples using cell microscopy or flow cytometry. Cell-based tests that can quantify levels of cell death are broadly useful for fundamental biomedical research and drug development. Early studies with PSVue-380 found that it was a selective stain for apoptotic cells,86 but the blue fluorescence emission wavelength (380/440 nm) was a technical limitation since it overlapped with cell background autofluorescence. We subsequently designed ZnDPA sensors using a receptor–linker–reporter approach that linked a ZnDPA complex (anion receptor) to a signaling unit (typically a fluorophore) (Table 1).88,89 The green emitting PSVue-480 was evaluated using cancer cells treated with the drug camptothecin to induce apoptosis. Simultaneous treatment with PSVue-480 and the nuclear stain 7-aminoactinomycin D (7AAD) allowed identification of early-apoptotic cells (with impermeable plasma membranes) due to selective staining with PSVue-480 and exclusion of 7AAD (Fig. 1A). Flow cytometry analysis of the same cell populations showed that approximately 30% of the cells were apoptotic or necrotic and thus stained with PSVue-480 (Fig. 1B).
Table 1.
ZnDPA imaging and affinity probes
|
|||||
|---|---|---|---|---|---|
|
| |||||
| Name | Reporter Structure | Uses and References |
Name | Reporter Structure | Uses and References |
|
PSVue-
480 |
|
imaging
cell death88,89,90 |
mSeek |
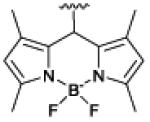
|
imaging
leishmania136 bacteria137 |
|
PSVue-
643 |
|
imaging
bacteria117 cell death105 |
mDestroy |
|
photoinactivation
bacteria137 |
|
PSVue-
794 |
|
imaging
cell death92,95 cancer therapy93,92 arthritis96,97 bacteria116 |
ZnDPA-
[111In] DOTA |
|
imaging
bacteria125 |
|
PSVue-
Biotin |
|
imaging
bacteria124,129 |
ZnDPA-
[111In] DTPA |
|
imaging
bacteria125 |
|
bis-
ZnDPA- [111In] DTPA |
|
imaging
bacteria125 |
|||
Fig. 1.

(A) Fluorescence micrographs of Jurkat cells treated with camptothecin (10 μM) for 3.5 h to induce apoptosis, then stained with PSVue-480 (green, 5 μM) and 7AAD (red, 500 ng/mL). (B) Flow cytometry histograms illustrating staining of Jurkat cells by PSVue-480 and 7AAD. A and B reprinted with permission from Ref. 89. © 2005 John Wiley and Sons. (C) X-ray and fluorescence overlay image of a rat prostate tumor model at 24 h postinjection of PSVue-794. Reprinted with permission from Ref. 92. © 2010 American Chemical Society. (D) Ex vivo epifluorescence image of a rat bearing two subcutaneous PAIII prostate tumors and dosed with PSVue-794. The right flank tumor received focal beam radiation therapy, and the left flank tumor was not treated. Reprinted with permission from Ref. 93. © 2011 Springer. (E) Whole body fluorescence image of a mouse with induced arthritis given a single dose of PSVue-794 48 hours prior to imaging. Arrows point to arthritic feet joints. Reprinted from open access Ref. 96. (F) Decay-corrected transaxial and coronal [18F]-ZnDPA PET images of a rat following acute myocardial infarction and 60 min after subsequent probe injection. The short red line indicates the location of the heart. Reprinted from open access Ref. 101.
Another pre-clinical application of ZnDPA probes is to enable molecular imaging of cell death in animal models of disease and disease treatment. Detecting dead and dying cells in a living organism is quite challenging because the cell death process is a time-dependent phenomenon. The amount of probe accumulation at cell death sites in a living animal depends on the rate and location of the cell death process as well as the rate of dead cell clearance by the animal's innate immune response. The challenge is compounded by the limitations of fluorescence imaging with visible light, which suffers large signal loss due to scattering/attenuation by the animal tissue. Thus, animal studies with green emitting PSVue-480 can only be conducted on superficial sites that can be directly illuminated, such as the interior of the eye. An example of this type of study was recently performed by Kwong and coworkers who determined that PSVue-480 could label retinal ganglion cells undergoing apoptotic degeneration induced by N-methyl-D-aspartate (NMDA).90 Adult rats were given intravitreal injections of NMDA followed by euthanasia at various time points. One hour before euthanasia, PSVue-480 was intravitreally injected followed by imaging of the treated eye. Fluorescence labeling was observed in the retinas at one hour and up to 24 hours following NMDA treatment. Targeting of exposed PS on the apoptotic retinal neurons occurred earlier than detection of DNA fragmentation, indicating the potential of ZnDPA probes for tracking the early stages of degenerating retinal neurons.
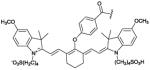
It is well-known that light penetration through skin and tissue is a maximum when the wavelength is in the near-infrared window of 650 and 900 nm.91 Therefore, in vivo imaging performance was greatly improved by developing the near-infrared ZnDPA probe PSVue-794 with an appended carbocyanine fluorophore. Cell death imaging studies focused initially on two in vivo tumor models that spontaneously developed foci of necrotic cells in the tumor cores, namely, immunocompetent Lobund-Wistar rats with PAIII prostate tumors, and athymic nude mice containing EMT-6 mammary tumors.92 Whole-body optical imaging of tumor-bearing animals dosed with PSVue-794 found that the probe selectively targeted the tumor core. Probe accumulation in the PAIII prostate tumors was 36-fold higher than an untargeted control fluorophore. Ex vivo biodistribution and histological analyses showed PSVue-794 targeting to the interior necrotic regions of the tumors (Fig. 1C). Similarly, PSVue-794 accumulation in the EMT-6 mammary tumors was 18-fold higher than control fluorophore.
These results led to the development of PSVue-794 as a commercial fluorescent probe for pre-clinical imaging,‡ and it is now finding increasing use in a broad range of applications. Several studies have employed PSVue-794 to monitor tumor cell death induced by different therapeutic treatments. Rats bearing PAIII prostate tumors were given a focal beam of radiation which induced rapid apoptosis and necrosis at the irradiation site.93 Subsequent dosing with PSVue-794 followed by whole-body imaging showed probe accumulation in a radiation-treated tumor over a non-treated tumor on the same animal (Fig. 1D). To simulate cell death generated by chemotherapeutic treatment, separate cohorts of healthy rats were dosed with dexamethasone, a glucocorticoid that induces selective cell death at the thymus, followed by dosing with PSVue-794 twenty-four hours later. Ex vivo imaging of organs showed thymus uptake of PSVue-794 was four times higher than a control fluorophore. Palmowski and coworkers conducted a comparative study of cell death imaging performance after antiangiogenic therapy and found that apoptosis was detected significantly better with the low molecular weight PSVue-794 than with the dye-labeled protein probe annexin V.94
Cell death detection due to physical injury was examined in a murine traumatic brain injury model. Traumatic brain injury is characterized as acute cranial tissue damage that leads to secondary processes such as cell death and blood-brain-barrier disruption. The ability of PSVue-794 to detect cranial cell death was tested using a cryolesion mouse model that conditionally mimics features of human brain injury.95 In short, the model involves brief contact of a cold rod to the head of a living, anesthetized mouse, which triggers localized cell death at the contact site. Whole-body fluorescence imaging of PSVue-794 permitted visualization of the cryolesion as well as longitudinal monitoring of healing process over a one week period. PSVue-794 has also been used for arthritis imaging96 and optical imaging of articular cartilage degeneration.97 Arthritis is a chronic inflammatory state caused by neutrophil and macrophage infiltration at skeletal joints, counterbalanced by apoptosis of the activated immune cells leading to termination of the inflammatory response.98 PSVue-794 was assessed for the non-invasive molecular imaging of rheumatoid arthritis via metabolic function in murine collagen-induced arthritis. DBA/1 mice were intravenously treated twice with chicken collagen type II in Freund’s adjuvant which generates skeletal inflammation and arthritis that was easily monitored at the mouse footpad. Fluorescent emission from arthritic and non-arthritic mice treated with PSVue-794 correlated reliably with the degree of footpad swelling and the manifestation of arthritis, illustrating a useful and economical alternative method for non-invasively assessing arthritis in murine models (Fig. 1E). One factor that contributes to arthritis is the degradation of cartilage, a flexible connective tissue at the joints. An early signal of cartilage degradation is the loss of negatively charged glycosaminoglycans (GAGs), which play an essential role in generating supportive pressure in the collagen matrix. The feasibility of PSVue-794 for optical imaging of cartilage degeneration was tested using mice with young and aged knee joints. Ex vivo imaging of cartilage treated with PSVue-794 showed high probe accumulation on young cartilage compared with aged cartilage indicating a change of the level of GAGs. Optical imaging of young and old mice further indicated that PSVue-794 demonstrated higher uptake and retention in young mice (high GAGs) than old mice (low GAGs) when administrated via local injection in mouse knee joints. In a surgically induced osteoarthritis model, dramatically higher PSVue-794 signal was observed in sham joints (high GAGs) than osteoarthritis joints, demonstrating the ability of PSVue-794 for visual detection of early degeneration of cartilage in living subjects.
The promising fluorescence imaging results in living animals prompted the design of radiolabeled ZnDPA complexes with greater signal tissue penetration and potential for clinical translation. Wyffels and coworkers labeled a ZnDPA targeting unit with gamma-emitting technetium-99m (99mTc) in two different forms, [99mTc(CO)3 ]3+ and [99mTc-HYNIC].99 The two 99mTc-labeled probes were tested in mice that had been treated with anti-Fas antibody which induced rapid and massive hepatic apoptosis. Single-photon emission tomography and computed tomography (SPECT/CT) imaging studies showed significantly greater accumulation of both probes in the livers of antibody-treated mice compared to mice given no treatment. Furthermore, the increased liver uptake of the [99mTc(CO)3 ]3+ conjugate was comparable to radiolabeled annexin V. Mice with an induced myocardial ischemia-reperfusion injury were used as a second model to demonstrate the sensitivity of the 99mTc-labeled tracers for apoptotic and necrotic tissue. Myocardial ischemia was produced by ligation of the left coronary artery for thirty minutes, followed by two hours of reperfusion. At the end of reperfusion, rats were injected intravenously with tracer followed by euthanasia and removal of organs two hours later for scintillation counting. Selective accumulation of both probes was observed in mice with myocardial ischemia-reperfusion injury, however, the absolute tracer uptake in the damaged myocardium was low and likely would have been undetectable by in vivo cardiac imaging.
Positron emission tomography (PET) imaging of cell death has been explored using ZnDPA covalently labeled with the positron-emitting isotope, 18F.100 Wang et al. reported in vivo PET imaging experiments using two different 18F-labeled ZnDPA probes and 18F-fluorodeoxyglucose (18F-FDG) in separate cohorts of hepatocellular carcinoma-bearing mice with and without cytotoxic doxorubicin treatment. 18F-FDG showed no significant uptake change before and after chemotherapy, indicating that 18F-FDG lacked specificity to monitor anticancer therapy. In contrast, one of the 18F-ZnDPA probes accumulated significantly in the doxorubicin-treated tumors compared to untreated control. Recently, a similar 18F-ZnDPA analog was synthesized and evaluated for noninvasive imaging of cardiomyocyte apoptosis following acute myocardial infarction.101 PET imaging showed significant cardiac accumulation of the probe in rats following infarction which was consistent with histological evidence of cardiac cell death (Figure 1F).
Translation of ZnDPA probes to the clinic for cell death imaging would be facilitated if the probes exhibited higher affinity and selectivity for dead and dying tissue. The simplest way to increase probe affinity is to attach multiple recognition elements to a single scaffold and create a multivalent probe (Scheme 5). This strategy was explored using an fluorescent near-infrared dye scaffold with two (bis-ZnDPA-SR) or four (tetra-ZnDPA-SR) appended ZnDPA units.102 Vesicle titration assays showed that tetra-ZnDPA-SR exhibited higher selectivity than bis-ZnDPA-SR for anionic PS/PC vesicles over zwitterionic PC vesicles. Both probes selectively targeted dead and dying cells, but there was a marked difference in the cell microscopy images. The tetra-ZnDPA-SR localized strongly at the cell plasma membrane, whereas the bis-ZnDPA-SR distributed throughout the cytosol. The fluorescent probes were tested in three animal models of cell death, and in each case there was more tetra-ZnDPA-SR accumulation at the site of cell death than bis-ZnDPA-SR supporting the general hypothesis that increasing the number of ZnDPA units improves the cell death targeting ability. Unfortunately, the multivalent probes showed a tendency to undergo cross-linking and self-aggregation, which produced undesired pharmacokinetic properties including high off-target organ accumulation. This led to us to pursue an alternative design strategy for enhancing PS affinity; that is, a single ZnDPA scaffold with appended functional groups that are capable of forming secondary noncovalent interactions with the PS headgroup (Scheme 6).103 A focused 25-member library of modified dipicolylamine units with different appended 2-amido substituents were synthesized and screened for affinity to PS-rich membranes using a rapid equilibrium dialysis assay.104 The screening identified a lead candidate with two lipophilic phenethyl ureido groups (PU-ZnDPA), which was converted into a deep-red fluorescent probe and compared with an unmodified ZnDPA probe (PSVue-643) in a series of FRET-based vesicle titration studies, cell microscopy experiments, and in vivo rat tumor biodistribution measurements. The cell microscopy showed that PU-ZnDPA possessed comparatively higher affinity for dead and dying cells. However, the in vivo experiments indicated undesirable high uptake of PU-ZnDPA in the liver, spleen and intestines. With the aim of improving in vivo pharmacokinetics, we subsequently examined molecular imaging probes based on phenoxide-bridged ZnDPA structures.105 More specifically, we evaluated a scaffold derived from L-tyrosine (Scheme 7), which had been employed previously for phospholipid translocation across model bilayer membranes and protein labeling.106-109 Monovalent (mono-T-ZnDPA) and bivalent (bis-T-ZnDPA) deep-red fluorescent probes were synthesized and compared to earlier PSVue-643 for targeting performance and in vivo biodistribution properties. Studies using liposomes showed that the bivalent probe bis-T-ZnDPA had a higher affinity for PS-rich membranes compared to the monovalent probe mono-T-ZnDPA, but bis-T-ZnDPA exhibited fluorescence self-quenching at the membrane surface, a feature that was also apparent in cell imaging experiments. The in vivo biodistribution of mono-T-ZnDPA was very impressive with relatively high targeting of dead and dying tissue and low off-target accumulation in other organs. An analogous, radioactive 111In-labelled version, mono-T-ZnDPA-[111In]DTPA was prepared and found to also exhibit a clean biodistribution profile making it a promising candidate for further development as a clinical imaging probe of cell death.
Scheme 5.
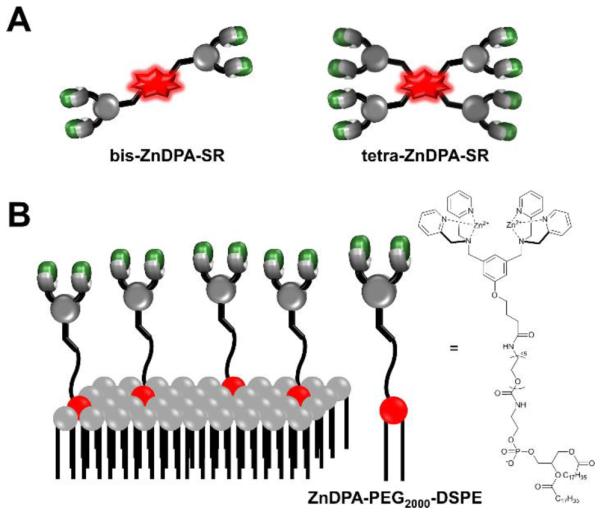
Multivalent ZnDPA probes (A) Bivalent and tetravalent probes containing near-infrared fluorescent squaraine rotaxane reporter groups. (B) Liposome-anchored multivalent ZnDPA probe.
Scheme 6.

(A) Strategy for improving ZnDPA affinity for PS (red) through covalent modification of the targeting group with urea groups (green). (B) Structure of lead compound discovered by screening a library of ureido modified ZnDPA receptors.
Scheme 7.

Structures of monovalent and bivalent T-ZnDPA probes built from L-tyrosine derived scaffold.
Molecular imaging of bacteria
There is a need for fluorescent molecular probes that can detect very small numbers of pathogenic bacterial cells in food, drinking water, or biomedical samples.110-112 The ability of ZnDPA probes to selectively target bacteria was first discovered using PSVue-380.113 Fluorescence microscopy of bacteria treated with PSVue-380 showed strong staining of both Gram-negative Escherichia coli and Gram-positive Staphylococcus aureus bacterial cells with a large enhancement in fluorescence emission intensity upon binding (Fig. 2A). The fluorescent probes appeared to preferentially bind the cell membrane over the intracellular DNA. Also, PSVue-380 could selectively stain bacterial cells in the complex biological medium of saliva without staining off-target mammalian cells (Fig. 2B). Ganesh and co-workers subsequently reported that PSVue-380 has high affinity for bacterial LPS, a primary component of the outer membrane of Gram-negative bacteria and a potent stimulant of the mammalian immune response.114 Studies of other ZnDPA probe designs have consistently demonstrated high affinity for the anionic surfaces of bacteria.115
Fig. 2.

(A) Epifluorescence microscopy of planktonic E. coli (−) after incubation with PSVue-380 (10 μM). (B) Saliva sample containing human epithelial cells (stained red) with an associated ‘clump’ of bacteria stained with PSVue-380 (blue). A and B reproduced with permission from Ref. 113 © Royal Society of Chemistry. (C) Fluorescence microscopy image of human A-549 cells and E. coli K12 (−) cell mixtures were stained with DAPI (1 μg/mL), and treated 15 min later with tetra-ZnDPA-SR. (D) Optical image of a mouse with a S. aureus infection in the left rear leg at 21 hours after intravenous injection of PSVue-794. Reprinted with permission from Ref. 116. © 2006 American Chemical Society. (E) Whole-body fluorescence and bioluminescence imaging of an athymic mouse containing a leg infection of bioluminescent S. enterica serovar typhimurium FL6 (−) and imaged 3 hours after dosage with tetra-ZnDPA-SR. Reprinted with permission from Ref. 119. © 2010 American Chemical Society. (F) SPECT/CT image of a mouse with a leg infection of S. pyogenes and imaged 20 hours post-administration of bis-ZnDPA-[111In]DTPA. Reproduced from with permission Ref. 125. © 2015 Springer.
The selective bacterial staining led us to explore non-invasive imaging of localized infection sites within living mice using PSVue-643 or PSVue-794.116,117 These studies employed a straightforward leg infection model in which a bolus of bacteria was injected into the mouse thigh and allowed to incubate for several hours, followed by injection of the molecular probe into the tail vein and whole-body imaging twenty-four hours later. Target-to-Non-Target (T/NT) ratios were measured by taking region of interest measurements at the infected and uninfected leg. The representative fluorescence image in Fig. 2D shows the distribution of PSVue-794 in a mouse with a leg infection of Gram-positive S. aureus.116, 117 The fluorescent probe cleared slowly from the bloodstream and the T/NT ratio reached four at 21 h post-injection of the probe. Histological samples taken from the infection site indicated that the PSVue-794 probe targeted the bacteria cells and not the dead and dying mammalian cells that surrounded the infection site. Improved fluorescence imaging probes with high bacterial affinity were designed using brighter and more photostable squaraine rotaxane dyes.118,119 These probes were more hydrophilic and washed out more rapidly from the blood clearance organs. Whole-animal optical imaging showed co-localization of the fluorescent ZnDPA probe signal with the bioluminescence signal from a genetically engineered strain of Salmonella enterica serovar typhimurium (S. enterica) (Fig. 2E). Furthermore, there was no accumulation of the ZnDPA probes in sites of sterile inflammation induced by injection of λ-carrageenan. These in vivo studies showed that fluorescent ZnDPA probes were not highly sensitive imaging agents (the localized infection site had to contain at least ~106 colony forming units for fluorescence visualization), but they were remarkably selective for bacterial cells.
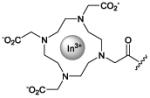
Collectively, these results have encouraged efforts to develop radiolabeled ZnDPA conjugates for clinical imaging. Clinical detection of bacterial infections is crucial for immunosuppressed patients and patients with prosthetic devices due to their increased vulnerability to relatively nonvirulent microorganisms.120 It has been recognized for some time that noninvasive imaging technology will greatly facilitate the process of infection diagnosis.121-123 Liu et al. first produced radiolabeled ZnDPA for nuclear imaging of infection in mice using a streptavidin protein that can bind biotinylated reagents.124 Biotinylated ZnDPA (PSVue-Biotin) and the biotinylated reporter 111In-DOTA were linked to the streptavidin. The nuclear probe was tested in mice injected intramuscularly with Streptococcus pyogenes (infection model) or with lipopolysaccharide (inflammation model). Cohorts were injected intravenously with the probe followed by periodic SPECT/CT imaging which displayed rapid and selective accumulation at the site of S. pyogenes infection. The 111In T/NT ratio was 2.8 fold higher in infection bearing animals compared to sterile inflammation animals, demonstrating that ZnDPA targeting can accurately distinguish bacterial invasion. Our lab subsequently prepared two monovalent radiolabeled tracers, ZnDPA-[111In]DTPA and ZnDPA-[111In]DOTA, each with a single ZnDPA targeting unit, and a bivalent tracer, bis-ZnDPA-[111In]DTPA, and tested them in the same S. Pyogenes infection model.125 All three [111In]-ZnDPA tracers selectively targeted the S. Pyogenes infection with the highest target selectivity observed with divalent bis-ZnDPA-[111In]DTPA. Clearance of the divalent tracer from the bloodstream was slower and primarily through the liver and the T/NT ratio rose to six after 20 h. The SPECT/CT imaging indicated the same large difference in tracer pharmacokinetics and higher accumulation of the divalent tracer at the site of infection (Fig. 2F). The tracer pharmacokinetics depended heavily on tracer molecular structure suggesting that it is possible to fine tune the structural properties for optimized in vivo imaging performance and clinical translation. Additionally, Matosziuk et al. designed a ZnDPA magnetic resonance contrast agent consisting of ZnDPA targeting unit conjugated to a gadolinium chelate. In vitro studies with S. aureus and E. coli show that the ZnDPA contrast agent efficiently labeled bacteria compared to a gadolinium control.126 The ZnDPA probe also significantly reduced the T1 relaxation time of labeled bacteria, thus enhancing magnetic resonance image contrast and illustrating the potential for visualizing bacterial infections using whole body MR imaging.
As stated above, clinical translation of ZnDPA imaging agents would be facilitated if the probes exhibited higher target affinity, and one design strategy is to create multivalent probes. To date, we have prepared and studied the bacteria binding properties of multivalent and dendritic probe architectures with four to thirty-two ZnDPA units, and found them to act as broad-spectrum bacterial agglutination agents.127 The multivalent probes rapidly cross-linked ten different strains of bacteria, regardless of Gram-type and cell morphology. A fluorescent probe with four ZnDPA units (tetra-ZnDPA-SR) was found to target an infection site in a living mouse. The therapeutic potential of multivalent ZnDPA probes as bacteria agglutination agents is discussed further in the next section. The potential of these probes as reporters of cell surface structure was raised by an early bacterial imaging study using CdSe/ZnS core/shell quantum dots coated with PSVue-Biotin.128 Bacterial staining with these extremely bright fluorescent imaging probes was observed to be sensitive to the cell surface topology. For example, there was no staining of smooth E. coli strains with wild type LPS and extended O-antigen polysaccharides which prevented access of the relatively large nanoparticles to the phosphorylated lipid A buried in the outer membrane. Conversely, there was strong staining of a rough E. coli mutant that lacked the O-antigen element.
Therapeutic applications using ZnDPA complexes
Intracellular delivery of macromolecular therapeutics is an important unsolved clinical challenge, particularly for RNA-based treatments (e.g., siRNA, miRNA or oligonucleotides) which are rapidly degraded by tissue enzymes. Recently, Choi et al. applied ZnDPA complexes for the delivery of siRNA by exploiting the selective affinity of ZnDPA for the phosphodiester groups on the siRNA backbone.129 ZnDPA complexes were covalently conjugated to hyaluronic acid-based nanoparticles to enable efficient tumor-targeted delivery of RNAs and small-molecule drugs.130 ZnDPA-coated nanoparticles were loaded with siRNA specific for a bioluminescence gene within cancer cells were and tested in a mouse colon cancer model. Bioluminescent intensities from tumors treated with RNA-delivering nanoparticles showed a substantial decrease in signal 24 h after injection, demonstrating substantial promise for drug delivery.
The bacterial targeting properties of ZnDPA-coated nanoparticles makes them an attractive platform for different types of therapeutic strategies. For example, iron oxide nanoparticles coated with ZnDPA units were designed for the physical removal of bacteria from the bloodstream. Bacterial sepsis is a serious clinical condition that can lead to multiple organ dysfunction and death despite timely treatment with antibiotics and fluid resuscitation. Lee et al. developed ZnDPA-conjugated magnetic nanoparticles and used them for highly selective and rapid separation of bacteria and potentially their endotoxins from whole blood.131 Custom magnetic microfluidic devices were found to remove E. coli bound to the nanoparticles at flow rates as high as 60 mL/h, resulting in almost 100% clearance (Fig. 3A). In principle, such devices could be adapted to clear bacteria from septicemic patients.
Fig. 3.

(A) Magnetophoretic separation of green-fluorescent E. coli (−) from whole blood after pre-treatment with iron oxide nanoparticles coated with ZnDPA using a surrounding magnet within a microfluidic device. Reprinted with permission from Ref. 131. © 2014 American Chemical Society. (B) Selective triggered drug release using ZnDPA association with anionic liposomes causing rapid leakage of encapsulated contents. Reproduced with permission from Ref. 134 © the Royal Society of Chemistry. (C) Representative photographs of mouse footpads infected with cutaneous L. Major 12 days after regimens of saline, antimonial, and ZnDPA. Reproduced with permission from Ref. 136 © American Society for Microbiology. (D) Agar plates with E. coli (−) bacteria treated with photosensitizer conjugated ZnDPA (mDestroy) in the presence and absence of light treatment.
Our group has pursued two different therapeutic ideas using nanoscale liposomes and ZnDPA complexes. In one approach, we prepared liposomes coated with PEG chains terminating with ZnDPA affinity units (Scheme 5B) and found that they selectively agglutinated bacterial cells in the presence of healthy human cells. PEGylated liposomes are known to collect in sites of bacterial infection within living subjects and additional bacteria-specific ligands on the liposome surface should conceivably enhance the accumulation.132, 133 While not bactericidal, the multivalent liposomes may have value as immobilization agents that sequester an infection and perhaps deliver antibiotic cargo. Our second idea is a two-step liposome delivery and triggered drug release strategy. In short, ZnDPA complexes are used to chemically trigger drug release from anionic liposomes that are pre-targeted to a site of disease (Fig. 3B). Proof-of-concept studies have shown that certain ZnDPA structures such as PU-ZnDPA-Trigger (Scheme 6) are very effective at releasing dyes134 or prodrugs135 from liposomes with membranes containing 5 mol% of anionic PS.
Not surprisingly, some ZnDPA compounds have been found to have antibiotic activity, especially against Gram-positive bacteria such as S. aureus with a minimum inhibitory concentration of 1 μg/ml. Preliminary mechanism studies suggest that the mode of action is depolarization of the bacterial membrane. An exciting new discovery is that ZnDPA compounds have selective activity against the trypanosomatid parasite L. Major, a causative agent of the neglected tropical disease cutaneous leishmaniasis.136 Modern treatment regimens rely on a small number of chemotherapeutics with serious side effects and there is a need for more effective alternatives. Fluorescence microscopy of L. major promastigotes treated with a fluorescently labeled ZnDPA probe indicated rapid accumulation of the probe within the axenic promastigote cytosol. Selective antileishmanial activity was observed with eight ZnDPA complexes against L. major axenic promastigotes with 50% effective concentrations (EC50) values in the range of 12.7 – 0.3 μM. In vivo treatment efficacy studies showed that a ZnDPA treatment regimen reduced the parasite burden nearly as well as the reference care agent, potassium antimony(III) tartrate, and with less necrosis in the local host tissue (Fig. 3C). The results demonstrate that ZnDPA coordination complexes are a promising new class of antileishmanial agents with potential for clinical translation.
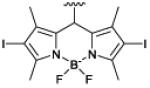
Another therapeutic concept is targeted photodynamic inactivation using photosensitizer appended with ZnDPA units. Photodynamic inactivation is an attractive antimicrobial strategy for blood sterilization and surface decontamination with the potential advantage of killing antibiotic-resistant bacteria with no apparent induction of resistance. We designed two structurally related optical probes that are conjugates of a ZnDPA unit and a BODIPY chromophore: one is a microbial targeted fluorescent imaging agent, mSeek, and the other is an oxygen photosensitizing analogue, mDestroy (Table 1).137 The fluorescent probe, mSeek, is not phototoxic and enabled detection of all tested bacteria at concentrations of ~100 CFU/mL for B. thuringiensis spores, ~1000 CFU/mL for S. aureus and ~10,000 CFU/mL for E. coli. The photosensitizer analogue, mDestroy, inactivated 99–99.99% of bacterial samples and selectively killed bacterial cells in the presence of mammalian cells (Fig. 3D). However, it is worth noting that mDestroy was ineffective against B. thuringiensis spores, a genetic and structural mimic of deadly anthrax spores.
Future Directions
Considering the simplicity of the ZnDPA structure, the binding selectivity for anionic cell surfaces within complex biological media is quite remarkable. One reason for this is that off-target association with polyphosphorylated biomolecules is low since most phosphorylated species are intracellular. As discussed above, imaging and therapeutic performance will be enhanced if anionic membrane affinity can be increased and further studies are warranted to achieve this goal. The development of two other desirable ZnDPA probe features is also worth future attention. One structural feature is fluorescent ZnDPA probes with ‘turn on’ emission that would allow ‘no wash’ in vitro assays or improved in vivo imaging contrast at the target site. Several of our studies have noted self-aggregation of fluorescent ZnDPA probes on the membrane surface and decreased emission intensity due to an aggregation-induced quenching effect.105, 138 Recently, a new fluorescent ZnDPA probe with ‘aggregation-induced emission’ was reported by Hu and coworkers for apoptosis detection.139 The deep-red probe is weakly emissive in aqueous media but turns on when it associates with the surfaces of early apoptotic mammalian cells. A near-infrared version of this type of probe would likely be very useful for in vivo imaging. A second desirable structural feature is the ability to cross the blood-brain barrier (BBB), which would facilitate efforts to image neurodegenerative disease. Non-invasive imaging of common pathophysiological conditions, such as Parkinson’s and Alzheimer’s disease, is greatly limited by the inability of contrast agents to cross the BBB.140 To date, brain imaging with ZnDPA probes has focused on disease models that have a compromised BBB which enables probe entry.95, 141 Nonspecific disruption of the BBB has been employed to facilitate entrance of BBB-impermeable probes into brain parenchyma, but these approaches induce uncontrolled neuronal injuries and expose the brain to circulating neuroactive and neurotoxic agents.140,144 ZnDPA probes have been reported to possess high affinity for neurofibrillary tangles of hyperphosphorylated tau proteins, a component of amyloid plaques which are an established biomarker of Alzheimer’s disease.142,143 Radiolabeled ZnDPA probes that can safely cross the BBB are likely to be excellent tools for quantifying and diagnosing tau protein-related neurodegenerative disorders. Recent community success at formulating small molecules and nanoparticles with BBB-permeation properties is reason for pursuit and optimism.144-147
Nuclear ZnDPA imaging probes should also be examined for real-time monitoring of tumorigenic cell death due to therapeutic intervention. Prescribing effective cancer treatments remains a substantial clinical challenge due to extensive heterogeneity among tumor sizes, cell type and growth behaviors from patient to patient.148 There is an urgent need for technologies that can accurately and rapidly assess therapeutic response to help guide physician decision-making for personalized patient care and to assist researchers in the assessment of experimental treatments in clinical trials. Therapeutic efficacy is universally assessed using the Response Evaluation Criteria in Solid Tumors (RECIST) which are guidelines that define when tumor burden improves ("respond"), stays the same ("stabilize"), or worsens ("progress") during treatment.59 These evaluations are based on size measurements before and after treatment using conventional CT or MRI anatomical imaging. While providing useful structural information, these modalities are very insensitive often requiring long intervals between imaging scans (months to years) to detect noticeable changes in tumor morphology, especially for slow-growing lesions.149 Furthermore, anatomical imaging methods cannot identify tumor status (viable, necrotic, fibrotic etc.) and are highly limited in characterizing responses in tumors that do not change in size during therapy. Consequently, the degree of response may be underestimated or overestimated leading to incorrect conclusions.150 Molecular imaging strategies are slowly becoming the standard in monitoring tumor response.151 For example, the PET tracer 18F-FDG, a molecular imaging agent for glucose uptake, plays an essential role in defining tumor response in malignant lymphoma by monitoring tumor cell vitality before and after treatment.152, 153 As a complement, ZnDPA probes could be employed to monitor levels of therapeutic tumor deterioration and concurrently assess off-target cell death in organs tissues susceptible to side effects such as the heart and liver. A dual imaging strategy can be envisioned where multiple probes simultaneously monitor tumorigenic death (ZnDPA) and active metabolism (18FDG), potentially offering a new methodology for promptly evaluating treatment response in solid tumors and surrounding tissue.
In terms of therapeutic applications, ZnDPA targeting of PS on dead and dying cancer cells may be a useful way to alter the immune status of tumor tissue. In general, macrophages quickly ingest apoptotic cells exposing PS and release an anti-inflammatory response, which can promote tumor growth. However, if apoptotic cells are not efficiently cleared, the surrounding tissue cells can enter secondary necrosis, a process where the plasma membrane becomes irreversibly permeable causing the release of damage-associated proteins involved in the heat shock response and DNA organization.154 The release of these proteins signals dendritic cells to acquire antigens from dead cells, which in turn induces a co-stimulatory immune response against them. Thus, it is advantageous to shift the clearance of dead and dying cells from macrophages to dendritic cells when developing treatments for cancer and other diseases. Munoz et al. tested this concept using annexin V to determine if blocking PS could make apoptotic cells immunogenic in vivo.155 They discovered that annexin V inhibited cell phagocytosis by monocyte-derived macrophages, thus triggering a change in the immunogenicity of apoptotic and necrotic cells. When incubated with apoptotic tumor cells, annexin V inhibited cell clearance from macrophages initiating secondary necrosis and subsequent identification and uptake by dendritic cells.156 Intravenous dosing with annexin V has been shown to reduce colorectal tumor growth, indicating PS-targeting may naturally induce tumor regression. Similar tumor regression activity has been observed with other PS-specific antibodies.157-159 Taken together, molecular targeting of PS on apoptotic cells may be an effective strategy for initiating a pro-inflammatory response towards tumor tissue, and ZnDPA complexes may be especially good candidates for this type of immunotherapy.
Acknowledgements
We are grateful for funding support from the NIH (RO1GM059078 to B.D.S. and T32GM075762 to D.R.R.), GAANN (K.J.C.) and technical support from the University of Notre Dame, the Notre Dame Integrated Imaging Facility, the Harper Cancer Research Institute Imaging and Flow Cytometry Core Facility, and the Freimann Life Sciences Center.
Biographies
Douglas R. Rice

Douglas Rice was born in Portland, Oregon. He received his BA in Biochemistry from Willamette University in 2010 and his PhD from the University of Notre Dame in 2015. His dissertation research focused on developing molecular imaging strategies for identifying cancer therapeutic efficacy and bacterial infection using targeted nanoparticles and small molecules. Currently, he is developing and studying novel therapeutic agents for parasitic diseases.
Kasey J. Clear

Kasey J. Clear was born in Niles, Michigan. He was awarded his B.S. in Chemistry in 2011 from Indiana University South Bend, and he completed his Ph.D. in 2016 at the University of Notre Dame. He is currently an Assistant Professor of Chemistry at Murray State University. His research interests include the design and study of synthetic receptors for biologically important anions.
Bradley D. Smith

Bradley D. Smith is Emil T. Hofman Professor of Chemistry and Biochemistry and Director of the Notre Dame Integrated Imaging Facility. His research interests are primarily in the fields of bioorganic and supramolecular chemistry applied to biological systems. A current topic is the creation of molecular probes for detecting and treating cancer and microbial infections.
Footnotes
The authors declare no financial conflict of interest.
Several fluorescent ZnDPA probes are commercially available from Molecular Targeting Technologies Incorporated (MTTI, www.mtarget.com) under the brand name PSVue®. MTTI has a license agreement with the University of Notre Dame, the employer of B. D. Smith.
- PS
- phosphatidylserine
- PG
- phosphatidylglycerol
- CL
- cardiolipin
- PC
- phosphatidylcholine
- PE
- phosphatidylethanolamine
- DPA
- zinc(II)-dipicolylamine
- DPA
- dipicolylamine
- 7AAD
- 7-aminoactinomycin
- NMDA
- N-methyl-D-aspartate
- GAG
- glycosaminoglycan
- SPECT
- single photon emission computed tomography
- PET
- positron emission tomography
- FDG
- fluorodeoxyglucose
- FRET
- Förster resonance energy transfer
- DOTA
- 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid
- DTPA
- diethylenetriaminepentaacetic acid
- LPS
- lipopolysaccharide
- BBB
- blood-brain barrier
References
- 1.van Meer G, Voelker DR, Feigenson GW. Nat. Rev. Mol. Cell. Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simons K, Sampaio JL. Cold Spring Harb. Perspect. Biol. 2011;3:a004697. doi: 10.1101/cshperspect.a004697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brandley BK, Schnaar RL. J. Leukocyte Biol. 1986;40:97–111. doi: 10.1002/jlb.40.1.97. [DOI] [PubMed] [Google Scholar]
- 4.Fadeel B, Xue D. Crit. Rev. Biochem. Mol. Biol. 2009;44:264–277. doi: 10.1080/10409230903193307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uchida Y, Hasegawa J, Chinnapen D, Inoue T, Okazaki S, Kato R, Wakatsuki S, Misaki R, Koike M, Uchiyama Y, Iemura S, Natsume T, Kuwahara R, Nakagawa T, Nishikawa K, Mukai K, Miyoshi E, Taniguchi N, Sheff D, Lencer WI, Taguchi T, Arai H. Proc. Natl. Acad. Sci. U. S. A. 2011;108:15846–15851. doi: 10.1073/pnas.1109101108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee SH, Meng XW, Flatten KS, Loegering DA, Kaufmann SH. Cell Death. Differ. 2013;20:64–76. doi: 10.1038/cdd.2012.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lentz BR. Prog. Lipid Res. 2003;42:423–438. doi: 10.1016/s0163-7827(03)00025-0. [DOI] [PubMed] [Google Scholar]
- 8.Leventis PA, Grinstein S. Annu. Rev. Biophys. 2010;39:407–427. doi: 10.1146/annurev.biophys.093008.131234. [DOI] [PubMed] [Google Scholar]
- 9.Schutters K, Reutelingsperger C. Apoptosis. 2010;15:1072–1082. doi: 10.1007/s10495-010-0503-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quigley HA, Nickells RW, Kerrigan LA, Pease ME, Thibault DJ, Zack DJ. Invest. Ophthalmol. Vis. Sci. 1995;36:774–786. [PubMed] [Google Scholar]
- 11.Okisaka S, Murakami A, Mizukawa A, Ito J. Jpn. J. Ophthalmol. 1997;41:84–88. doi: 10.1016/s0021-5155(97)00013-0. [DOI] [PubMed] [Google Scholar]
- 12.Galvao J, Davis BM, Cordeiro MF. Curr. Opin. Pharmacol. 2013;13:123–127. doi: 10.1016/j.coph.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 13.Eda S, Sherman IW. Cell. Physiol. Biochem. 2002;12:373–384. doi: 10.1159/000067908. [DOI] [PubMed] [Google Scholar]
- 14.Yasin Z, Witting S, Palascak MB, Joiner CH, Rucknagel DL, Franco RS. Blood. 2003;102:365–370. doi: 10.1182/blood-2002-11-3416. [DOI] [PubMed] [Google Scholar]
- 15.Porreca E, Marchisio M, Di Nisio M, Moretta V, Lanuti P, Pierdomenico L, Cuccurullo F, Miscia S. Thromb. Res. 2009;124:502–504. doi: 10.1016/j.thromres.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 16.Connor J, Bucana C, Fidler IJ, Schroit AJ. Proc. Natl. Acad. Sci. U. S. A. 1989;86:3184–3188. doi: 10.1073/pnas.86.9.3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schroder-Borm H, Bakalova R, Andra J. FEBS Lett. 2005;579:6128–6134. doi: 10.1016/j.febslet.2005.09.084. [DOI] [PubMed] [Google Scholar]
- 18.Utsugi T, Schroit AJ, Connor J, Bucana CD, Fidler IJ. Cancer Res. 1991;51:3062–3066. [PubMed] [Google Scholar]
- 19.Woehlecke H, Pohl A, Alder-Baerens N, Lage H, Herrmann A. Biochem. J. 2003;376:489–495. doi: 10.1042/BJ20030886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zwaal RF, Comfurius P, Bevers EM. Cell. Mol. Life Sci. 2005;62:971–988. doi: 10.1007/s00018-005-4527-3. [DOI] [PubMed] [Google Scholar]
- 21.Zwaal RF, Schroit AJ. Blood. 1997;89:1121–1132. [PubMed] [Google Scholar]
- 22.Sugimura M, Donato R, Kakkar VV, Scully MF. Blood Coagul. Fibrinolysis. 1994;5:365–373. [PubMed] [Google Scholar]
- 23.Rao LV, Tait JF, Hoang AD. Thromb. Res. 1992;67:517–531. doi: 10.1016/0049-3848(92)90013-z. [DOI] [PubMed] [Google Scholar]
- 24.Huynh ML, Fadok VA, Henson PM. J. Clin. Invest. 2002;109:41–50. doi: 10.1172/JCI11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amara A, Mercer J. Nat. Rev. Microbiol. 2015;13:461–469. doi: 10.1038/nrmicro3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maruri-Avidal L, Weisberg AS, Moss B. J. Virol. 2013;87:12313–12326. doi: 10.1128/JVI.02137-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lorizate M, Sachsenheimer T, Glass B, Habermann A, Gerl MJ, Krausslich HG, Brugger B. Cell. Microbiol. 2013;15:292–304. doi: 10.1111/cmi.12101. [DOI] [PubMed] [Google Scholar]
- 28.Fairn GD, Schieber NL, Ariotti N, Murphy S, Kuerschner L, Webb RI, Grinstein S, Parton RG. J. Cell. Biol. 2011;194:257–275. doi: 10.1083/jcb.201012028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mercer J, Helenius A. Ann. N. Y. Acad. Sci. 2010;1209:49–55. doi: 10.1111/j.1749-6632.2010.05772.x. [DOI] [PubMed] [Google Scholar]
- 30.Vanlandschoot P, Leroux-Roels G. Trends Immunol. 2003;24:144–147. doi: 10.1016/s1471-4906(03)00026-7. [DOI] [PubMed] [Google Scholar]
- 31.Callahan MK, Popernack PM, Tsutsui S, Truong L, Schlegel RA, Henderson AJ. J. Immunol. 2003;170:4840–4845. doi: 10.4049/jimmunol.170.9.4840. [DOI] [PubMed] [Google Scholar]
- 32.Richard AS, Zhang A, Park SJ, Farzan M, Zong M, Choe H. Proc. Natl. Acad. Sci. U. S. A. 2015;112:14682–14687. doi: 10.1073/pnas.1508095112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wanderley JL, Benjamin A, Real F, Bonomo A, Moreira ME, Barcinski MA. Braz. J. Med. Biol. Res. 2005;38:807–812. doi: 10.1590/s0100-879x2005000600001. [DOI] [PubMed] [Google Scholar]
- 34.Wassef MK, Fioretti TB, Dwyer DM. Lipids. 1985;20:108–115. doi: 10.1007/BF02534216. [DOI] [PubMed] [Google Scholar]
- 35.Wanderley JL, Thorpe PE, Barcinski MA, Soong L. Parasite Immunol. 2013;35:109–119. doi: 10.1111/pim.12019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Glew RH, Saha AK, Das S, Remaley AT. Microbiol. Rev. 1988;52:412–432. doi: 10.1128/mr.52.4.412-432.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turco SJ, Descoteaux A. Annu. Rev. Microbiol. 1992;46:65–94. doi: 10.1146/annurev.mi.46.100192.000433. [DOI] [PubMed] [Google Scholar]
- 38.Weingartner A, Kemmer G, Muller FD, Zampieri RA, Gonzaga dos Santos M, Schiller J, Pomorski TG. PLoS One. 2012;7 doi: 10.1371/journal.pone.0042070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nguewa PA, Fuertes MA, Valladares B, Alonso C, Perez JM. Trends Parasitol. 2004;20:375–380. doi: 10.1016/j.pt.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 40.Wanderley JL, Moreira ME, Benjamin A, Bonomo AC, Barcinski MA. J. Immunol. 2006;176:1834–1839. doi: 10.4049/jimmunol.176.3.1834. [DOI] [PubMed] [Google Scholar]
- 41.Santos TA, Jde A. Porte., Damasceno-Sa JC, Caldas LA, Souza W, Damatta RA, Seabra SH. PLoS One. 2011;6:e27867. doi: 10.1371/journal.pone.0027867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Montoya JG, Liesenfeld O. Lancet. 2004;363:1965–1976. doi: 10.1016/S0140-6736(04)16412-X. [DOI] [PubMed] [Google Scholar]
- 43.Czuczman MA, Fattouh R, van Rijn JM, Canadien V, Osborne S, Muise AM, Kuchroo VK, Higgins DE, Brumell JH. Nature. 2014;509:230–234. doi: 10.1038/nature13168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ratledge C, Wilkinson SG. Microbial Lipids Volume One. Academic Press Inc.; San Diego, CA: 1988. [Google Scholar]
- 45.Rosch JW, Hsu FF, Caparon MG. J. Bacteriol. 2007;189:801–806. doi: 10.1128/JB.01549-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haest CW, de Gier J, den Kamp JO, Bartels P, van Deenen LL. Biochim. Biophys. Acta. 1972;255:720–733. doi: 10.1016/0005-2736(72)90385-9. [DOI] [PubMed] [Google Scholar]
- 47.Raetz CR. Microbiol. Rev. 1978;42:614–659. doi: 10.1128/mr.42.3.614-659.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lugtenberg B, Van Alphen L. Biochim. Biophys. Acta. 1983;737:51–115. doi: 10.1016/0304-4157(83)90014-x. [DOI] [PubMed] [Google Scholar]
- 49.Osborn MJ, Gander JE, Parisi E, Carson J. J. Biol. Chem. 1972;247:3962–3972. [PubMed] [Google Scholar]
- 50.Kotra LP, Golemi D, Amro NA, Liu GY, Mobashery S. J. Am. Chem. Soc. 1999;121:8707–8711. [Google Scholar]
- 51.Raetz CRH, Whitfield C. Annu. Rev. Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Raetz CRH, Reynolds CM, Trent MS, Bishop RE. Annu. Rev. Biochem. 2007;76:295–329. doi: 10.1146/annurev.biochem.76.010307.145803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ghosal S, Leighton TJ, Wheeler KE, Hutcheon ID, Weber PK. Appl. Environ. Microbiol. 2010;76:3275–3282. doi: 10.1128/AEM.02485-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Silhavy TJ, Kahne D, Walker S. Cold Spring Harb. Perspect. Biol. 2010;2:a000414. doi: 10.1101/cshperspect.a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ebenhan T, Gheysens O, Kruger HG, Zeevaart JR, Sathekge MM. BioMed Res. Int. 2014;2014:867381. doi: 10.1155/2014/867381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kersemans V, Cornelissen B. Pharmaceuticals. 2010;3:600–620. doi: 10.3390/ph3030600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zaro JL, Shen WC. Front. Chem. Sci. Eng. 2015;9:407–427. [Google Scholar]
- 58.An FF, Cao W, Liang XJ. Adv. Healthcare Mater. 2014;3:1162–1181. doi: 10.1002/adhm.201300600. [DOI] [PubMed] [Google Scholar]
- 59.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J. Eur. J. Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 60.Brown KL, Hancock RE. Curr. Opin. Immunol. 2006;18:24–30. doi: 10.1016/j.coi.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 61.DeRose P, Thorpe PE, Gerber DE. Immunotherapy. 2011;3:933–944. doi: 10.2217/imt.11.87. [DOI] [PubMed] [Google Scholar]
- 62.Boersma HH, Kietselaer BL, Stolk LM, Bennaghmouch A, Hofstra L, Narula J, Heidendal GA, Reutelingsperger CP. J. Nucl. Med. 2005;46:2035–2050. [PubMed] [Google Scholar]
- 63.Hou J, Fu Y, Zhou J, Li W, Xie R, Cao F, Gilbert GE, Shi J. Vox Sang. 2011;100:187–195. doi: 10.1111/j.1423-0410.2010.01375.x. [DOI] [PubMed] [Google Scholar]
- 64.Fang W, Wang F, Ji S, Zhu X, Meier HT, Hellman RS, Brindle KM, Davletov B, Zhao M. Nucl. Med. Biol. 2007;34:917–923. doi: 10.1016/j.nucmedbio.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 65.Murphy MP, Smith RA. Annu. Rev. Pharmacol. Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- 66.Simoes S, Filipe A, Faneca H, Mano M, Penacho N, Duzgunes N, de Lima MP. Expert Opin. Drug. Deliv. 2005;2:237–254. doi: 10.1517/17425247.2.2.237. [DOI] [PubMed] [Google Scholar]
- 67.Zheng H, Wang F, Wang Q, Gao J. J. Am. Chem. Soc. 2011;133:15280–15283. doi: 10.1021/ja205911n. [DOI] [PubMed] [Google Scholar]
- 68.Xiong C, Brewer K, Song S, Zhang R, Lu W, Wen X, Li C. J. Med. Chem. 2011;54:1825–1835. doi: 10.1021/jm101477d. [DOI] [PubMed] [Google Scholar]
- 69.Marcus Y. Ion Properties. Marcel Dekker; New York ; Basel: 1997. [Google Scholar]
- 70.Biniecki S, Kabzinska S. Ann. Pharm. Fr. 1964;22:685–687. [PubMed] [Google Scholar]
- 71.Burdette SC, Walkup GK, Spingler B, Tsien RY, Lippard SJ. J. Amer. Chem. Soc. 2001;123:7831–7841. doi: 10.1021/ja010059l. [DOI] [PubMed] [Google Scholar]
- 72.Chang CJ, Lippard SJ. Neurodegenerative Diseases and Metal Ions. John Wiley & Sons, Ltd; 2006. pp. 321–370. [Google Scholar]
- 73.Zhang XA, Lovejoy KS, Jasanoff A, Lippard SJ. Proc. Natl. Acad. Sci. U. S. A. 2007;104:10780–10785. doi: 10.1073/pnas.0702393104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.O'Neil EJ, Smith BD. Coord. Chem. Rev. 2006;250:3068–3080. [Google Scholar]
- 75.Ojida A, Mito-Oka Y, Inoue MA, Hamachi I. J. Am. Chem. Soc. 2002;124:6256–6258. doi: 10.1021/ja025761b. [DOI] [PubMed] [Google Scholar]
- 76.Ojida A, Mito-oka Y, Sada K, Hamachi I. J. Am. Chem. Soc. 2004;126:2454–2463. doi: 10.1021/ja038277x. [DOI] [PubMed] [Google Scholar]
- 77.Nonaka H, Tsukiji S, Ojida A, Hamachi I. J. Am. Chem. Soc. 2007;129:15777–15779. doi: 10.1021/ja074176d. [DOI] [PubMed] [Google Scholar]
- 78.Ojida A, Hamachi I. Bull. Chem. Soc. Jpn. 2006;79:35–46. [Google Scholar]
- 79.Ojida A, Park S, Mito-oka Y, Hamachi I. Tetrahedron Lett. 2002;43:6193–6195. [Google Scholar]
- 80.Rhee HW, Choi HY, Han K, Hong JI. J. Am. Chem. Soc. 2007;129:4524–4525. doi: 10.1021/ja070026r. [DOI] [PubMed] [Google Scholar]
- 81.Ngo HT, Liu X, Jolliffe KA. Chem. Soc. Rev. 2012;41:4928–4965. doi: 10.1039/c2cs35087d. [DOI] [PubMed] [Google Scholar]
- 82.Smith B, editor. Synthetic Receptors for Biomolecules: Design Principles and Applications. Royal Society of Chemistry; Cambridge: 2015. [Google Scholar]
- 83.Minami T, Minamiki T, Tokito S. Chem. Lett. 2016;45:371–373. [Google Scholar]
- 84.Williams FJ, Fiedler D. ACS Chem. Biol. 2015;10:1958–1963. doi: 10.1021/acschembio.5b00256. [DOI] [PubMed] [Google Scholar]
- 85.Conway JH, Fiedler D. Angew. Chem. 2015;54:3941–3945. doi: 10.1002/anie.201411232. [DOI] [PubMed] [Google Scholar]
- 86.Koulov AV, Stucker KA, Lakshmi C, Robinson JP, Smith BD. Cell Death. Differ. 2003;10:1357–1359. doi: 10.1038/sj.cdd.4401315. [DOI] [PubMed] [Google Scholar]
- 87.Koulov AV, Hanshaw RG, Stucker KA, Lakshmi C, Smith BD. Israel J. Chem. 2005;45:373–379. [Google Scholar]
- 88.Lakshmi C, Hanshaw RG, Smith BD. Tetrahedron. 2004;60:11307–11315. [Google Scholar]
- 89.Hanshaw RG, Lakshmi C, Lambert TN, Johnson JR, Smith BD. ChemBioChem. 2005;6:2214–2220. doi: 10.1002/cbic.200500149. [DOI] [PubMed] [Google Scholar]
- 90.Kwong JM, Hoang C, Dukes RT, Yee RW, Gray BD, Pak KY, Caprioli J. Invest. Ophthalmol. Vis. Sci. 2014;55:4913–4921. doi: 10.1167/iovs.13-13346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Smith AM, Mancini MC, Nie S. Nat. Nanotechnol. 2009;4:710–711. doi: 10.1038/nnano.2009.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cui SB. Act. Math.Sci. 2006;26:781–796. [Google Scholar]
- 93.Smith BA, Xiao S, Wolter W, Wheeler J, Suckow MA, Smith BD. Apoptosis. 2011;16:722–731. doi: 10.1007/s10495-011-0601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Palmowski K, Rix A, Lederle W, Behrendt FF, Mottaghy FM, Gray BD, Pak KY, Palmowski M. Eur. Radiol. 2014;24:363–370. doi: 10.1007/s00330-013-3014-8. [DOI] [PubMed] [Google Scholar]
- 95.Smith BA, Xie BW, van Beek ER, Que I, Blankevoort V, Xiao S, Cole EL, Hoehn M, Kaijzel EL, Lowik CW, Smith BD. ACS Chem. Neurosci. 2012;3:530–537. doi: 10.1021/cn3000197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chan MM, Gray BD, Pak KY, Fong D. Arthritis Res. Ther. 2015;17:50. doi: 10.1186/s13075-015-0565-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hu X, Wang Q, Liu Y, Liu H, Qin C, Cheng K, Robinson W, Gray BD, Pak KY, Yu A, Cheng Z. Biomaterials. 2014;35:7511–7521. doi: 10.1016/j.biomaterials.2014.05.042. [DOI] [PubMed] [Google Scholar]
- 98.Chan MM, Moore AR. J. Immunol. 2010;184:6418–6426. doi: 10.4049/jimmunol.0903816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wyffels L, Gray BD, Barber C, Moore SK, Woolfenden JM, Pak KY, Liu Z. Bioorg. Med. Chem. 2011;19:3425–3433. doi: 10.1016/j.bmc.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang H, Tang X, Tang G, Huang T, Liang X, Hu K, Deng H, Yi C, Shi X, Wu K. Apoptosis. 2013;18:1017–1027. doi: 10.1007/s10495-013-0852-4. [DOI] [PubMed] [Google Scholar]
- 101.Sun T, Tang G, Tian H, Hu K, Yao S, Su Y, Wang C. Oncotarget. 2015;6:30579–30591. doi: 10.18632/oncotarget.5769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Smith BA, Harmatys KM, Xiao S, Cole EL, Plaunt AJ, Wolter W, Suckow MA, Smith BD. Mol. Pharm. 2013;10:3296–3303. doi: 10.1021/mp300720k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hanshaw RG, Stahelin RV, Smith BD. Chemistry. 2008;14:1690–1697. doi: 10.1002/chem.200701589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Plaunt AJ, Harmatys KM, Wolter WR, Suckow MA, Smith BD. Bioconjugate Chem. 2014;4:727–737. doi: 10.1021/bc500003x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Clear KJ, Harmatys KM, Rice DR, Wolter WR, Suckow MA, Wang Y, Rusckowski M, Smith BD. Bioconjugate Chem. 2016;27:363–375. doi: 10.1021/acs.bioconjchem.5b00447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jiang H, O'Neil EJ, DiVittorio KM, Smith BD. Org. Lett. 2005;7:3013–3016. doi: 10.1021/ol0510421. [DOI] [PubMed] [Google Scholar]
- 107.Nonaka H, Fujishima S.-h., Uchinomiya S.-h., Ojida A, Hamachi I. J. Am. Chem. Soc. 2010;132:9301–9309. doi: 10.1021/ja910703v. [DOI] [PubMed] [Google Scholar]
- 108.Ojida A, Honda K, Shinmi D, Kiyonaka S, Mori Y, Hamachi I. J. Am. Chem. Soc. 2006;128:10452–10459. doi: 10.1021/ja0618604. [DOI] [PubMed] [Google Scholar]
- 109.Honda K, Nakata E, Ojida A, Hamachi I. Chem. Commun. 2006:4024–4026. doi: 10.1039/b608684e. [DOI] [PubMed] [Google Scholar]
- 110.Swaminathan B, Feng P. Annu. Rev. Microbiol. 1994;48:401–426. doi: 10.1146/annurev.mi.48.100194.002153. [DOI] [PubMed] [Google Scholar]
- 111.Noble RT, Weisberg SB. J. Water Health. 2005;3:381–392. doi: 10.2166/wh.2005.051. [DOI] [PubMed] [Google Scholar]
- 112.Ahmed A, Rushworth JV, Hirst NA, Millner PA. Clin. Microbiol. Rev. 2014;27:631–646. doi: 10.1128/CMR.00120-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Leevy WM, Johnson JR, Lakshmi C, Morris J, Marquez M, Smith BD. Chem. Commun. 2006:1595–1597. doi: 10.1039/b517519d. [DOI] [PubMed] [Google Scholar]
- 114.Ganesh V, Bodewits K, Bartholdson SJ, Natale D, Campopiano DJ, Mareque-Rivas JC. Angew. Chem., Int. Ed. 2009;48:356–360. doi: 10.1002/anie.200804168. [DOI] [PubMed] [Google Scholar]
- 115.DiVittorio KM, Leevy WM, O'Neil EJ, Johnson JR, Vakulenko S, Morris JD, Rosek KD, Serazin N, Hilkert S, Hurley S, Marquez M, Smith BD. ChemBioChem. 2008;9:286–293. doi: 10.1002/cbic.200700489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Leevy WM, Gammon ST, Jiang H, Johnson JR, Maxwell DJ, Jackson EN, Marquez M, Piwnica-Worms D, Smith BD. J. Am. Chem. Soc. 2006;128:16476–16477. doi: 10.1021/ja0665592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.White AG, Gray BD, Pak KY, Smith BD. Bioorg. Med. Chem. Lett. 2012;22:2833–2836. doi: 10.1016/j.bmcl.2012.02.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lee JJ, White AG, Baumes JM, Smith BD. Chem. Commun. 2010;46:1068–1069. doi: 10.1039/b924350j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.White AG, Fu N, Leevy WM, Lee JJ, Blasco MA, Smith BD. Bioconjugate Chem. 2010;21:1297–1304. doi: 10.1021/bc1000998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rubin RH, Fischman AJ. Clin. Infect. Dis. 1996;22:414–423. doi: 10.1093/clinids/22.3.414. [DOI] [PubMed] [Google Scholar]
- 121.Goldsmith SJ, Vallabhajosula S. Semin. Nucl. Med. 2009;39:2–10. doi: 10.1053/j.semnuclmed.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 122.Bunschoten A, Welling MM, Termaat MF, Sathekge M, van Leeuwen FW. Bioconjugate Chem. 2013;24:1971–1989. doi: 10.1021/bc4003037. [DOI] [PubMed] [Google Scholar]
- 123.Sasser TA, Van Avermaete AE, White A, Chapman S, Johnson JR, Van Avermaete T, Gammon ST, Leevy WM. Curr. Top. Med. Chem. 2013;13:479–487. doi: 10.2174/1568026611313040008. [DOI] [PubMed] [Google Scholar]
- 124.Liu X, Cheng D, Gray BD, Wang Y, Akalin A, Rusckowski M, Pak KY, Hnatowich DJ. Nucl. Med. Biol. 2012;39:709–714. doi: 10.1016/j.nucmedbio.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 125.Rice DR, Plaunt AJ, Turkyilmaz S, Smith M, Wang Y, Rusckowski M, Smith BD. Mol. Imaging Biol. 2015;17:204–213. doi: 10.1007/s11307-014-0758-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Matosziuk LM, Harney AS, Macrenaris KW, Meade TJ. Eur. J. Inorg. Chem. 2012;2012:2099–2107. doi: 10.1002/ejic.201101362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Xiao S, Abu-Esba L, Turkyilmaz S, White AG, Smith BD. Theranostics. 2013;3:658–666. doi: 10.7150/thno.6811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Leevy WM, Lambert TN, Johnson JR, Smith BD. Chem. Comm. 2008;20:2331–2333. doi: 10.1039/b803590c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Choi KY, Silvestre OF, Huang X, Min KH, Howard GP, Hida N, Jin AJ, Carvajal N, Lee SW, Hong JI, Chen X. ACS Nano. 2014;8:4559–4570. doi: 10.1021/nn500085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Choi KY, Silvestre OF, Huang X, Hida N, Liu G, Ho DN, Lee S, Lee SW, Hong JI, Chen X. Nat. Protoc. 2014;9:1900–1915. doi: 10.1038/nprot.2014.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lee JJ, Jeong KJ, Hashimoto M, Kwon AH, Rwei A, Shankarappa SA, Tsui JH, Kohane DS. Nano Lett. 2014;14:1–5. doi: 10.1021/nl3047305. [DOI] [PubMed] [Google Scholar]
- 132.Azzopardi EA, Ferguson EL, Thomas DW. J. Antimicrob. Chemother. 2013;68:257–274. doi: 10.1093/jac/dks379. [DOI] [PubMed] [Google Scholar]
- 133.Gao W, Thamphiwatana S, Angsantikul P, Zhang L. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2014;6:532–547. doi: 10.1002/wnan.1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Plaunt AJ, Courbanou MB, Cuison KD, Harmatys KM, Smith BD. Chem. Commun. 2012;48:8123–8125. doi: 10.1039/c2cc32962j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Plaunt AJ, Harmatys KM, Hendrie KA, Musso AJ, Smith BD. RSC Adv. 2014;4:57983–57990. doi: 10.1039/C4RA10340H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rice DR, Vacchina P, Norris-Mullins B, Morales MA, Smith BD. Antimicrob. Agents Chemother. 2016;60:2932–2940. doi: 10.1128/AAC.00410-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rice DR, Gan H, Smith BD. Photochem. Photobiol. Sci. 2015;14:1271–1281. doi: 10.1039/c5pp00100e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Suzuki Y, Yokoyama K. J. Am. Chem. Soc. 2005;127:17799–17802. doi: 10.1021/ja054739q. [DOI] [PubMed] [Google Scholar]
- 139.Hu Q, Gao M, Feng G, Chen X, Liu B. ACS Appl. Mater. Interfaces. 2015;7:4875–4882. doi: 10.1021/am508838z. [DOI] [PubMed] [Google Scholar]
- 140.Pardridge WM. NeuroRx. 2005;2:3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chu C, Huang X, Chen CT, Zhao Y, Luo JJ, Gray BD, Pak KY, Dun NJ. Mol. Imaging. 2013;12:8–16. [PubMed] [Google Scholar]
- 142.Ojida A, Sakamoto T, Inoue MA, Fujishima SH, Lippens G, Hamachi I. J. Am. Chem. Soc. 2009;131:6543–6548. doi: 10.1021/ja9008369. [DOI] [PubMed] [Google Scholar]
- 143.Kim HY, Sengupta U, Shao P, Guerrero-Munoz MJ, Kayed R, Bai M. Am. J. Nucl. Med. Mol. Imaging. 2013;3:102–117. [PMC free article] [PubMed] [Google Scholar]
- 144.Mikitsh JL, Chacko AM. Perspect. Med. Chem. 2014;6:11–24. doi: 10.4137/PMC.S13384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Koffie RM, Farrar CT, Saidi L-J, William CM, Hyman BT, Spires-Jones TL. Proc. Natl. Acad. Sci. U. S. A. 2011;108:18837–18842. doi: 10.1073/pnas.1111405108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bacskai BJ, Klunk WE, Mathis CA, Hyman BT. J. Cereb. Blood. Flow. Metab. 2002;22:1035–1041. doi: 10.1097/00004647-200209000-00001. [DOI] [PubMed] [Google Scholar]
- 147.Ono M, Saji H. Int. J. Mol. Imaging. 2011;2011 doi: 10.1155/2011/543267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Fisher R, Pusztai L, Swanton C. Br. J. Cancer. 2013;108:479–485. doi: 10.1038/bjc.2012.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.El Sharouni SY, Kal HB, Battermann JJ. Br. J. Cancer. 2003;89:2184–2189. doi: 10.1038/sj.bjc.6601418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Birchard KR, Hoang JK, Herndon JE, Jr., Patz EF., Jr. Cancer. 2009;115:581–586. doi: 10.1002/cncr.24060. [DOI] [PubMed] [Google Scholar]
- 151.van Persijn van Meerten EL, Gelderblom H, Bloem JL. Eur. Radiol. 2010;20:1456–1467. doi: 10.1007/s00330-009-1685-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Weber WA. J. Nucl. Med. 2009;50(Suppl 1):1S–10S. doi: 10.2967/jnumed.108.057174. [DOI] [PubMed] [Google Scholar]
- 153.Wahl RL, Jacene H, Kasamon Y, Lodge MA. J. Nucl. Med. 2009;50(Suppl 1):122S–150S. doi: 10.2967/jnumed.108.057307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zitvogel L, Kepp O, Kroemer G. Cell. 2010;140:798–804. doi: 10.1016/j.cell.2010.02.015. [DOI] [PubMed] [Google Scholar]
- 155.Munoz LE, Franz S, Pausch F, Furnrohr B, Sheriff A, Vogt B, Kern PM, Baum W, Stach C, von Laer D, Brachvogel B, Poschl E, Herrmann M, Gaipl US. J. Leukocyte Biol. 2007;81:6–14. doi: 10.1189/jlb.0306166. [DOI] [PubMed] [Google Scholar]
- 156.Frey B, Schildkopf P, Rodel F, Weiss EM, Munoz LE, Herrmann M, Fietkau R, Gaipl US. J. Immunotoxicol. 2009;6:209–216. doi: 10.3109/15476910903204058. [DOI] [PubMed] [Google Scholar]
- 157.Huang X, Bennett M, Thorpe PE. Cancer Res. 2005;65:4408–4416. doi: 10.1158/0008-5472.CAN-05-0031. [DOI] [PubMed] [Google Scholar]
- 158.Beck AW, Luster TA, Miller AF, Holloway SE, Conner CR, Barnett CC, Thorpe PE, Fleming JB, Brekken RA. Int. J. Cancer. 2006;118:2639–2643. doi: 10.1002/ijc.21684. [DOI] [PubMed] [Google Scholar]
- 159.He J, Yin Y, Luster TA, Watkins L, Thorpe PE. Clin. Cancer Res. 2009;15:6871–6880. doi: 10.1158/1078-0432.CCR-09-1499. [DOI] [PubMed] [Google Scholar]