

ABSTRACT
Familial hypocalciuric hypercalcemia (FHH) is a genetically heterogeneous disorder with three variants, FHH1 to FHH3. FHH1 is caused by loss‐of‐function mutations of the calcium‐sensing receptor (CaSR), a G‐protein coupled receptor that predominantly signals via G‐protein subunit alpha‐11 (Gα11) to regulate calcium homeostasis. FHH2 is the result of loss‐of‐function mutations in Gα11, encoded by GNA11, and to date only two FHH2‐associated Gα11 missense mutations (Leu135Gln and Ile200del) have been reported. FHH3 is the result of loss‐of‐function mutations of the adaptor protein‐2 σ‐subunit (AP2σ), which plays a pivotal role in clathrin‐mediated endocytosis. We describe a 65‐year‐old woman who had hypercalcemia with normal circulating parathyroid hormone concentrations and hypocalciuria, features consistent with FHH, but she did not have CaSR and AP2σ mutations. Mutational analysis of the GNA11 gene was therefore undertaken, using leucocyte DNA, and this identified a novel heterozygous GNA11 mutation (c.161C>T; p.Thr54Met). The effect of the Gα11 variant was assessed by homology modeling of the related Gαq protein and by measuring the CaSR‐mediated intracellular calcium (Ca2+ i) responses of HEK293 cells, stably expressing CaSR, to alterations in extracellular calcium (Ca2+ o) using flow cytometry. Three‐dimensional modeling revealed the Thr54Met mutation to be located at the interface between the Gα11 helical and GTPase domains, and to likely impair GDP binding and interdomain interactions. Expression of wild‐type and the mutant Gα11 in HEK293 cells stably expressing CaSR demonstrate that the Ca2+ i responses after stimulation with Ca2+ o of the mutant Met54 Gα11 led to a rightward shift of the concentration‐response curve with a significantly (p < 0.01) increased mean half‐maximal concentration (EC50) value of 3.88 mM (95% confidence interval [CI] 3.76–4.01 mM), when compared with the wild‐type EC50 of 2.94 mM (95% CI 2.81–3.07 mM) consistent with a loss‐of‐function. Thus, our studies have identified a third Gα11 mutation (Thr54Met) causing FHH2 and reveal a critical role for the Gα11 interdomain interface in CaSR signaling and Ca2+ o homeostasis. © 2016 The Authors. Journal of Bone and Mineral Research published by Wiley Periodicals, Inc. on behalf of American Society for Bone and Mineral Research (ASBMR).
Keywords: DISORDERS OF CALCIUM/PHOSPHATE METABOLISM, PTH/VIT D/FGF23, PARATHYROID‐RELATED DISORDERS, CELL/TISSUE SIGNALING – ENDOCRINE PATHWAYS
Introduction
Familial hypocalciuric hypercalcemia (FHH) is characterized by lifelong elevations of serum calcium concentrations in association with normal or mildly raised serum parathyroid hormone (PTH) concentrations in 80% of patients and low urinary calcium excretion (urinary calcium‐to‐creatinine clearance ratio <0.01) in 80% of patients.1, 2 FHH may be inherited as an autosomal dominant condition, and it is a genetically heterogeneous disorder with three recognized variants, FHH1‐3. FHH1 (OMIM #145980) is caused by loss‐of‐function mutations of the calcium‐sensing receptor (CaSR), a G‐protein coupled receptor (GPCR)3 that initiates activation of the G‐protein subunit αq/11 (Gαq/11) family, leading to enhancement of phospholipase C (PLC) activity4 and elevation of inositol 1,4,5‐trisphosphate (IP3) with rapid increase in intracellular calcium (Ca2+ i) concentrations.5, 6 These signal transduction events allow the parathyroid CaSR to respond to small fluctuations in the prevailing extracellular calcium concentration ([Ca2+]o) by inducing alterations in PTH secretion through mechanisms that likely involve effects on PTH mRNA stability7 and PTH granule exocytosis from the apical pole of parathyroid cells.8 Moreover, the kidney CaSR is considered to influence urinary calcium excretion by modulating expression of claudin proteins that mediate the paracellular reabsorption of calcium in the renal thick ascending limb.9, 10 FHH2 (OMIM #145981) is the result of loss‐of‐function mutations in the G‐protein subunit‐α11 (Gα11), encoded by GNA11, and to date only two FHH2‐associated Gα11 missense mutations have been reported (Fig. 1).11 These two FHH2‐causing Gα11 mutations comprise a Leu135Gln missense substitution and an in‐frame isoleucine deletion at codon 200 (Ile200del), which are located in the Gα‐subunit helical and GTPase domains, respectively.11 Both of these FHH2‐causing Gα11 mutations, which are predicted to disrupt G‐protein activation, have been shown to impair CaSR signal transduction.11 FHH3 (OMIM #600740) is caused by loss‐of‐function mutations of the adaptor protein‐2 σ‐subunit (AP2σ), encoded by the AP2S1 gene. AP2σ has a pivotal role in clathrin‐mediated endocytosis of GPCRs such as the CaSR, and to date more than 50 FHH3 patients with AP2σ mutations, which are all missense mutations involving the Arg15 residue (Arg15Cys, Arg15His and Arg15Leu), have been reported.12, 13, 14, 15, 16 Approximately 65% of FHH patients will have a CaSR mutation, 5% an AP2σ mutation, <1% a Gα11 mutation, and the remaining ∼30% of FHH patients are considered to have involvement of a genetic abnormality that remains to be identified. Here we report the identification of a novel Gα11 mutation in a patient with FHH in whom CaSR and AP2σ mutations had been previously excluded.
Figure 1.
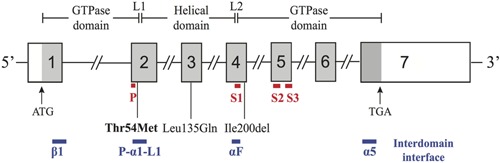
Schematic representation of the genomic organization of the human GNA11 gene showing the locations of FHH2‐causing mutations. The GNA11 gene consists of 7 exons with the start (ATG) and stop (TGA) codons located in exons 1 and 7, respectively. The GTPase domain (encoded by exon 1, 5' portion of exon 2, 3' portion of exon 4 and exons 5 to 7) is connected to the helical domain (encoded by the 3' portion of exon 2, exon 3, and 5' portion of exon 4) by the linker 1 (L1) and linker 2 (L2) peptides. The locations of the P‐loop (P) (red line), three flexible switch regions (S1 to S3) (red line), and the interdomain interface (comprising portions of the α1, αF, α5, β1, P‐loop, and L1 peptide motifs) (blue line) are shown below the GNA11 exons. The previously reported loss‐of‐function Leu135Gln and Ile200del mutations11 are located in the helical and GTPase domains, respectively, whereas the Thr54Met mutation (bold), identified by this study, is located at the interdomain interface. Coding regions are shaded gray and untranslated regions are represented by open boxes.
Materials and Methods
Case report
The patient, a 65‐year‐old woman of Indian origin, presented with poor mobility and recurrent falls. She underwent investigation and was found to have hypercalcemia (serum adjusted‐calcium concentration = 2.77 mmol/L, normal range = 2.20–2.60 mmol/L) in association with normal serum concentrations of phosphate (0.85 mmol/L; normal range = 0.70–1.40 mmol/L), creatinine (45 µmol/L; normal range = 40–130 µmol/L), PTH (5.9 pmol/L; normal range = 1.0–7.0 pmol/L), and serum 25‐hydroxyvitamin D (52 nmol/L; normal >50 nmol/L). Urinary calcium‐to‐creatinine clearance ratio was low at 0.01 (normal >0.02). These findings are consistent with a diagnosis of FHH, although there appeared to be an absence of a family history of hypercalcemia, based on the patient not having knowledge of any relatives who suffered from symptomatic hypercalcemia and the relatives not being available for medical assessment. DNA sequence analyses of the CASR and AP2S1 genes had not identified any abnormalities. Informed consent was obtained for the study using protocols approved by the Multi‐Centre Research Ethics Committee (UK) (MREC/02/2/93).
DNA sequence analysis
DNA sequence analyses of GNA11 exons 1–7 and their adjacent splice sites (NM_002067) (Fig. 1) was performed using leucocyte DNA and gene‐specific primers (Sigma‐Aldrich, St. Louis, MO, USA), as previously reported.11 Publicly accessible databases, including dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/), 1000 genomes (http://browser.1000genomes.org), the National Heart, Lung and Blood Institute (NHLBI) Exome Sequencing Project (http://evs.gs.washington.edu/EVS/, EVS data release ESP6500SI) representing the exomes of approximately 6500 individuals, and the Exome Aggregation Consortium (ExAC) (exac.broadinstitute.org) representing exomes of 60,706 unrelated individuals, were examined for the presence of sequence variants, and any potential pathogenic sequence abnormality identified within the patient DNA was confirmed by restriction endonuclease analyses, as described.12
Protein sequence alignment and three‐dimensional modeling of Gα11 structure
Protein sequences of Gα11 paralogs were aligned using ClustalOmega (http://www.ebi.ac.uk/Tools/msa/clustalo/).17 Gα11 three‐dimensional modeling was undertaken using the reported three‐dimensional structure of Gαq in complex with the small molecule inhibitor YM‐254890 (Protein Data Bank accession no. 3AH8).18 The Gαq protein, which shares 90% identity at the amino acid level with Gα11,11 was used because crystal structures of Gα11 are not available. Molecular modeling was performed using The PyMOL Molecular Graphics System (Version 1.2r3pre, Schrödinger, LL Pymol).11
Cell culture and transfection
Wild‐type and mutant GNA11 (pBI‐CMV2‐GNA11) expression constructs were generated as described,11 and transiently transfected into HEK293 cells stably expressing CaSR (HEK293‐CaSR)12 using Lipofectamine 2000 (Life Technologies, Carlsbad, CA, USA). The bidirectional pBI‐CMV2 cloning vector was used because it facilitated the co‐expression of Gα11 and GFP,11, 12 and site‐directed mutagenesis was used to generate the mutant GNA11 construct using the Quikchange Lightning Site‐directed Mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA) and gene‐specific primers (Sigma‐Aldrich), as described.19 Cells were maintained in DMEM‐Glutamax media (Thermo‐Fisher, Waltham, MA, USA) with 10% fetal bovine serum (Gibco, Thermo‐Fisher) and 400 μg/mL geneticin (Thermo‐Fisher) at 37°C, 5% CO2. Successful transfection was confirmed by visualizing GFP fluorescence using an Eclipse E400 fluorescence microscope with a Y‐FL Epifluorescence attachment and a triband 4,6‐diamidino‐2‐phenylindole‐FITC‐Rhodamine filter, and images captured using a DXM1200C digital camera and NIS Elements software (Nikon, Tokyo, Japan).11, 12, 13 The expression of Gα11 and CaSR proteins was also determined by Western blot analyses using anti‐Gα11 (Santa Cruz Biotechnology, Dallas, TX, USA), anti‐GFP (Santa Cruz), anti‐calnexin (Millipore, Billerica, MA, USA) or anti‐CaSR (AbCam, Cambridge, UK) antibodies. The Western blots were visualized using an Immuno‐Star Western C kit (Bio‐Rad, Hercules, CA, USA) on a Bio‐Rad Chemidoc XRS+ system.11
Intracellular calcium measurements
The Ca2+ i responses of HEK293‐CaSR cells expressing wild‐type or mutant Gα11 proteins were assessed by a flow cytometry‐based assay, as reported.11, 12, 13 In brief, HEK293‐CaSR cells were cultured in T75 flasks and transiently transfected 24 hours later with 16 μg DNA.11 Forty‐eight hours after transfection, the cells were detached, resuspended in calcium (Ca2+)‐ and magnesium (Mg2+)‐free Hanks' buffered saline solution (HBSS), and loaded with 1 μg/mL Indo‐1‐acetoxymethylester (Indo‐1‐AM) for 1 hour at 37°C. After removal of free dye, cells were resuspended in Ca2+‐ and Mg2+‐free HBSS and maintained at 37°C. Transfected cells, in suspension, were stimulated by sequentially adding Ca2+ to the Ca2+‐ and Mg2+‐free HBSS to increase the [Ca2+]o in a stepwise manner from 0 to 15 mM and then analyzed on a MoFlo modular flow cytometer (Beckman Coulter, Indianapolis, IN, USA) by simultaneous measurements of GFP expression (at 525 nm), Ca2+ i‐bound Indo‐1AM (at 410 nm), and free Indo‐1AM (ie, not bound to Ca2+ i) (at 485 nm), using a JDSU Xcyte UV laser (Coherent Radiation, Santa Clara, CA, USA), on each cell at each [Ca2+]o, as described11, 12. The peak mean fluorescence ratio of the Ca2+ i transient response after each individual stimulus was measured using Cytomation Summit software (Beckman Coulter) and expressed as a normalized response, as described.11, 12 Nonlinear regression of concentration‐response curves was performed with GraphPad Prism (GraphPad, La Jolla, CA, USA) using the normalized response at each [Ca2+]o for each separate experiment for the determination of EC50 (ie, [Ca2+]o required for 50% of the maximal response) and Hill coefficient values. The maximal signaling response was measured as a fold‐change of the peak transient Ca2+ i response to the basal Ca2+ i response measured at 0 mM [Ca2+]o. The maximal signaling responses for mutant Gα11 proteins were expressed as a percentage of the wild‐type Gα11 protein maximal signaling response. The mean EC50 and Hill coefficients obtained from four separate transfection experiments were used for statistical comparison by using the F‐test, and alterations in maximal signaling responses assessed using the Mann‐Whitney U test.
Results
Identification of a novel Thr54Met Gα11 mutation in an FHH proband
DNA sequence analyses of the GNA11 coding regions and adjacent splice sites (Fig. 1) identified a heterozygous C‐to‐T transition at nucleotide c.161, in the FHH patient (Figure 2A). This C‐to‐T transition (ACG to ATG) resulted in a missense substitution, Thr54Met, of the Gα11 protein (Fig. 2 B). The sequence alteration also led to the gain of an NspI and loss of a BsiHKAI restriction endonuclease site (Fig. 2 B), which were used to confirm the presence of the mutation in the patient (Fig. 2 C, D). Bioinformatic analyses using SIFT and MutationTasting software20, 21 predicted the variant to be damaging and likely disease‐causing (SIFT score 0, MutationTasting score 0.99). In addition, the absence of this DNA sequence abnormality in >6500 exomes from the NHLBI‐ESP cohort and >60,700 exomes from the ExAC cohort, together with evolutionary conservation of the Thr54 residue in vertebrate Gα‐subunit paralogs (Fig. 3 A), indicated that the Thr54Met abnormality likely represented a pathogenic GNA11 mutation rather than a benign polymorphic variant.
Figure 2.
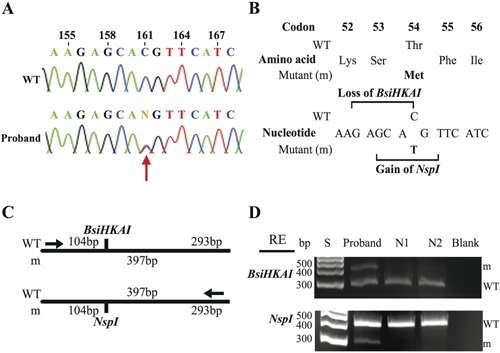
Identification of a Thr54Met GNA11 mutation in a FHH proband. (A) DNA sequence analyses revealed a heterozygous C‐to‐T transition at nucleotide c.161 (red arrow) within exon 2 of GNA11. (B) This sequence abnormality was predicted to lead to a missense amino acid substitution of Thr to Met at codon 54, resulting in the loss of a BsiHKAI restriction endonuclease site (GAGCA/C) and gain of an NspI (GCATG/T) restriction endonuclease site. (C) Restriction maps showing that BsiHKAI digestion would result in two products of 104 bp and 293 bp from the wild‐type (WT) sequence but not the mutant (m) sequence. In contrast, NspI digestion results in two products of 104 bp and 293 bp from the m sequence but not the WT sequence. (D) Restriction endonuclease (RE) digest of PCR products of exon 2 of GNA11 demonstrating the heterozygous C‐to‐T transition and confirming its absence in two unrelated unaffected individuals (N1 and N2). S = size marker. For BsiHKAI RE digest the mutant product (397 bp) and WT product (293 bp) are shown; the WT product at 104 bp is not shown. For NspI RE digest the WT product (397 bp) and mutant product (293 bp) are shown; the mutant product at 104 bp is not shown.
Figure 3.
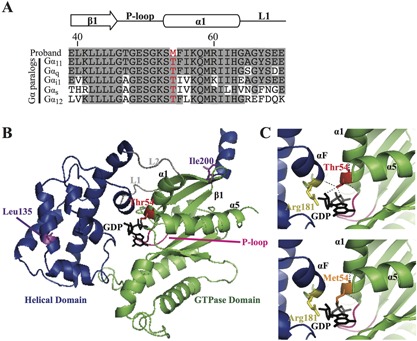
Predicted effects of the Thr54Met mutation on the Gα11 protein. (A) Multiple protein sequence alignment of residues contributing to the interdomain interface of Gα‐subunit paralogs. The wild‐type Thr54 (T) and mutant Met54 (M) residues are shown in red. Conserved residues are shaded gray. (B) Overall three‐dimensional structure of the Gα11 protein. The helical (blue) and GTPase (green) domains are connected by the L1 and L2 peptides (gray). The GTPase domain contains the P‐loop (pink), which binds GDP (black) at the interdomain interface (comprising parts of α1, α5, αF, P‐loop, L1, and β1 peptide motifs). The reported Leu135Gln and Ile200del mutations11 are shown in purple, and the Thr54Met mutation is shown in red. (C) Close‐up views of the Gα11 interdomain interface, which show the wild‐type Thr54 residue, located in the GTPase domain, form polar contacts (hatched black lines) with GDP and the Arg181 residue (yellow) of the helical domain αF helix. Substitution of the wild‐type Thr54 residue with the mutant Met54 residue (orange) is predicted to disrupt these polar contacts, thereby impairing interdomain interactions and the binding of GDP.
Structural characterization of the Thr54Met Gα11 mutant protein
The Thr54Met mutation is located within the Gα11 α‐1 helix (Fig. 1 and Fig. 3 A, B), which comprises part of the interface at which the GTPase and helical domains interact to bind GDP and GTP.22 In contrast, the previously reported FHH2‐causing Iledel200 and Leu135Gln mutations,11 which affect the GTPase and helical domains of Gα11, respectively, are situated away from the guanine‐nucleotide binding site (Fig. 1 and Fig. 3 B). The Thr54 Gα11 residue is located next to the phosphate‐binding loop (P‐loop) (Fig. 3 A), which is a highly conserved nucleotide‐binding peptide motif that plays a critical role in binding GDP.23, 24, 25 Three‐dimensional homology modeling of the Gα11 protein revealed the wild‐type Thr54 residue to form polar contacts with the ribose and β‐phosphate moieties of GDP within the interdomain interface (Fig. 3 B, C) and to interact with the α‐F helix of the helical domain, which also mediates GDP binding (Fig. 3 C).22 These findings are consistent with the reported role of the α‐1 helix as a structural hub that mediates interactions between the GTPase and helical domains to ensure GDP binding, thereby maintaining the Gα‐subunit in an inactive conformation.26 The Gα11 Met54 mutant is predicted to disrupt these interdomain contacts and alter GDP binding (Fig. 3 C).
Functional characterization of the Thr54Met Gα11 mutant protein
To determine the effects of the predicted changes in Gα11 structure (Fig. 3 B, C) on CaSR‐mediated signaling, Ca2+ i responses to alterations in [Ca2+]o were assessed in HEK293‐CaSR cells that were transiently transfected with either the pBI‐CMV2 empty vector or pBI‐CMV2 expressing the wild‐type (Thr54) or mutant (Met54) Gα11 proteins. The Ca2+ i responses of cells expressing the Met54 Gα11 mutant were also compared with cells transiently transfected with the reported FHH2‐associated Gln135 Gα11 mutant protein.11 Expression of CaSR, Gα11 and GFP was confirmed by fluorescence microscopy and/or Western blot analyses (Fig. 4 A, B). Calnexin was used as a loading control in Western blot analyses, and Gα11 expression was demonstrated to be similar in cells transiently transfected with wild‐type or mutant Gα11 proteins and greater than that of cells transfected with the empty pBI‐CMV2 vector (Fig. 4 B). The Ca2+ i responses in wild‐type and mutant Gα11‐expressing cells were shown to increase in a dose‐dependent manner after stimulation with increasing concentrations of Ca2+ o between 0–15 mM. However, exposure to a significantly greater [Ca2+]o was required to achieve half‐maximal (EC50) Ca2+ i responses for cells expressing either the Met54 or Gln135 mutant Gα11 proteins compared with wild‐type‐expressing cells. (Fig. 4 C, D). Thus, the Met54 or Gln135 mutant‐expressing cells showed rightward shifts in the concentration‐response curves, with significantly elevated mean EC50 values (p < 0.01) of 3.88 mM (95% confidence interval [CI] 3.76–4.01 mM) and 3.65 mM (95% CI 3.57–3.74 mM), respectively, compared with 2.94 mM (95% CI 2.81–3.07 mM) for wild‐type expressing cells and consistent with the Gα11 mutants leading to an impairment of CaSR signal transduction (Fig. 4 C, D). The Hill coefficients did not significantly differ between wild‐type and mutant Gα11‐expressing cells (Fig. 4 E). However, cells expressing the Met54 mutant had significantly reduced maximal signaling responses compared with cells expressing either wild‐type or Gln135 mutant Gα11 proteins (p < 0.05) (Fig. 4 F).
Figure 4.
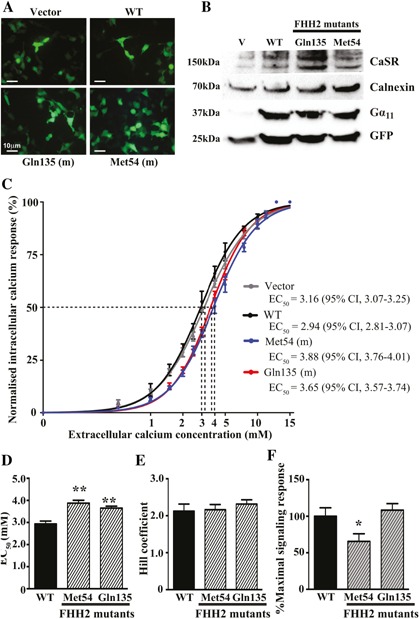
Functional characterization of wild‐type and FHH2‐associated mutant Gα11 proteins. (A) Fluorescence microscopy of HEK293 cells stably expressing CaSR (HEK293‐CaSR) and transiently transfected with wild‐type (WT) or FHH2‐associated mutant (Met54 and Gln135) pBI‐CMV2‐GNA11‐GFP constructs, or with vector only. GFP expression in these cells indicates successful transfection and expression by these constructs. Scale bar = 10 µm. (B) Western blot analyses of whole‐cell lysates using antibodies to CaSR, calnexin, Gα11, and GFP. Transient transfection of WT or FHH2‐associated mutant constructs resulted in overexpression of Gα11 when normalized to calnexin expression. (C) Concentration‐response curves showing normalized Ca2+ i response to changes in [Ca2+]o of HEK293‐CaSR cells transfected with WT or FHH2‐associated Gα11 mutants. The Ca2+ i responses are shown as the mean ± SEM of 4 independent transfections. The FHH2‐associated Gα11 mutants (Met54 and Gln135) led to a rightward shift of the concentration‐response curves (blue and red, respectively) when compared with WT Gα11 (black), which harbors Thr and Leu residues at codons 54 and 135, respectively. (D) The Met54 and Gln135 mutants (shaded bars) were associated with significantly increased EC50 values compared with cells expressing WT Gα11 (black bar). (E) The Hill coefficients of the wild‐type and mutant Gα11 proteins were similar (ie, not significantly different). (F) The Met54 mutant was associated with a significantly reduced maximal signaling response compared with WT and mutant Gln135 Gα11 proteins. *p < 0.05, **p < 0.01.
Discussion
Our studies have identified a novel heterozygous germline GNA11 mutation in a patient with FHH, which resulted in an impairment of Ca2+ i signaling similar to the loss‐of‐function previously reported for the FHH2‐associated Leu135Gln and Ile200del GNA11 mutations.11 The Thr54Met mutation represents only the third loss‐of‐function GNA11 mutation to be reported, and thus these findings provide further support for a critical role of the Gα11 protein in parathyroid gland function and Ca2+ o homeostasis, and highlight the importance of GNA11 gene analyses in FHH patients that do not harbor CASR or AP2S1 mutations. The Thr54Met, Leu135Gln, and Ile200del loss‐of‐function GNA11 mutations are all associated with a mild FHH phenotype characterized by serum adjusted‐calcium concentrations <2.80 mmol/L, and these clinical findings are in keeping with our in vitro studies that have shown FHH2‐associated mutations to induce only minor disturbances of CaSR signal transduction.11 Indeed, the FHH2 mutants were associated with around a 30% increase in the EC50 values of HEK293‐CaSR cells used in this study, whereas CaSR mutations leading to FHH1 generally cause a >50% increase in the EC50 value.3, 27 The milder shift in the Ca2+ o set point of cells expressing FHH2‐associated Gα11 mutants indicates that the CaSR may promote Ca2+ i signaling by Gα11‐independent mechanisms, such as via the related Gαq protein. Indeed, reported studies that selectively ablated Gαq and/or Gα11 in the parathyroid glands of mice have highlighted the importance of both of these G‐proteins for CaSR function,28, 29 and in the setting of FHH1, CaSR mutations likely impair Ca2+ i responses via both Gα11 and Gαq, thus leading to a greater loss‐of‐function than Gα11 mutations that cause FHH2.
The Gα11‐subunit consists of a Ras‐like GTPase domain that binds GDP and GTP, and a smaller helical domain that acts as a clasp to secure these bound guanine‐nucleotides.30 Three‐dimensional modeling indicated the Thr54Met mutation to be located at the interdomain interface, which represents a highly conserved and critical region containing the P‐loop motif that binds GDP23, 24 and also facilitates interactions between the helical and GTPase domains that maintain Gα‐subunits in an inactive GDP‐bound conformation.22 The Thr54Met mutation likely alters GDP binding, but in contrast to the other reported Gα11 mutations (Arg60Cys, Arg60Leu, and Arg181Gln),11, 31, 32 which are also located at the interdomain interface (Fig. 3 B, C) and cause Gα11 gain‐of‐function that is associated with the clinical disorder of autosomal dominant hypocalcemia type‐2 (ADH2), the Thr54Met Gα11 mutation causes loss‐of‐function and FHH2. Thus, it seems that mutations involving the Gα11 interdomain interface may result in Gα11 loss‐of‐function or gain‐of‐function. Crystal structures of Gα11 proteins are not available to evaluate the structure‐function effects of the Thr54Met mutation at the interdomain interface; however, the introduction of the mutant Met54 residue may sterically impair G‐protein function, as highlighted by a previous crystallography study of a loss‐of‐function G‐protein alpha‐i (Gαi) P‐loop mutation, which revealed the mutant Gαi residue to sterically hinder conformational changes of the flexible “switch” regions during Gα‐subunit activation.33 Moreover, interdomain interface mutations that disrupt guanine‐nucleotide binding may also result in an “empty‐pocket” mutant Gα‐subunit that exerts dominant‐negative effects by binding and sequestering partner GPCRs.34 These findings may also help to provide an explanation for the observed differences in the maximal signaling responses of the FHH2‐causing Met54 and Gln135 Gα11 mutants (Fig. 4 F). Thus, the Met54 Gα11 mutant, but not the Gln135 Gα11 mutant, led to a significant reduction in the maximal signaling response of CaSR‐expressing cells (Fig. 4 F), even though both Gα11 mutants increase the Ca2+ o set point of CaSR‐expressing cells to a similar degree, as illustrated by their EC50 values (Fig. 4 D). The maximal signaling response of a GPCR is influenced by the ability of the receptor to couple with its cognate G‐protein,35 and thus it is possible that the Met54 Gα11 mutant impairs coupling and/or dissociation of Gα11 from the CaSR by influencing guanine‐nucleotide binding at the interdomain interface,34 whereas the Gln135 Gα11 mutant, which is located in the Gα11 helical domain and not predicted to influence CaSR‐Gα11 coupling, may potentially diminish CaSR signal transduction by influencing the interaction of Gα11 with downstream effectors.36
In conclusion, we have identified a novel Gα11 loss‐of‐function mutation, Thr54Met, that causes FHH2 and which provides new insights into the critical role of the Gα11 interdomain interface in CaSR signaling.
Disclosures
All authors state that they have no conflicts of interest.
Acknowledgments
This work was supported by the United Kingdom Medical Research Council (MRC) program grant G1000467/2010 (to CMG, MAN, and RVT), a Wellcome Trust Senior Investigator Award (to RVT); National Institute for Health Research (NIHR) Oxford Biomedical Research Centre Programme (to RVT); and NIHR Senior Investigator Award (to RVT). The GO Exome Sequencing Project of the National Heart, Lung, and Blood Institute and its ongoing studies produced and provided exome variant calls for comparison (the Lung GO Sequencing Project [HL‐102923], the Women's Health Initiative Sequencing Project [HL‐102924], the Broad GO Sequencing Project [HL‐102925], the Seattle GO Sequencing Project [HL‐102926], and the Heart GO Sequencing Project [HL‐103010]).
Authors' roles: Study design: CMG and RVT; study conduct: CMG, TC, and NR; data collection: AQ and FMH; data analyses and interpretation: CMG, FMH, and RVT; manuscript preparation: CMG, TC, FMH, NR, AQ, MAN, and RVT.
References
- 1. Hannan FM, Thakker RV. Calcium‐sensing receptor (CaSR) mutations and disorders of calcium, electrolyte and water metabolism. Best Pract Res Clin Endocrinol Metab. 2013; 27(3):359–71. [DOI] [PubMed] [Google Scholar]
- 2. Eastell R, Brandi ML, Costa AG, D'Amour P, Shoback DM, Thakker RV. Diagnosis of asymptomatic primary hyperparathyroidism: proceedings of the Fourth International Workshop. J Clin Endocrinol Metab. 2014; 99(10):3570–9. [DOI] [PubMed] [Google Scholar]
- 3. Hannan FM, Nesbit MA, Zhang C, et al. Identification of 70 calcium‐sensing receptor mutations in hyper‐ and hypo‐calcaemic patients: evidence for clustering of extracellular domain mutations at calcium‐binding sites. Hum Mol Genet. 2012; 21(12):2768–78. [DOI] [PubMed] [Google Scholar]
- 4. Wu DQ, Lee CH, Rhee SG, Simon MI. Activation of phospholipase C by the alpha subunits of the Gq and G11 proteins in transfected Cos‐7 cells. J Biol Chem. 1992; 267(3):1811–7. [PubMed] [Google Scholar]
- 5. Hofer AM, Brown EM. Extracellular calcium sensing and signalling. Nat Rev Mol Cell Biol. 2003; 4(7):530–8. [DOI] [PubMed] [Google Scholar]
- 6. Breitwieser GE, Gama L. Calcium‐sensing receptor activation induces intracellular calcium oscillations. Am J Physiol Cell Physiol. 2001; 280(6):C1412–21. [DOI] [PubMed] [Google Scholar]
- 7. Kumar R, Thompson JR. The regulation of parathyroid hormone secretion and synthesis. J Am Soc Nephrol. 2011; 22(2):216–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Quinn SJ, Kifor O, Kifor I, Butters RR Jr, Brown EM. Role of the cytoskeleton in extracellular calcium‐regulated PTH release. Biochem Biophys Res Commun. 2007; 354(1):8–13. [DOI] [PubMed] [Google Scholar]
- 9. Gong Y, Hou J. Claudin‐14 underlies Ca(+)(+)‐sensing receptor‐mediated Ca(+)(+) metabolism via NFAT‐microRNA‐based mechanisms. J Am Soc Nephrol. 2014; 25(4):745–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Riccardi D, Brown EM. Physiology and pathophysiology of the calcium‐sensing receptor in the kidney. Am J Physiol Renal Physiol. 2010; 298(3):F485–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nesbit MA, Hannan FM, Howles SA, et al. Mutations affecting G‐protein subunit alpha11 in hypercalcemia and hypocalcemia. N Engl J Med. 2013; 368(26):2476–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nesbit MA, Hannan FM, Howles SA, et al. Mutations in AP2S1 cause familial hypocalciuric hypercalcemia type 3. Nat Genet. 2013; 45(1):93–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hannan FM, Howles SA, Rogers A, et al. Adaptor protein‐2 sigma subunit mutations causing familial hypocalciuric hypercalcaemia type 3 (FHH3) demonstrate genotype‐phenotype correlations, codon bias and dominant‐negative effects. Hum Mol Genet. 2015; 24(18):5079–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tenhola S, Hendy GN, Valta H, et al. Cinacalcet treatment in an adolescent with concurrent 22q11.2 deletion syndrome and familial hypocalciuric hypercalcemia type 3 caused by AP2S1 mutation. J Clin Endocrinol Metab. 2015; 100(7):2515–8. [DOI] [PubMed] [Google Scholar]
- 15. Fujisawa Y, Yamaguchi R, Satake E, et al. Identification of AP2S1 mutation and effects of low calcium formula in an infant with hypercalcemia and hypercalciuria. J Clin Endocrinol Metab. 2013; 98(12):E2022–7. [DOI] [PubMed] [Google Scholar]
- 16. Hendy GN, Canaff L, Newfield RS, et al. Codon Arg15 mutations of the AP2S1 gene: common occurrence in familial hypocalciuric hypercalcemia cases negative for calcium‐sensing receptor (CASR) mutations. J Clin Endocrinol Metab. 2014; 99(7):E1311–5. [DOI] [PubMed] [Google Scholar]
- 17. Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011; 7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nishimura A, Kitano K, Takasaki J, et al. Structural basis for the specific inhibition of heterotrimeric Gq protein by a small molecule. Proc Natl Acad Sci USA. 2010; 107(31):13666–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Newey PJ, Gorvin CM, Cleland SJ, et al. Mutant prolactin receptor and familial hyperprolactinemia. N Engl J Med. 2013; 369(21):2012–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009; 4(7):1073–81. [DOI] [PubMed] [Google Scholar]
- 21. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep‐sequencing age. Nat Methods. 2014; 11(4):361–2. [DOI] [PubMed] [Google Scholar]
- 22. Sun D, Flock T, Deupi X, et al. Probing Galphai1 protein activation at single‐amino acid resolution. Nat Struct Mol Biol. 2015; 22(9):686–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: conserved structure and molecular mechanism. Nature. 1991; 349(6305):117–27. [DOI] [PubMed] [Google Scholar]
- 24. Wittinghofer A, Vetter IR. Structure‐function relationships of the G domain, a canonical switch motif. Annu Rev Biochem. 2011; 80:943–71. [DOI] [PubMed] [Google Scholar]
- 25. Barbacid M. Ras genes. Annu Rev Biochem. 1987; 56:779–827. [DOI] [PubMed] [Google Scholar]
- 26. Flock T, Ravarani CN, Sun D, et al. Universal allosteric mechanism for Galpha activation by GPCRs. Nature. 2015; 524(7564):173–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pearce SH, Bai M, Quinn SJ, Kifor O, Brown EM, Thakker RV. Functional characterization of calcium‐sensing receptor mutations expressed in human embryonic kidney cells. J Clin Invest. 1996; 98(8):1860–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pi M, Chen L, Huang M, Luo Q, Quarles LD. Parathyroid‐specific interaction of the calcium‐sensing receptor and G alpha q. Kidney Int. 2008; 74(12):1548–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wettschureck N, Lee E, Libutti SK, Offermanns S, Robey PG, Spiegel AM. Parathyroid‐specific double knockout of Gq and G11 alpha‐subunits leads to a phenotype resembling germline knockout of the extracellular Ca2+‐sensing receptor. Mol Endocrinol. 2007; 21(1):274–80. [DOI] [PubMed] [Google Scholar]
- 30. Oldham WM, Hamm HE. Heterotrimeric G protein activation by G‐protein‐coupled receptors. Nat Rev Mol Cell Biol. 2008; 9(1):60–71. [DOI] [PubMed] [Google Scholar]
- 31. Li D, Opas EE, Tuluc F, et al. Autosomal dominant hypoparathyroidism caused by germline mutation in GNA11: phenotypic and molecular characterization. J Clin Endocrinol Metab. 2014; 99(9):E1774–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mannstadt M, Harris M, Bravenboer B, et al. Germline mutations affecting Galpha11 in hypoparathyroidism. N Engl J Med. 2013; 368(26):2532–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bosch DE, Willard FS, Ramanujam R, et al. A P‐loop mutation in Galpha subunits prevents transition to the active state: implications for G‐protein signaling in fungal pathogenesis. PLoS Pathog. 2012; 8(2):e1002553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yu B, Gu L, Simon MI. Inhibition of subsets of G protein‐coupled receptors by empty mutants of G protein alpha subunits in g(o), G(11), and G(16). J Biol Chem. 2000; 275(1):71–6. [DOI] [PubMed] [Google Scholar]
- 35. Shim JY, Ahn KH, Kendall DA. Molecular basis of cannabinoid CB1 receptor coupling to the G protein heterotrimer Galphaibetagamma: identification of key CB1 contacts with the C‐terminal helix alpha5 of Galphai. J Biol Chem. 2013; 288(45):32449–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu W, Northup JK. The helical domain of a G protein alpha subunit is a regulator of its effector. Proc Natl Acad Sci USA. 1998; 95(22):12878–83. [DOI] [PMC free article] [PubMed] [Google Scholar]