

Abstract
The auxiliary voltage‐gated calcium channel subunit β4 supports targeting of calcium channels to the cell membrane, modulates ionic currents and promotes synaptic release in the central nervous system. β4 is abundant in cerebellum and its loss causes ataxia. However, the type of calcium channels and cerebellar functions affected by the loss of β4 are currently unknown. We therefore studied the structure and function of Purkinje cells in acute cerebellar slices of the β4 −/− ataxic (lethargic) mouse, finding that loss of β4 affected Purkinje cell input, morphology and pacemaker activity. In adult lethargic cerebellum evoked postsynaptic currents from parallel fibres were depressed, while paired‐pulse facilitation and spontaneous synaptic currents were unaffected. Because climbing fibre input was spared, the parallel fibre/climbing fibre input ratio was reduced. The dendritic arbor of adult lethargic Purkinje cells displayed fewer and shorter dendrites, but a normal spine density. Accordingly, the width of the molecular and granular layers was reduced. These defects recapitulate the impaired cerebellar maturation observed upon Cav2.1 ataxic mutations. However, unlike Cav2.1 mutations, lethargic Purkinje cells also displayed a striking decrease in pacemaker firing frequency, without loss of firing regularity. All these deficiencies appear in late development, indicating the importance of β4 for the normal differentiation and function of mature Purkinje cells networks. The observed reduction of the parallel fibre input, the altered parallel fibre/climbing fibre ratio and the reduced Purkinje cell output can contribute to the severe motor impairment caused by the loss of the calcium channel β4 subunit in lethargic mice.
Keywords: ataxia, cerebellar cortex, development, electrophysiology, neuron
Introduction
In cerebellum, presynaptic P/Q‐type calcium channels are made up of the pore‐forming CaV2.1 α1 subunit and auxiliary subunits, among which β4 and α2δ‐2 are the most highly represented (Buraei & Yang, 2010; Schlick et al., 2010). Consistent with their joint function in cerebellar neurons, mutation of any one of these calcium channel subunits in mice and humans leads to similar forms of ataxia and epilepsy (Dung & Swigart, 1971; Fletcher et al., 1996; Burgess et al., 1997; Escayg et al., 2000; Barclay et al., 2001; Brodbeck et al., 2002; Khan & Jinnah, 2002; Pietrobon, 2005; Dolphin, 2012; Noebels, 2012). Although it is well established that dysfunctions of CaV2.1 or α2δ‐2 affect the morphology, synaptic input and pacemaker activity of cerebellar Purkinje neurons (Zwingman et al., 2001; Matsushita et al., 2002; Kodama et al., 2006; Walter et al., 2006; Lonchamp et al., 2009), the specific cerebellar functions of the β4 subunit and their causal link to the ataxic phenotype in β4 loss‐of‐function mutations remain elusive.
Calcium channel β subunits associate with CaV1 and CaV2 α1 subunits. They modulate their current properties and are important for proper membrane expression and subcellular targeting of the channels (Buraei & Yang, 2010; Etemad et al., 2014; Campiglio & Flucher, 2015). Consequently β4 subunits are important for calcium‐induced neurotransmitter release at synaptic terminals of central nervous system (CNS) neurons (Caddick et al., 1999; Lin et al., 1999; Wittemann et al., 2000). Furthermore, new channel‐independent functions of β4 in gene transcription regulation have recently emerged, suggesting potential roles of this protein in regulation of neuronal development and physiology (Tadmouri et al., 2012; Etemad et al., 2014). Previous studies in the ataxic β4 −/− mouse model (lethargic) suggested that the function of β4 in CNS neurons might be redundant and that the lack of β4 is largely compensated for by increased expression of other β isoforms (McEnery et al., 1998). This notion is consistent with the observation that calcium currents in Purkinje cell (PC) somas are unaffected by loss of β4 (Burgess & Noebels, 1999a). However, a redundancy of β4 is at odds with the prominent motor impairment in lethargic mice (Burgess et al., 1997; Khan & Jinnah, 2002) and the functional compensation, which occurs at the PC soma (Burgess et al., 1999), might not necessarily extend to dendrites, axons and synapses (Wittemann et al., 2000; Obermair et al., 2010; Etemad et al., 2014) .
Here we applied electrophysiological recordings from PCs in acute cerebellar slices of β4 −/− lethargic mice to test the hypothesis that the calcium channel β4 subunit is necessary for normal function of cerebellar cortex networks, where physiological defects upon loss of β4 may contribute to the ataxic phenotype in lethargic mice. We reasoned that, because β4 is an essential subunit of P/Q‐type calcium channels, the deficiencies in PC networks in lethargic mice will resemble those previously reported in CaV2.1 mutant mice, unless compensated for by redundant β functions. On the other hand, β4‐specific defects will expose essential roles of β4 in other CaV isoforms and channel‐independent functions of β4 in the development of cerebellar cortex networks.
Materials and methods
Animal handling
All animal procedures were performed according to the guidelines of ethical approval by the European Parliament Directive (Council Directive 2010/63EU and Council of 22 September 2010) for the protection of animal used for scientific purposes and were approved by the Austrian Ministry BMWFW, TWG 2012. Animals were kindly provided by the laboratory of Professor V. Flockerzi, Universität Saarland (Germany). For all animal experiments, the 3Rs system was followed (replacement, reduction and refinement). Experiments were carried out on animals of either sex. To ease the access to food and further promote the fitness and comfort of lethargic mice, dry food pellets and hydrating gel was placed on the cage floor and provided ad libitum. All mice were bred at least for ten generations into 129/SvJ inbreed background. Wildtype and lethargic (β4−/−) mice were obtained by homozygous mating. The numbers of animals used for the experiments were: 16 juvenile [postnatal day (P)07–P20] wildtype, 20 juvenile lethargic, 34 adult (P35–P65) wildtype and 20 adult lethargic mice.
Electrophysiology
After killing the mice by cervical dislocation and after decapitation, scalp and skull bones were gently removed with the help of fine scissors and tweezers. After this, the mouse cerebellum was acutely dissected and immediately chilled (~ 0 °C) in artificial cerebrospinal fluid containing (in mm): NaCl, 125; NaHCO3, 26; glucose, 10; KCl, 2.5; NaHPO4, 1.25; CaCl2, 2; MgCl2, 1; pH was adjusted to 7.4 with a saturating carbogen mix (95/5% CO2/O2). Parasagittal cerebellar slices were cut along the vermis, with a vibratome (Leica VT1200S). After dissection, slices were stored in artificial cerebrospinal fluid at 21.5–22.5 °C (room temperature) for up to 5 h.
Recordings were obtained from visually identified PCs from the anterior lobe, mostly within lobules III and IV. Electrophysiological data were acquired with an Axon 900A amplifier and the software pclamp (Axon Instruments, Union City, CA, USA); sampling rate was 20 kHz. Traces were filtered at 1–4 kHz and analysed with Origin, Microsoft Excel and graphpad‐prism. Series resistances (R s) were typically about 10 MΩ. R s compensation was applied at 60–70%. Recordings with R s drift > 10% were discarded. The internal pipette solution contained (in mm): K‐gluconate, 132; EGTA/KOH, 1; MgCl2, 2; NaCl, 2; Hepes/KOH, 10; Mg‐ATP, 2; GTP, 0.5; pH was adjusted to 7.2 (Lonchamp et al., 2009). Pipette resistance was about 2 MΩ for whole‐cell measurements and 3–7 MΩ for cell‐attached recordings. Parallel fibre (PF) volleys were evoked by unipolar stimulation electrode (glass pipette of about 1 mΩ) in the molecular layer beneath the pial surface. Climbing fibre (CF) responses were evoked by similar stimulation in the granular layer. In the representative recordings of evoked synaptic responses, the stimulation artefacts were removed for clarity of representation. The optimal age range for electrophysiological recording was determined after preliminary excitatory postsynaptic current (ePSC) and inhibitory postsynaptic current (iPSC) measurement between P06 and P20, where the largest increase in PSC frequency occurred before P12 (data not shown). ePSC and iPSC on young mice were sampled at room temperature from mice between P12 and P20, with the same distribution between β4 +/+ and β4 −/− (P14.5 ± 1.0 and P15.0 ± 0.9, respectively). Pacemaker firing was measured at 34–36 °C. Action potential frequency in young mice was determined in cell‐attached experiments at P14–P15. Action potential frequency in adult mice was recorded between postnatal weeks 5 and 8 (P43 ± 1 for β4 +/+ and P44 ± 1 for β4 −/−); cells from the same mice were also used for morphological analysis. In adult populations no correlation existed between ages and firing frequency (R = 0.3, P = 0.5 for β4+/+ and R = −0.6, P = 0.1 for β4−/−). Evoked postsynaptic currents were recorded at P15 and P55–P65 (P60). Drugs were applied at the following concentrations (mm): (±)‐trans‐1‐amino‐1,3‐dicarboxycyclopentane (trans‐ACPD; Tocris, Bristol, UK) 0.05; SR 95531 hydrobromide (gabazine; Tocris) 0.01; and 6,7 dinitroquinoxaline‐2,3‐dione (DNQX; Tocris) 0.01, dl‐AP‐5 (AP‐5; Tocris) 0.05.
Morphological analysis
During whole‐cell patch clamp recordings, Purkinje neurons were dialysed with biocytin via patch‐pipette (Benedetti et al., 2011). After 10–20 min the pipette was gently retracted, slices were transferred to 4% sucrose/paraformaldehyde in 0.1 m phosphate buffer, and stored at 4 °C for 24–96 h before staining. Then, slices were thoroughly rinsed in phosphate‐buffered saline, incubated for 1 h in 0.2% Triton‐X100 at room temperature, rinsed and again with phosphate buffer. To block endogenous biotin, slices were treated with the Endogenous Biotin‐Blocking Kit (Molecular Probes, Carlsbad, CA, USA). Streptavidin conjugated with Alexa Fluor 568 (Molecular Probes) was added (1:1000 in phosphate‐buffered saline) and incubated overnight at 4 °C with shaking. Finally, slices were thoroughly rinsed in phosphate buffer and mounted with Vectashield (Vector Laboratories, Burlingame, CA, USA). Labelled neurons were imaged using a confocal microscope (SP5; Leica, Wetzlar, Germany) using a 20× air objective and leica las af software. For morphological analysis, vertical projections of z‐stacks were processed with the software las af (Leica) and image j. For Scholl analysis, intersections were counted between dendritic branches and concentric circles, starting at 6 μm from the cell soma and at radial increments of 3 μm, up to 250 μm (maximal radial dendrite extension). Dendritic tracking was performed with the ‘Neuron J’ plugin to image j (courtesy of Dr Erik Meijering, Erasmus University Medical Center Rotterdam). Counting of synaptic spines was performed on higher‐magnification scans (63× glycerol objective) from non‐overlapping portions of proximal (< 100 μm from soma) or distal (> 100 μm from soma) dendrites. The number of spines was then normalized for the length of the analysed dendrite.
Data analysis and statistics
Statistical analysis was performed with the software graphpad‐prism 5.03. Statistical significance in the comparison among groups was determined as follows. (i) Normality of data distribution was determined for each group with the Kolmogorov–Smirnoff test. (iia) In case of a normal distribution, a t‐test or analysis of variance (anova) + Bonferroni test were used for comparison between, respectively, two or multiple groups. (iib) In case of a non‐normal distribution, Mann–Whitney test or Kruskal–Wallis + Dunn's test were used for comparison between, respectively, two or multiple groups (see Figs 5 and 6). (iii). Two‐way anova was used to compare differences in PF–PC PSC in the presence or absence of β4 at each stimulation intensity (see Fig. 2). The type of test employed to analyse each set of data is specified in the figures and tables. Bar graphs represent mean ± standard error of the mean (SEM). Scatter dot plots represent single data points. Plots in which symbols are connected by lines (Figs 1H and I, and 3F) represent paired sets of data. Box plots (Fig. 2) represent mean, interquartile range and 5–95% of range. Numerical P values are reported in the tables, where F and P values are reported for repeated measurements (F; P). Significance is indicated as: *P < 0.05, **P < 0.001, ***P < 0.0001. The number of independent samples (different cells) is reported in parentheses in the figures and tables. For each type of experiment, measurements were carried out on three or more β4+/+ and three or more β4−/− mice, except for the measurements of pacemaker firing in thge presence of DNQX/gabazine (Fig. 5) which were carried out on two animals.
Figure 5.
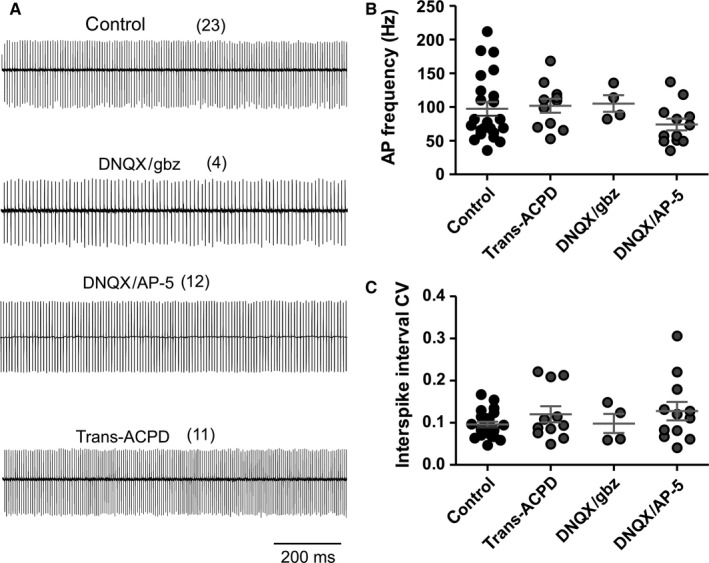
Pacemaker firing of wildtype adult Purkinje neurons in control conditions and after blockage of fast synaptic neurotransmission. (A) Representative cell‐attached recording from PCs in untreated control slices, and in the presence of: 10 μm 6,7 dinitroquinoxaline‐2,3‐dione and 10 μm SR 95531 (DNQX/gbz); 10 μm DNQX and 50 μm AP‐5 (DNQX/AP‐5); or 50 μm trans‐ACPD. (B, C) Average pacemaker firing frequency (B) and coefficient of variation of the inter‐spike interval (C) in untreated neurons and after pharmacological treatment. Differences between action potential firing in the presence of drugs and untreated controls were analysed with unpaired t‐test for normally distributed samples (frequencies) or Mann–Whitney test for non‐normally distributed samples (CV). Numbers of independent samples are indicated in parentheses in the figure.
Figure 6.
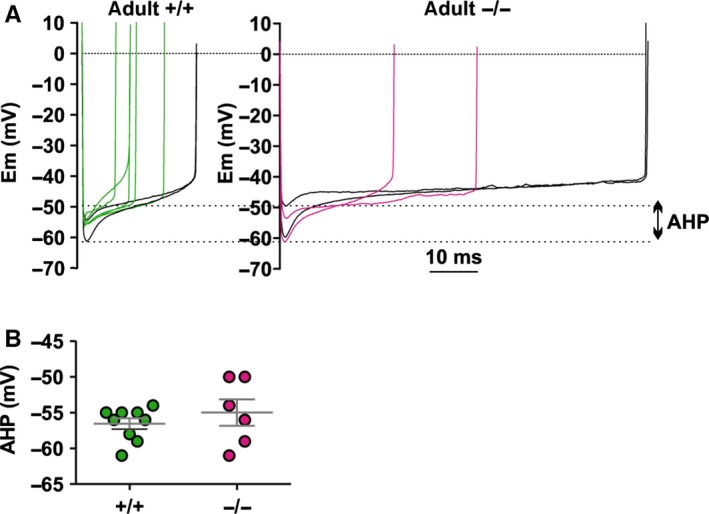
Whole‐cell current clamp recordings of afterhyperpolarizations (AHPs) in adult PCs. (A) Representative action potential inter‐spikes from regularly firing cells in adult wildtype (+/+) and lethargic (−/−) mice; dotted lines indicate the range of AHP. Note that action potentials with the same inter‐spike duration produce extremely different amplitudes of AHP (highlighted in black). (B) Scatter plot of average AHP in +/+ and −/− pacemaker firing neurons. Each mark represents an individual average AHP. Differences between AHP were analysed with unpaired t‐test.
Figure 2.
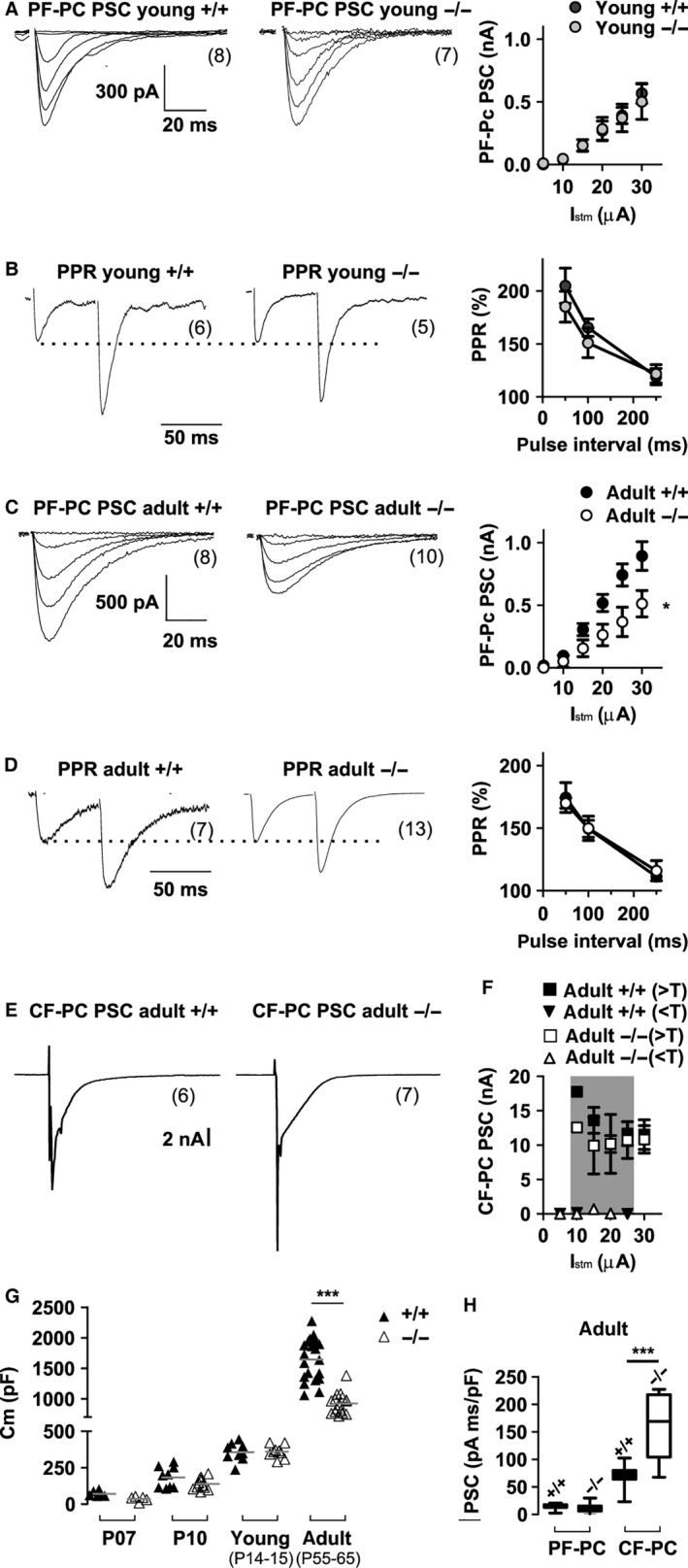
Evoked postsynaptic currents (PSCs) and paired pulse facilitation in cerebellar Purkinje neurons upon stimulation of parallel fibres and climbing fibres in wildtype (+/+) and lethargic (−/−) mice. (A–D) Representative PF volley recordings in juvenile (A, B) and adult (C, D) mice; *P < 0.05; two‐way anova. (A, C) PSC amplitude in responses to increasing stimulus intensity displays a significantly higher amplitude in adult +/+ compared to −/− mice (c). (B, D) PPF upon repetitive PF stimulation shows that the PPRs are not different in lethargic mice compared to age‐matched controls (mean ± SEM). (E) Representative PSCs upon CF stimulation. (F) CF–PC PSC all‐or‐none responses display constant amplitude at increasing stimulus intensities above effective threshold (> T, squares) and below threshold (< T, triangles). (G) Differences in whole cell capacitance (Cm) in adult PCs reveals a significantly decreased PC size in −/− mice. (H) Integral of PF–PC and CF–PC PSCs normalized to whole cell capacitance demonstrates that relative to cell size current density of PF–PC is identical in genotypes, but CF–PC current density is significantly increased in −/− mice. Box‐plots show mean, inter‐quartile distribution and 10–90% of the data range. ‘Young’ mice were P14–P15, while ‘adult’ mice were P55–P65. Differences between PF–PC PSCs were measured at any stimulation intensity with two‐way anova test; other differences between multiple populations (G, H) were measured with one‐way anova test followed by Bonferroni post‐test as detailed in Table 2. *P < 0.05; ***P < 0.0001. Numbers of independent samples are indicated in parentheses in the figure.
Figure 1.
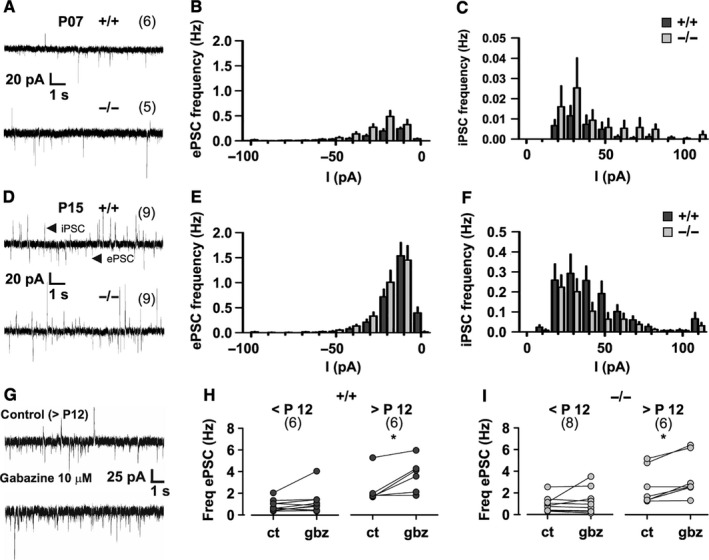
Spontaneous excitatory and inhibitory postsynaptic currents (ePSCs and iPSCs) in young Purkinje neurons from wildtype (+/+) and lethargic (−/−) mice. (A, D) Representative recordings from P07 and P15 mice show ePSCs and iPSCs as inward and outward current spikes, respectively. (B, C, E, F) Graphs depicting ePSC and iPSC amplitude and frequency in P07 (B, C) and P15 mice (E, F). (G) iPSCs and ePSCs in control conditions, and in the presence of gabazine; note the absence of iPSCs after gabazine application. (H, I) Mean ePSC frequency in paired recordings before and after gabazine application in +/+ (H) and −/− (I) mice. Note the significant increase in ePSC frequency in both +/+ and −/− mice after P12. Bar graphs indicate mean ± SEM; anova tests were used for comparison of amplitude and frequency as detailed in Table 1. A paired t‐test was used in H and I; *P < 0.05. Numbers of independent samples are indicated in parentheses in the figure.
Figure 3.
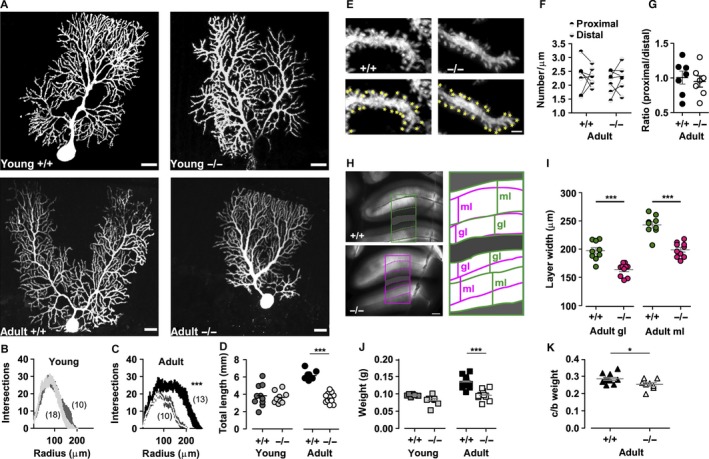
Morphological analysis of Purkinje neurons, granular and molecular layers. (A) Purkinje neurons of juvenile and adult wildtype (+/+) and lethargic (−/−) mice, perfused with biocytin during patch‐clamp recording and subsequently fluorescently labelled and analysed with confocal microscopy. Scale bars = 20 μm. (B, C) Graphs of the number of radial intersections of dendrites at increasing distance from the soma (Scholl analysis) in young (B) and adult (C) +/+ and −/− mice. Note the significantly diminished branching of adult −/− PCs. (D) Total dendritic length is significantly increased in dendrites in adult +/+ PCs. (E) High‐magnification images of PC dendrite segments in +/+ and −/− mice with dendritic spines highlighted by yellow marks (lower panels). Scale bar = 1.5 μm. (F) Average spine density in proximal and distal dendritic segments in adult +/+ and −/− neurons; paired measurements in each cell are connected by lines. (G) Ratio between proximal and distal spine density. (H) Brightfield images of acute cerebellar slices from adult +/+ and −/− mice. Scale bar = 250 μm. Masks outlining the granular and molecular layers are expanded and superimposed in the right panel. (I) Average width of adult +/+ and adult −/− granular and molecular layers. Note that both granular and molecular layers are significantly narrower in the cerebellar cortex of −/− mice. (J) Cerebellar weight of young +/+ (dark grey) and young −/− (light grey), adult +/+ (black) and adult −/− (white) mice. (K) Ratio between cerebellum and brain weight (c/b) in adult +/+ (black) and adult −/− (white) mice. Differences in dendritic branching (B, C) were analysed with two‐way anova and numbers of independent samples are indicated in parentheses. Differences between multiple populations were analysed with one‐way anova followed by Bonferroni post‐test (D, F, J, I). A Mann–Whitney test was used in the comparison between adult cerebellum/brain weight ratio and proximal/distal spine ratio (G, K). *P < 0.05, ***P < 0.0001.
Results
Loss of β4 does not reduce the spontaneous synaptic input of Purkinje neurons
The β4 subunit is known to support CaV2.1 calcium channel activity and thus promotes synaptic transmission in CNS thalamic neurons and in cultured hippocampal neurons (Caddick et al., 1999; Wittemann et al., 2000). Therefore, loss of β4 in lethargic (β4 −/−) mice is expected to severely affect the activity of CaV2.1 in cerebellar neurotransmission (Lonchamp et al., 2009; Galliano et al., 2013). To test this possibility we performed patch clamp recordings of ePSC and iPSC in PCs of acute cerebellar slices. In the whole‐cell configuration the membrane potential of PCs was held at −40 mV in 1‐week‐old mice (P07), or at −70 mV in 2‐week‐old mice (P15). At P07 (Fig. 1A) postsynaptic currents principally comprised ePSCs, while iPSCs were very rare (Fig. 1B and C). At P15 (Fig. 1D) ePSCs and iPSCs occurred at increased frequency (Fig. 1E and F; P > 0.04). Importantly, at both ages we found no difference in the frequency of ePSCs or iPSCs between lethargic and age‐matched control cerebellar slices (Table 1). Current amplitude was also not impaired by the lack of β4; while the ePSC amplitude was slightly increased in lethargic mice (Table 1), the iPSC amplitude was not changed.
Table 1.
Postsynaptic current (PSC) frequency and amplitude
| Frequency (Hz) | n | Amplitude (pA) | n | Postnatal days | |||
|---|---|---|---|---|---|---|---|
| ePSC | iPSC | ePSC | iPSC | ||||
| +/+P07 | 0.8 ± 0.2 | 0.15 ± 0.07 | 6; 6 | 25 ± 1 | 29 ± 4 | 6; 4 | 7.0 ± 0.0 |
| −/−P07 | 0.6 ± 0.1 | 0.13 ± 0.07 | 5; 5 | 35 ± 2 | 54 ± 17 | 5; 3 | 6.8 ± 0.2 |
| P * | 0.4 | 0.9 | 0.009 | 0.15 | |||
| anova: F; P | – | – | 19.73; < 0.0001 | 2.1; 0.1 | |||
| Bonferroni | – | – | < 0.001 | – | |||
| +/+P15 | 3.0 ± 0.6 | 1.3 ± 0.4 | 9; 9 | 18 ± 1 | 49 ± 6 | 9; 9 | 15.0 ± 0.8 |
| −/−P15 | 3.2 ± 0.5 | 0.8 ± 0.2 | 9; 9 | 25 ± 1 | 35 ± 5 | 9; 9 | 16.0 ± 0.9 |
| P * | 1.0 | 0.4 | 0.0005 | 0.1 | |||
| anova: F; P | – | – | 19.73; < 0.0001 | 2.1; 0.1 | |||
| Bonferroni | – | – | < 0.05 | – | |||
Mean ± SEM are reported. *P values refer to unpaired t‐test. anova (F; P) values refer to multiple comparisons between groups of different age, and Bonferroni post‐test (P) amongst the same data. –, not tested; n, number of samples.
Because excitatory input from granule cells to PCs is subject to tonic GABAergic inhibition (Hamann et al., 2002), we wondered whether this inhibitory activity could have masked differences in excitatory synaptic activity. Therefore, in a subset of experiments ePSCs were recorded in the presence of the selective GABA receptor blocker gabazine (Fig. 1G). In cerebellar slices from animals older than 12 days, gabazine (10 μm) completely blocked the iPSCs and concomitantly led to a significant increase of ePSC frequency (paired t‐test; P = 0.03, β4 +/+; P = 0.01, β4 −/−). Younger cerebellar slices (< P12) were not affected (P = 0.2, β4 +/+; P = 0.4, β4 −/−). Notably, the effect of gabazine was not different between wildtype (Fig. 1H) and lethargic neurons (Fig. 1I). Together these data demonstrate that loss of β4 does not impair the spontaneous synaptic transmission onto PCs. Considering the importance of P/Q‐type calcium channels for presynaptic function, this suggests that the lack of β4 in cerebellar synapses of lethargic mice may be compensated for by other β isoforms, as previously suggested (McEnery et al., 1998; Burgess et al., 1999).
Loss of β4 in adult lethargic mice specifically affects PF volley but not paired‐pulse facilitation (PPF)
Although β4 loss did not impair spontaneous synaptic activity, it might affect PF input via postsynaptic mechanisms, similar to what was previously observed in Cav2.1 mutations (Matsushita et al., 2002; Kodama et al., 2006). We therefore measured postsynaptic currents evoked by synchronous synaptic release from PFs (PF–PC PSCs). Electrical pulses increasing from 5 to 30 μA were locally delivered in the molecular layer. In both young and adult PCs these stimuli triggered PF–PC PSCs of increasing amplitudes (Fig. 2A and C; Table 2). In cerebellum of young lethargic mice the amplitudes of the evoked PF–PC PSCs were comparable to those recorded in wildtype controls. As PPF is a measure of presynaptic calcium signalling and regulation of release probability, we also recorded PSCs in response to two consecutive stimuli of equal strength (Fig. 2B). In young wildtype and lethargic mice the paired‐pulse ratios (PPRs) of PF–PC synapses were not significantly different. Thus, in young mice the function of PF–PC synapses and total synaptic input from PFs is apparently not affected by the lack of the calcium channel β4 subunit.
Table 2.
PF–PC PSC amplitude, paired pulse ratios and CF–PC PSC amplitude; see also Fig. 2
| PF–PC PSC (STM = 30 μA) | PF–PC PPR | CF‐Pc PSC | ||||
|---|---|---|---|---|---|---|
| nA | nA/pF | ʃPSC/Cm (pA.ms)/pF | nA | ʃPSC/Cm (pA.ms)/pF | ||
| +/+ young (P14–15) | 0.6 ± 0.1 (n = 8) | 209 ± 21 (n = 7) | ||||
| −/− young (P14–15) | 0.6 ± 0.1 (n = 6) | 185 ± 14 (n = 5) | ||||
| P * | 0.7 | 0.7 | ||||
| +/+ adult (P55–65) | 0.9 ± 0.1 (n = 8) | 0.54 ± 0.05 (n = 8) | 15 ± 2 (n = 8) | 182 ± 10 (n = 7) | 11.5 ± 2.2 (n = 6) | 69 ± 9 (n = 6) |
| −/− adult (P55‐65) | 0.5 ± 0.1 (n = 10) | 0.61 ± 0.15 (n = 10) | 12 ± 3 (n = 10) | 170 ± 4 (n = 13) | 10.8 ± 2.0 (n = 7) | 158 ± 24 (n = 7) |
| P * | 0.03 | 0.7 | 0.5 | 0.4 | 0.9 | 0.009 |
| anova: F; P | – | – | 34; < 0.0001 | – | – | 34; < 0.0001 |
| Bonferroni | – | – | ns | – | – | < 0.0001 |
|
A‐2 (S): F; P
A‐2 (T): F; P |
5.3; 0.03 58; < 0.001 |
– | – | – | – | – |
Mean ± SEM are reported. *P values refer to unpaired t‐test. anova (F; P) values refer to multiple comparison between all evoked responses (PF–PC PSCs and CF–PC PSCs) normalized for cell capacitance in adult mice followed by Bonferroni post‐test. ns, not significant; –, not tested. Two‐way anova (F; P) analyses the differences between PF–PC PSC amplitudes in the presence or absence of β4 (A2‐S) at each stimulation intensity (A2‐T; intensity = 5–30 μA).
In contrast, evoked PF–PC PSCs were significantly decreased in adult lethargic cerebellum (Fig. 2C). Under the assumption that axonal excitability, axonal density and probability of PF–PC connection are not altered, changes in the PF–PC PSC in lethargic mice would imply altered synaptic transmission. Here PSC amplitudes evoked by 30‐μA pulses were about half the size of PSCs in age‐matched wildtype controls (−/−, 0.5 ± 0.1 nA; +/+, 0.9 ± 0.1 nA; P = 0.03). Nevertheless, PPRs in adult lethargic mice were still unaltered (Fig. 2D; P = 0.4). Thus, the loss of β4 dramatically decreased the total synaptic input from PFs in adult but not in juvenile lethargic mice. However, the unaltered release probability indicates that this dysfunction does not arise from an impeded presynaptic function in PF–PC synapses.
Because these results suggested a postsynaptic mechanism affecting excitatory neurotransmission in PCs of adult lethargic mice, we next examined synaptic input from CFs. Electrical stimuli in the granular layer typically evoked large all‐or‐none postsynaptic currents with effective stimulation thresholds between 10 and 25 μA (Fig. 2E and F). In the cerebellum of wildtype and lethargic mice, the amplitudes of CF–PC PSCs were not significantly different from each other (Fig. 2F; Table 2, P = 0.9), equal to 10.5 ± 2.1 and 10.8 ± 2.0 nA, respectively, at a stimulus intensity of 30 μA. This indicates that, contrary to PF input, β4 is not critically involved in shaping the synaptic input of CFs to PCs.
In our whole‐cell recordings, we noticed that the cell capacitance of adult β4 −/− PCs was significantly smaller than that of adult β4 +/+ neurons (−/−, 925 ± 42 pF; +/+, 1643 ± 70 pF; P < 0.0001; Fig. 2G). No difference was detected at P15 (P = 0.8) and in younger mice (P07, P10). When PF–PC PSCs were normalized to the capacitance of each respective neuron, the resulting current densities were not significantly different (P = 0.7) between lethargic and wildtype mice. Similarly, the integrals of PF–PC PSC/whole cell capacitance of both genotypes were not significantly different (P = 0.5; Fig. 2H; Table 2). Thus, in lethargic mice the reduction of the PF–PC input matches the decrease in PC size. Integrals of CF–PC PSCs instead did not correspond to the decreased PC cell capacitance. Consequently, the density of CF–PC PSCs was significantly increased in the smaller lethargic PCs compared to wildtype controls (Fig. 2H; P < 0.01), and the resulting CF–PC PSC/PF–PC PSC ratio was smaller in lethargic mice. Furthermore, this decrease in PC capacitance and the associated decrease of PF–PC input in adult lethargic mice suggests that the loss of the calcium channel β4 subunit affects the late differentiation of the dendritic compartment of PCs.
Loss of β4 affects PC dendritic morphology in adult mice
To further examine the differentiation of PCs dendritic compartment, we analysed the dendritic shape and length in wildtype and lethargic PCs. The neurons were dialysed with biocytin via the patch pipette during whole‐cell recordings in acute cerebellar slices. Streptavidin staining of the labelled cells revealed the structure of the complete dendritic arbor (Fig. 3A). Using Scholl analysis we quantified dendritic branching and extension. The overall appearance, size and branching of juvenile lethargic PC dendrites was comparable to that of controls (Fig. 3A and B; F = 0.4, P = 0.5, n: +/+ = 18, n: −/− = 11). By contrast, in adult lethargic mice dendritic branches were fewer and shorter than in age‐matched wildtype controls (Fig. 3B and C; F = 22.3, P = 0.0001 n: +/+ = 10, n: −/− = 11). However, weeping‐willow‐like structures (Zwingman et al., 2001), or missing portions of the dendritic arbor (Kodama et al., 2006) were not observed. Dendrites were also counted within three concentric annular areas centred at the soma (proximal = 0–70 μm; medial = 70–120 μm; distal = 120–200 μm). The greatest differences occurred in the distal (P < 0.0001) and medial circle (P = 0.01). In the proximal circle, the number of dendrites was comparable in cerebella of lethargic and wildtype mice (P = 0.1). Total dendrite lengths were equal in young lethargic and wildtype mice (−/−, 3.6 ± 0.2 mm; +/+, 3.8 ± 0.4 mm; P = 0.7; Fig. 3D). In adults, however, lethargic dendrites were significantly shorter than in control PCs (−/−, 3.1 ± 0.1 mm; +/+, 5.4 ± 0.1 mm; P < 0.0001; unpaired t‐test). In agreement with the electrophysiological data described above, these results reveal a severely retarded differentiation of distal PC dendrites in the absence of β4.
If this deficiency in the PC dendrites in lethargic cerebellum arises from a diminished number of PFs and sparser synaptic contacts, a decrease in spine density would also be expected (Oda et al., 2011). Therefore, synaptic spines were counted in non‐overlapping dendritic segments of wildtype and lethargic PCs in proximal and distal dendrites (Fig. 3E). Spine density in β4 +/+ and β4 −/− neurons was comparable in proximal and distal dendrites (proximal: +/+, 2.3 ± 0.2/μm; −/−, to 2.1/μm; P = 0.6; distal: +/+, 2.3 ± 0.1/μm; −/−, 2.3 ± 0.2/μm; P = 0.8. Fig. 3F). Also the ratios of spine density in proximal to distal spines were not significantly different in the two genotypes (+/+, 1.01 ± 0.09; −/−, 0.95 ± 0.08; P = 0.6. Fig. 3G). Evidently, although the size of the distal dendritic arbor is substantially decreased in lethargic PCs, the density of synaptic spines on the remaining dendritic branches is unchanged.
The reduced size of the dendritic arbor is expected to result in a reduced width of the molecular layer. Furthermore, a decreased synaptic input from PFs may originate from a smaller number of granule cells. To examine these possibilities, we measured the widths of the molecular and granular cell layers in wildtype and lethargic acute cerebellar slices (Fig. 3H). Indeed, the widths of both the molecular and the granular cell layers were significantly reduced in lethargic mice (molecular layer: +/+, 240 ± 6 μm; −/−, 200 ± 6 μm; granular layer: +/+, 200 ± 4 μm; −/−, 160 ± 4 μm; P < 0.0001; Fig. 3I). Thus, the lack of β4 not only affects the size of the dendritic arbor of PCs and the thickness of the molecular layer, it probably also causes a reduction in the number of granule cells. This probably results in reduced PF input to PCs. Changes in the thickness of the molecular and granular cell layers were accompanied by a significant weight reduction of the cerebellum (P < 0.0001) and by a significant decrease in the proportion of cerebellar to whole‐brain weights (P = 0.029) of adult lethargic mice compared to wildtype (Fig. 3K and J), indicative of widespread effects of β4 loss on brain development.
Lack of the calcium channel β4 subunit impairs the development of high PC pacemaker frequency but not of firing regularity
Finding that the lack of β4 in lethargic mice specifically reduces the PF input and dendritic morphology of cerebellar PCs, we reasoned that this might impair the output of these neurons. Therefore, spontaneous PC pacemaker activity was recorded in acute cerebellar slices from young and adult mice. At P15, locomotion is acquired and the cerebellar network is not fully developed (Altman, 1972). At 1 month of age, locomotion and the cerebellar networks are mature. Figure 4A shows tonic action potential firing of young and adult PCs from cell‐attached recordings in acute cerebellar slices of wildtype and lethargic mice. Firing occurred at relatively low frequencies in young mice (P14–15) of both genotypes (+/+, 50 ± 5 Hz; −/−, to 47 ± 5 Hz; P = 0.7; Fig. 4B). Firing frequencies increased to 97 ± 10 Hz in wildtype adults (P35–60). However, in adult lethargic mice PC firing frequencies failed to increase (38 ± 5 Hz; Fig. 4A and B) and were equal to values measured in the young animals. Accordingly, the age‐related increase in firing frequency was highly significant only in the wildtype mouse cerebellum (P = 0.002), but not in the lethargic mouse cerebellum (P = 0.2). Furthermore, the firing frequency of wildtype adults was significantly higher than that of lethargic adults (P < 0.0001). Thus, in the absence of β4, functional maturation of the PC pacemaker firing frequency fails after P15. As Purkinje axons are the only cortical efferent, low PC firing rates correspond to decreased cerebellar cortical output.
Figure 4.
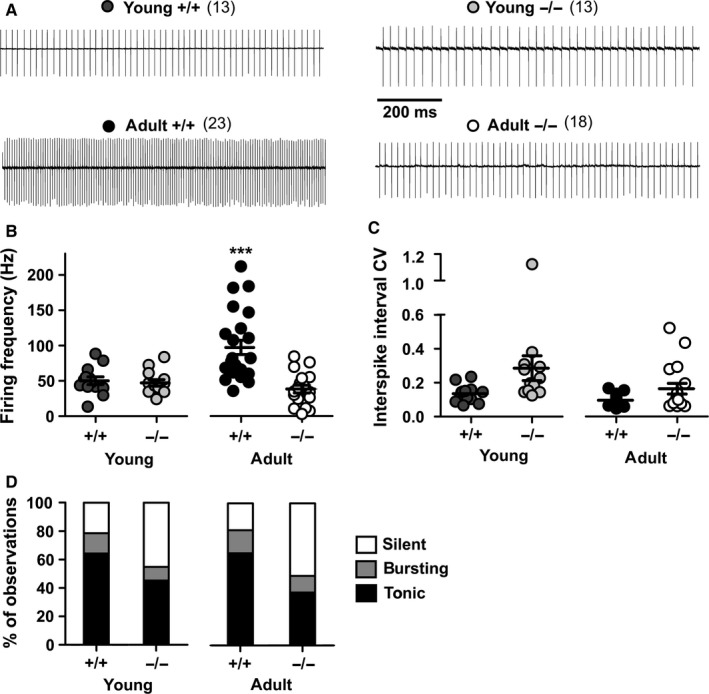
Pacemaker firing of Purkinje neurons in young and adult wildtype (+/+) and lethargic (−/−) mice. (A) Representative cell‐attached recordings; vertical deflection of baseline represents individual action potentials. (B, C) Average pacemaker firing frequency (B) and coefficient of variation of the inter‐spike interval (C) in young and adult +/+ and −/− PCs. Each mark in the scatter plots represents individual average firing properties. Bars indicate mean ± SEM. Note that the frequency of adult wildtype mice is significantly higher than that of lethargic mice. (D) Bar graph indicating the probability and the proportion between independent observations of silent, bursting and tonic firing cells in young and adult +/+ and −/− mice. Differences between firing frequencies were analysed with one‐way anova followed by Bonferroni post‐test and differences between CV (not normally distributed) were analysed with Kruskal–Wallis test, followed by Dunn's post‐test. ***P < 0.0001. Numbers of independent samples are indicated in parentheses in the figure.
To also quantify the regularity of PC action potential firing in lethargic and wildtype mice, we calculated the coefficient of variation (CV) of the inter‐spike intervals (Fig. 4C). In young neurons CV was increased from 0.13 ± 0.01 in β4 +/+ to 0.28 ± 0.07 in β4 −/− neurons (P = 0.007, unpaired t‐test). However, after a Bonferroni post‐test, the difference between CV of young mice or between CV of adult mice was no longer significant (Table 3). In adults, the firing regularity had improved in both wildtype and lethargic mouse neurons (+/+, P = 0.02; −/−, P = 0.008; unpaired t‐test); the difference in CV between the two genotypes was not statistically significant (0.09 ± 0.01 and 0.16 ± 0.03, respectively; P = 0.2). Together these analyses demonstrate that in adult lethargic cerebellum the firing rate of PCs is substantially reduced without a concomitant loss of firing regularity. In wildtype and lethargic mice we observed tonic firing, bursting and silent PCs. In young and in adult wildtype mice, the tonic firing PCs accounted for 65% and silent PCs for 20% of the whole cell population. In lethargic mice, tonic firing was instead limited to 45% of the young and 37% of the adult PC cell population. This was coincident with an increased number of silent PCs to 45 and 50% in young and adult lethargic mice, respectively. In conclusion, not only were the tonic firing rates were reduced but also the number of active neurons was diminished by the loss of β4, which could contribute to depression of cortical output. The burst firing mode occurred rarely and the proportion of bursting neurons in wildtype and lethargic mice (10–15% of the total cell population) was virtually the same at any age.
Table 3.
Pacemaker firing frequency and coefficient of variation
| Frequency (Hz) | CV | n | |
|---|---|---|---|
| +/+ young (P14–15) | 50 ± 5 | 0.135 ± 0.014 | 13 |
| −/− young (P14–15) | 47 ± 5 | 0.285 ± 0.073 | 13 |
| P | 0.7* | 0.007† | |
| +/+ adult (P35–60) | 97 ± 10 | 0.096 ± 0.006 | 23 |
| −/− adult (P35–60) | 38 ± 5 | 0.164 ± 0.031 | 18 |
| P | < 0.0001* | 0.2† | |
| P young vs. adult +/+ | 0.002* | 0.02† | |
| P young vs. adult −/− | 0.2* | 0.008† | |
| Multiple comparison (one‐way anova = * or K‐W = †) F; P | 13.05*; < 0.0001 | 21.6 < 0.0001† | |
| Post‐hoc (young +/+ vs. −/−) | ns* | ns† | |
| Post‐hoc (adult +/+ vs. −/−) | < 0.0001* | ns† | |
| Post‐hoc (young +/+ vs. adult +/+) | < 0.01* | ns† | |
| Post‐hoc test (young −/− vs. adult −/−) | ns* | P < 0.5† |
Mean ± SEM are reported. P (*) values refer to unpaired t‐test. Multiple comparison refers to one‐way anova (P; F) and Bonferroni post‐test. The coefficient of variation (CV) of −/− populations was not normally distributed, and therefore significance was tested with Mann–Whitney test and Kruskal–Wallis test (K‐W statistic; P) followed by Dunn's multiple comparison test (†). ns, not significant; –, not tested.
High pacemaker firing rates are not dependent on tonic glutamatergic neurotransmission
To examine whether the decreased PC tonic firing rates might be related to the observed reduction of PF–PC, we recorded spontaneous firing of PCs in cerebellar slices of wildtype and lethargic mice upon block of tonic neurotransmission (Fig. 5A). We used bath application of DNQX and gabazine, DNQX and AP‐5, or DNQX and trans‐ACPD for 15–60 min to block α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid (AMPA), N‐methyl‐d‐aspartate (NMDA), gamma‐aminobutyric acid (GABA)A and metabotropic glutamate receptors. However, none of these conditions affected the firing rates or firing regularity of PCs in wildtype (Fig. 5B, Table 4) or lethargic cerebellum. Thus, altered synaptic input is probably not involved in the short‐ to medium‐term modulation of PC pacemaker firing frequency and therefore unlikely to be the cause for the differences in firing frequency observed in adult wildtype and lethargic mice. However, these experiments do not exclude possible long‐term effects of altered neurotransmission in lethargic mice and a critical involvement of β4 in other potential modulatory pathways.
Table 4.
Pacemaker firing frequency and coefficient of variation after pharmacological blockage of glutamate and/or GABA receptors; see also Fig. 5
| Frequency (Hz) | CV | P | n | |
|---|---|---|---|---|
| +/+DNQX/gbz | 105 ± 12 | 0.098 ± 0.022 | 0.3; 0.9 | 4 |
| +/+DNQX/AP‐5 | 74 ± 9 | 0.128 ± 0.021 | 0.15; 0.4 | 12 |
| +/+ trans‐ACPD | 102 ± 10 | 0.120 ± 0.019 | 0.2; 0.5 | 11 |
Average ± SEM are reported. P values refer to unpaired t‐test. These experiments were carried out in adult mice (P35–60).
Afterhyperpolarization is not affected by the loss of β4
Reduction of PC pacemaker rates occurs in an ataxia mutation of the BK calcium‐activated potassium channel (Sausbier et al., 2004). This is associated with a prominent decrease in potassium currents during afterhyperpolarization (AHP). If sparser pacemaker firing in lethargic PCs involved BK potassium currents, this would be revealed by smaller AHP amplitudes. However, whole cell recordings from regularly firing wildtype or lethargic neurons showed normal AHP with negative peaks of −56.6 ± 0.7 and −55.0 ± 1.9 mV, respectively (Fig. 6; P = 0.4). Furthermore, longer inter‐spike intervals among our samples were not necessarily associated with smaller AHP. Therefore, small AHP derived from loss of BK potassium currents does not contribute to the lethargic phenotype.
Discussion
Loss of the calcium channel β4 subunit causes motor impairment (Khan & Jinnah, 2002) with striking similarities between the lethargic (β4 −/−) mouse and mouse models with Cav2.1‐related forms of ataxia (Fletcher et al., 1996; Burgess & Noebels, 1999a,b; Barclay et al., 2001; Guida et al., 2001; Pietrobon, 2002). However, past investigations of physiological and pathophysiological roles of β4 in the brain yielded conflicting results. Depending on the brain region or experimental system used β4 was found either to be essential for normal neuronal function (Caddick et al., 1999; Lin et al., 1999; Wittemann et al., 2000), potentially relevant (McEnery et al., 1998), or even entirely redundant with other β isoforms (Burgess et al., 1999). Apparently the context of the brain region and the specific neuronal network is critical for understanding the role of the β4 subunit in the etiology of the lethargic phenotype (Caddick et al., 1999). Because of the importance of the cerebellar cortex in motor control we examined here (i) whether the loss of β4 recapitulates the effects of loss of Cav2.1 on cerebellar function and morphology, (ii) which functions of PC networks are primarily impaired by the loss of β4 and (iii) how this may contribute to the ataxic phenotype in lethargic mice. The present slice electrophysiology study demonstrates that the lack of the calcium channel β4 subunit causes a decreased late development in cerebellar cortical networks, characterized by reduced PF–PC input, shorter PC dendrites and depressed PC output (Fig. 7). This is the first study addressing the physiological role of β4 and its involvement in the etiology of ataxia in the context of native PC networks in acute cerebellar slices.
Figure 7.
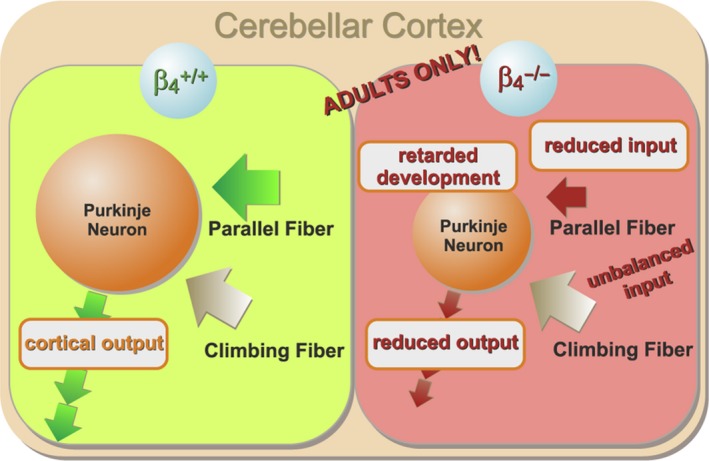
Changes in cerebellar cortical network caused by the lack of β4 in adult lethargic mice. In the absence of β4 the input from PFs is reduced, whereas the input from CFs is spared. Thus, the PC inputs are unbalanced. The size of PCs (dendritic branching) and the width of the granular cell layer are reduced. The firing frequency of PCs and thus cerebellar output is reduced. All these deficits affect only adult and not juvenile lethargic mice, consistent with a developmental retardation of cerebellar cortical networks in lethargic mice.
Does loss of the β4 subunit affect known functions of Cav2.1 in the cerebellum?
Because the β4 subunit is the primary partner of CaV2.1 in cerebellum and loss‐of‐function mutations of both calcium channel subunits result in ataxia (Burgess et al., 1997; Burgess & Noebels, 1999a; Escayg et al., 2000; Guida et al., 2001; Pietrobon, 2002; Buraei & Yang, 2010; Schlick et al., 2010), it is reasonable to expect that loss of either one of these proteins affects the same brain functions. Indeed, the loss of β4 in the lethargic mouse recapitulates a number of impairments of cerebellar function and morphology previously observed in the ataxic Cav2.1 mouse models rocker, tottering and purky (Zwingman et al., 2001; Matsushita et al., 2002; Kodama et al., 2006; Erickson et al., 2007; Mark et al., 2011). These common impairments encompass a retarded cerebellar development, smaller PC dendrites, PF depression, the resulting imbalance between PF and CF input to PCs, and the late onset of the disease phenotype. Moreover, both the lethargic and the rocker mice show a normal synaptic density in PC dendrites (Kodama et al., 2006), although this was not observed in several other ataxic models (Kashiwabuchi et al., 1995; Kurihara et al., 1997; Oda et al., 2011). This similarity indicates that altered function of cerebellar P/Q‐type calcium channels may contribute to the lethargic phenotype. Importantly, it further demonstrates that β4 is not functionally redundant, as its loss cannot be fully compensated for by other β isoforms.
On the other hand, other cerebellar functions are differently affected by the loss of Cav2.1 and β4. For example general presynaptic loss of Cav2.1 affects the spontaneous synaptic activity (Lonchamp et al., 2009; Galliano et al., 2013) and alters release probability (Maejima et al., 2013). However, in the lethargic mouse cerebellum we found that spontaneous synaptic activity and PPF of PF–PC PSCs were not reduced. In theory, the lack of a presynaptic phenotype might indicate compensation for the lost β4 function in PF–PC synapses. However, only complete knockout of Cav2.1 in cerebellar granule cells affected presynaptic PF–PC function (Lonchamp et al., 2009; Galliano et al., 2013; Maejima et al., 2013), whereas partial loss of Cav2.1 function did not show a presynaptic phenotype (Matsushita et al., 2002; Kodama et al., 2006). Apparently the safety margin with regard to calcium influx in this synapse is very high (Matsushita et al., 2002), so that a possibly small reduction of P/Q‐type channels caused by the lack of β4 may have no functional consequence. By contrast, the spontaneous ePSC amplitude in juvenile lethargic mice was increased. While this together with unaltered PPR does not suggest impairment of presynaptic P/Q‐type channels, the slight increase in the lethargic PSC amplitude may relate to more subtle changes in the formation of presynaptic channel complexes (Iwasaki et al., 2000) upon loss of β4. The situation in the cerebellum is probably different from that in other brain regions as the loss of β4 resulted in reduced presynaptic function in cultured hippocampal neurons (Wittemann et al., 2000; Xie et al., 2007), as well as in whole‐brain synaptosome preparations (Lin et al., 1999). Moreover, even though the PF PSC was decreased in the absence of β4, the unaltered PF PPR and the presence of all‐or‐none CF responses suggest that unlike Cav2.1−/−, β4 −/− has little relevance for heterosynaptic competition between PFs and CFs or for homosynaptic competition among CFs (Miyazaki et al., 2004).
Notably, the effects of the loss of β4 in lethargic mice were not limited to deficiencies of cerebellar functions previously observed in Cav2.1 mutants. We also observed a dramatically decreased PC pacemaker frequency, while the regularity of pacemaker firing was maintained. A similar deficiency of PC pacemaker activity was not observed upon Cav2.1 mutation (leaner) nor in closely related (ducky) forms of ataxia where both controls and mutants had average firing rates between 75 and 115 Hz (Sausbier et al., 2004; Walter et al., 2006). In contrast, lethargic PCs had firing rates of about 40 Hz, which is significantly reduced compared to wildtype controls which fired at about 100 Hz. In lethargic mice, the β4‐specific dysfunction of PC firing suggests a critical function of this protein in the context of another type of calcium channel that cannot be compensated for by alternative β isoforms (McEnery et al., 1998; Burgess et al., 1999). Alternatively, reduced cerebellar PC firing rates as well as lower number of constitutively active neurons could result from a loss of calcium‐activated BK potassium currents (Sausbier et al., 2004). However, in β4 −/− mice, AHP was not affected. Even so, prolonged inter‐spike intervals and reduced cortical output may contribute to the motor impairment of lethargic as well as of BK−/− mice.
Overall, this study reveals both common and specific functions of β4 and Cav2.1 in cerebellar networks. Considering the role of β4 as an auxiliary calcium channel subunit, the common deficiencies involving PF–PC PSCs and PC development suggest a role of the β4 isoform in modulating PSCs, pacemaker activity and dendritic maturation. In contrast, the absence of a phenotype in CF–PC synapse function and in PF–PC release probability indicates that for neurotransmitter release and synaptic competition β subunit functions may be redundant. Furthermore, the absence of β4‐specific defects of PC pacemaker firing regularity confirms an earlier study showing the redundancy of β4 at the PC soma (Burgess et al., 1999). Conversely, β4‐specific defects of PC pacemaker frequency suggest an essential function of β4 with a hitherto unidentified calcium channel isoform other than Cav2.1. Finally, considering the role of β4b as a regulator of gene transcription (Etemad et al., 2014), it is possible that loss of β4 may affect cerebellar development through calcium channel‐independent pathways. Taken together, the similarities and differences between β4 −/− and Cav2.1 mutants suggests that Cav2.1‐related and unrelated dysfunctions may coexist in the lethargic cerebellum.
How does the loss of β4 affect the network activity in the cerebellar cortex of lethargic mice?
The observed reduction of the evoked PF input in cerebellar PCs is one of the most striking functional consequences of the loss of β4. In lethargic mice the cerebellar granular layer was thinner. This may imply a reduced number of granule cells and consequently fewer PFs. More importantly, the distal dendritic arbor of PCs in lethargic mice had a reduced height and width and with slightly sparser branching (Fig. 3; Fig. S1). According to our experimental paradigm, electrical stimulation of a given PF bundle would recruit a smaller dendritic area of lethargic PCs than in controls. This contributes to limiting the evoked PF–PC PSCs in lethargic mice. However, given the unchanged synaptic density (Fig. 3) and only mildly reduced distal dendritic density (Fig. S1) the morphological changes cannot fully account for the large reduction in the PF–PC PSC recorded in lethargic mice.
This explanation for the reduced PF volley is also challenged by previous research suggesting that other presynaptic and postsynaptic factors might contribute to aberrant neurotransmission upon loss of Cav2.1 or β4 (Caddick et al., 1999; Lin et al., 1999; Wittemann et al., 2000; Kodama et al., 2006; Xie et al., 2007). However, in contrast to defects of neurotransmission found in Cav2.1‐related pathologies (Pietrobon, 2002, 2005) and to what was observed upon loss of β4 in other neuronal systems (Lin et al., 1999; Wittemann et al., 2000; Xie et al., 2007), we found unaltered PF–PC release probability, suggesting that the loss of β4 in cerebellum did not severely affect presynaptic neurotransmitter release. This along with unaltered spine density of PF–PC synapses suggests a normal presynaptic physiology in the cerebellum of lethargic mice. The decreased PF–PC PSC could therefore be caused by specific impairment of postsynaptic function (Kodama et al., 2006) or directly related to the reduced distal branching of PC dendrites. Given the abundance of Cav2.1 channels in PC dendrites and their function in the amplification of postsynaptic currents (De Schutter & Bower, 1994), a potential reduction of dendritic Cav2.1 is expected to alter processing of the PF input. As postsynaptic enrichment of Cav2.1 channels is an age‐dependent phenomenon and follows a somato‐dendritic gradient (Indriati et al., 2013), a loss of postsynaptic Cav2.1 would well agree with the observed reduction of distal dendritic tree (Fig. S1), the decreased PF–PC input and the late onset of the lethargic phenotype. Specifically, dendritic spikes (Llinas et al., 1968; Llinas & Sugimori, 1980) may be affected by the postsynaptic loss of Cav2.1 (Tank et al., 1988) in lethargic mice, which in turn would impair the processing and integration of PF and CF inputs (Rancz & Hausser, 2006; Davie et al., 2008), and long‐ and short‐term synaptic plasticity (Kreitzer & Regehr, 2001; Golding et al., 2002; Holthoff et al., 2004; Rancz & Hausser, 2006).
A reduced PC firing rate in adult lethargic mice was another striking effect of the loss of the β4 subunit in cerebellar cortex. Because PF and CF input have antagonistic effects on PC excitability and pacemaker rates (Shibuki & Kimura, 1997; Smith & Otis, 2003; Cerminara & Rawson, 2004), the skewed proportion between PFs and CFs observed here in lethargic mice might dictate reduced rates of pacemaker firing in PCs. However, PC firing frequency was not affected by the acute block of tonic neurotransmission, thus questioning a causal link between reduced PF input and reduced PC pacemaker activity in the lethargic cerebellum.
How do defects in the cerebellar network contribute to the motor impairment in lethargic mice?
Our slice electrophysiology analysis of the cerebellar network in lethargic mice revealed deficiencies at the input and output side of PCs, both of which could contribute to ataxia. Motor learning relies on the plasticity of PCs and their role in integrating the inputs from PFs and CFs (Ito et al., 1982; Hansel & Linden, 2000; Ito, 2001). In lethargic mice evoked PF PSCs are specifically reduced, which could impair motor learning (Galliano et al., 2013). In the presynaptic compartment this could be caused by a paucity of granule cells from which the PFs originate, and in the postsynaptic compartment by smaller medial and distal PC dendrites onto which PFs project. Yet, the CF input and proximal PC dendrites are unaffected by the loss of β4. The resulting imbalance between PF and CF input onto PCs by itself could account for impaired plasticity, learning and coordination (Ito et al., 1982; Hansel & Linden, 2000; Ito, 2001; Galliano et al., 2013). Similarly, defective dendritic processing of both PF and CF inputs could lead to impaired network plasticity (Kreitzer & Regehr, 2001; Golding et al., 2002; Holthoff et al., 2004; Rancz & Hausser, 2006). Ultimately, cerebellar dysfunctions must be reflected in altered PC tonic output. As PCs represent the sole cerebellar cortical output, the observed slowed PC pacemaker firing will result in inefficient inhibition of the deep cerebellar nuclei (Gauck & Jaeger, 2000). Similarly reduced tonic output has previously been described in other mouse models with impaired motor coordination (Sausbier et al., 2004). Thus, reduced tonic firing of cerebellar PCs could be a major cause of the motor impairment generated by the loss of the calcium channel β4 subunit in lethargic mice.
Conflict of interest
The authors declare no competing financial interests.
Supporting information
Fig. S1. Substantially reduced size and modestly reduced density of the distal dendritic arbor in lethargic Purkinje neurons.
Acknowledgements
We thank Drs J. Striessnig, A. Lieb and G. Obermair for practical support and valuable scientific discussion, Dr M. Offterdinger for valuable support in the Biooptic facility of the Medical University of Innsbruck and Dr E. Meijering, Biomedical Imaging Group, Erasmus University Medical Center Rotterdam, for providing the image analysis plugin ‘Neuron J’. This work was supported by the Austrian Science Fund (FWF) grants F44010 (SFB‐F44 – Cell Signaling in chronic CNS disorders), F44060, P23479 and P27031 (to B.E.F.), the Medical University Innsbruck (ST712600S441) and the Tiroler Wissenschaftsfond 2013 (UNI‐0404/1419 to B.B.).
Abbreviations
- ACPD
1‐amino‐1,3‐dicarboxycyclopentane
- AHP
afterhyperpolarization
- Cav
voltage‐activated calcium channel
- CF
climbing fibre
- CNS
central nervous system
- CV
coefficient of variation
- ePSC
excitatory postsynaptic current
- iPSC
inhibitory postsynaptic current
- PC
Purkinje cell
- PF
parallel fibre
- PPF
paired pulse facilitation
- PPR
paired pulse ratio
References
- Altman, J. (1972) Postnatal development of the cerebellar cortex in the rat. I. The external germinal layer and the transitional molecular layer. J. Comp. Neurol., 145, 353–397. [DOI] [PubMed] [Google Scholar]
- Barclay, J. , Balaguero, N. , Mione, M. , Ackerman, S.L. , Letts, V.A. , Brodbeck, J. , Canti, C. , Meir, A. et al (2001) Ducky mouse phenotype of epilepsy and ataxia is associated with mutations in the Cacna2d2 gene and decreased calcium channel current in cerebellar Purkinje cells. J. Neurosci., 21, 6095–6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti, B. , Matyash, V. & Kettenmann, H. (2011) Astrocytes control GABAergic inhibition of neurons in the mouse barrel cortex. J. Physiol., 589, 1159–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodbeck, J. , Davies, A. , Courtney, J.M. , Meir, A. , Balaguero, N. , Canti, C. , Moss, F.J. , Page, K.M. et al (2002) The ducky mutation in Cacna2d2 results in altered Purkinje cell morphology and is associated with the expression of a truncated alpha 2 delta‐2 protein with abnormal function. J. Biol. Chem., 277, 7684–7693. [DOI] [PubMed] [Google Scholar]
- Buraei, Z. & Yang, J. (2010) The b subunit of voltage‐gated Ca2+ channels. Physiol. Rev., 90, 1461–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess, D.L. & Noebels, J.L. (1999a) Single gene defects in mice: the role of voltage‐dependent calcium channels in absence models. Epilepsy Res., 36, 111–122. [DOI] [PubMed] [Google Scholar]
- Burgess, D.L. & Noebels, J.L. (1999b) Voltage‐dependent calcium channel mutations in neurological disease. Ann. N.Y. Acad. Sci., 868, 199–212. [DOI] [PubMed] [Google Scholar]
- Burgess, D.L. , Jones, J.M. , Meisler, M.H. & Noebels, J.L. (1997) Mutation of the Ca2+ channel beta subunit gene Cchb4 is associated with ataxia and seizures in the lethargic (lh) mouse. Cell, 88, 385–392. [DOI] [PubMed] [Google Scholar]
- Burgess, D.L. , Biddlecome, G.H. , McDonough, S.I. , Diaz, M.E. , Zilinski, C.A. , Bean, B.P. , Campbell, K.P. & Noebels, J.L. (1999) Beta subunit reshuffling modifies N‐ and P/Q‐type Ca2+ channel subunit compositions in lethargic mouse brain. Mol. Cell Neurosci., 13, 293–311. [DOI] [PubMed] [Google Scholar]
- Caddick, S.J. , Wang, C. , Fletcher, C.F. , Jenkins, N.A. , Copeland, N.G. & Hosford, D.A. (1999) Excitatory but not inhibitory synaptic transmission is reduced in lethargic (Cacnb4 lh) and tottering (Cacna1atg) mouse thalami. J. Neurophysiol., 81, 2066–2074. [DOI] [PubMed] [Google Scholar]
- Campiglio, M. & Flucher, B.E. (2015) The role of auxiliary subunits for the functional diversity of voltage‐gated calcium channels. J. Cell. Physiol., 230, 2019–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerminara, N.L. & Rawson, J.A. (2004) Evidence that climbing fibers control an intrinsic spike generator in cerebellar Purkinje cells. J. Neurosci., 24, 4510–4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davie, J.T. , Clark, B.A. & Hausser, M. (2008) The origin of the complex spike in cerebellar Purkinje cells. J. Neurosci., 28, 7599–7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Schutter, E. & Bower, J.M. (1994) Simulated responses of cerebellar Purkinje cells are independent of the dendritic location of granule cell synaptic inputs. Proc. Natl. Acad. Sci. USA, 91, 4736–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin, A.C. (2012) Calcium channel auxiliary alpha2delta and beta subunits: trafficking and one step beyond. Nat. Rev. Neurosci., 13, 542–555. [DOI] [PubMed] [Google Scholar]
- Dung, H.C. & Swigart, R.H. (1971) Experimental studies of ‘lethargic’ mutant mice. Tex. Rep. Biol. Med., 29, 273–288. [PubMed] [Google Scholar]
- Erickson, M.A. , Haburcak, M. , Smukler, L. & Dunlap, K. (2007) Altered functional expression of Purkinje cell calcium channels precedes motor dysfunction in tottering mice. Neuroscience, 150, 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escayg, A. , De Waard, M. , Lee, D.D. , Bichet, D. , Wolf, P. , Mayer, T. , Johnston, J. , Baloh, R. et al (2000) Coding and noncoding variation of the human calcium‐channel beta4‐subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am. J. Hum. Genet., 66, 1531–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etemad, S. , Obermair, G.J. , Bindreither, D. , Benedetti, A. , Stanika, R. , Di Biase, V. , Burtscher, V. , Koschak, A. et al (2014) Differential neuronal targeting of a new and two known calcium channel beta4 subunit splice variants correlates with their regulation of gene expression. J. Neurosci., 34, 1446–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher, C.F. , Lutz, C.M. , O'Sullivan, T.N. , Shaughnessy, J.D. Jr , Hawkes, R. , Frankel, W.N. , Copeland, N.G. & Jenkins, N.A. (1996) Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell, 87, 607–617. [DOI] [PubMed] [Google Scholar]
- Galliano, E. , Gao, Z. , Schonewille, M. , Todorov, B. , Simons, E. , Pop, A.S. , D'Angelo, E. , van den Maagdenberg, A.M. et al (2013) Silencing the majority of cerebellar granule cells uncovers their essential role in motor learning and consolidation. Cell Rep., 3, 1239–1251. [DOI] [PubMed] [Google Scholar]
- Gauck, V. & Jaeger, D. (2000) The control of rate and timing of spikes in the deep cerebellar nuclei by inhibition. J. Neurosci., 20, 3006–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding, N.L. , Staff, N.P. & Spruston, N. (2002) Dendritic spikes as a mechanism for cooperative long‐term potentiation. Nature, 418, 326–331. [DOI] [PubMed] [Google Scholar]
- Guida, S. , Trettel, F. , Pagnutti, S. , Mantuano, E. , Tottene, A. , Veneziano, L. , Fellin, T. , Spadaro, M. et al (2001) Complete loss of P/Q calcium channel activity caused by a CACNA1A missense mutation carried by patients with episodic ataxia type 2. Am. J. Hum. Genet., 68, 759–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann, M. , Rossi, D.J. & Attwell, D. (2002) Tonic and spillover inhibition of granule cells control information flow through cerebellar cortex. Neuron, 33, 625–633. [DOI] [PubMed] [Google Scholar]
- Hansel, C. & Linden, D.J. (2000) Long‐term depression of the cerebellar climbing fiber–Purkinje neuron synapse. Neuron, 26, 473–482. [DOI] [PubMed] [Google Scholar]
- Holthoff, K. , Kovalchuk, Y. , Yuste, R. & Konnerth, A. (2004) Single‐shock LTD by local dendritic spikes in pyramidal neurons of mouse visual cortex. J. Physiol., 560, 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indriati, D.W. , Kamasawa, N. , Matsui, K. , Meredith, A.L. , Watanabe, M. & Shigemoto, R. (2013) Quantitative localization of Cav2.1 (P/Q‐type) voltage‐dependent calcium channels in Purkinje cells: somatodendritic gradient and distinct somatic coclustering with calcium‐activated potassium channels. J. Neurosci., 33, 3668–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito, M. (2001) Cerebellar long‐term depression: characterization, signal transduction, and functional roles. Physiol. Rev., 81, 1143–1195. [DOI] [PubMed] [Google Scholar]
- Ito, M. , Sakurai, M. & Tongroach, P. (1982) Climbing fibre induced depression of both mossy fibre responsiveness and glutamate sensitivity of cerebellar Purkinje cells. J. Physiol., 324, 113–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki, S. , Momiyama, A. , Uchitel, O.D. & Takahashi, T. (2000) Developmental changes in calcium channel types mediating central synaptic transmission. J. Neurosci., 20, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwabuchi, N. , Ikeda, K. , Araki, K. , Hirano, T. , Shibuki, K. , Takayama, C. , Inoue, Y. , Kutsuwada, T. et al (1995) Impairment of motor coordination, Purkinje cell synapse formation, and cerebellar long‐term depression in GluR delta 2 mutant mice. Cell, 81, 245–252. [DOI] [PubMed] [Google Scholar]
- Khan, Z. & Jinnah, H.A. (2002) Paroxysmal dyskinesias in the lethargic mouse mutant. J. Neurosci., 22, 8193–8200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama, T. , Itsukaichi‐Nishida, Y. , Fukazawa, Y. , Wakamori, M. , Miyata, M. , Molnar, E. , Mori, Y. , Shigemoto, R. et al (2006) A CaV2.1 calcium channel mutation rocker reduces the number of postsynaptic AMPA receptors in parallel fiber‐Purkinje cell synapses. Eur. J. Neurosci., 24, 2993–3007. [DOI] [PubMed] [Google Scholar]
- Kreitzer, A.C. & Regehr, W.G. (2001) Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron, 29, 717–727. [DOI] [PubMed] [Google Scholar]
- Kurihara, H. , Hashimoto, K. , Kano, M. , Takayama, C. , Sakimura, K. , Mishina, M. , Inoue, Y. & Watanabe, M. (1997) Impaired parallel fiber→Purkinje cell synapse stabilization during cerebellar development of mutant mice lacking the glutamate receptor delta2 subunit. J. Neurosci., 17, 9613–9623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, F. , Barun, S. , Lutz, C.M. , Wang, Y. & Hosford, D.A. (1999) Decreased 45Ca2+ uptake in P/Q‐type calcium channels in homozygous lethargic (Cacnb4lh) mice is associated with increased beta3 and decreased beta4 calcium channel subunit mRNA expression. Brain Res. Mol. Brain Res., 71, 1–10. [DOI] [PubMed] [Google Scholar]
- Llinas, R. & Sugimori, M. (1980) Electrophysiological properties of in vitro Purkinje cell dendrites in mammalian cerebellar slices. J. Physiol., 305, 197–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas, R. , Nicholson, C. , Freeman, J.A. & Hillman, D.E. (1968) Dendritic spikes and their inhibition in alligator Purkinje cells. Science, 160, 1132–1135. [DOI] [PubMed] [Google Scholar]
- Lonchamp, E. , Dupont, J.L. , Doussau, F. , Shin, H.S. , Poulain, B. & Bossu, J.L. (2009) Deletion of Cav2.1(α1A) subunit of Ca2+‐channels impairs synaptic GABA and glutamate release in the mouse cerebellar cortex in cultured slices. Eur. J. Neurosci., 30, 2293–2307. [DOI] [PubMed] [Google Scholar]
- Maejima, T. , Wollenweber, P. , Teusner, L.U. , Noebels, J.L. , Herlitze, S. & Mark, M.D. (2013) Postnatal loss of P/Q‐type channels confined to rhombic‐lip‐derived neurons alters synaptic transmission at the parallel fiber to purkinje cell synapse and replicates genomic Cacna1a mutation phenotype of ataxia and seizures in mice. J. Neurosci., 33, 5162–5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark, M.D. , Maejima, T. , Kuckelsberg, D. , Yoo, J.W. , Hyde, R.A. , Shah, V. , Gutierrez, D. , Moreno, R.L. et al (2011) Delayed postnatal loss of P/Q‐type calcium channels recapitulates the absence epilepsy, dyskinesia, and ataxia phenotypes of genomic Cacna1a mutations. J. Neurosci., 31, 4311–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita, K. , Wakamori, M. , Rhyu, I.J. , Arii, T. , Oda, S. , Mori, Y. & Imoto, K. (2002) Bidirectional alterations in cerebellar synaptic transmission of tottering and rolling Ca2+ channel mutant mice. J. Neurosci., 22, 4388–4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEnery, M.W. , Copeland, T.D. & Vance, C.L. (1998) Altered expression and assembly of N‐type calcium channel alpha1B and beta subunits in epileptic lethargic (lh/lh) mouse. J. Biol. Chem., 273, 21435–21438. [DOI] [PubMed] [Google Scholar]
- Miyazaki, T. , Hashimoto, K. , Shin, H.S. , Kano, M. & Watanabe, M. (2004) P/Q‐type Ca2+ channel alpha1A regulates synaptic competition on developing cerebellar Purkinje cells. J. Neurosci., 24, 1734–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noebels, J.L. (2012) The voltage‐gated calcium channel and absence epilepsy In Noebels J.L., Avoli M., Rogawski M.A., Olsen R.W. & Delgado‐Escueta A.V. (Eds), Jasper's Basic Mechanisms of the Epilepsies. National Center for Biotechnology Information, Bethesda, MD, ISBN‐10: 0199746540; ISBN‐13: 978‐0199746545; pp. 1–16. [PubMed] [Google Scholar]
- Obermair, G.J. , Schlick, B. , Di Biase, V. , Subramanyam, P. , Gebhart, M. , Baumgartner, S. & Flucher, B.E. (2010) Reciprocal interactions regulate targeting of calcium channel beta subunits and membrane expression of alpha1 subunits in cultured hippocampal neurons. J. Biol. Chem., 285, 5776–5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda, S.I. , Lee, K.J. , Arii, T. , Imoto, K. , Hyun, B.H. , Park, I.S. , Kim, H. & Rhyu, I.J. (2011) Differential regulation of Purkinje cell dendritic spines in rolling mouse Nagoya (tg/tg), P/Q type calcium channel (α1A/Cav2.1) mutant. Anat. Cell Biol., 43, 211–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrobon, D. (2002) Calcium channels and channelopathies of the central nervous system. Mol. Neurobiol., 25, 31–50. [DOI] [PubMed] [Google Scholar]
- Pietrobon, D. (2005) Function and dysfunction of synaptic calcium channels: insights from mouse models. Curr. Opin. Neurobiol., 15, 257–265. [DOI] [PubMed] [Google Scholar]
- Rancz, E.A. & Hausser, M. (2006) Dendritic calcium spikes are tunable triggers of cannabinoid release and short‐term synaptic plasticity in cerebellar Purkinje neurons. J. Neurosci., 26, 5428–5437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sausbier, M. , Hu, H. , Arntz, C. , Feil, S. , Kamm, S. , Adelsberger, H. , Sausbier, U. , Sailer, C.A. et al (2004) Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2+‐activated K+ channel deficiency. Proc. Natl. Acad. Sci. USA, 101, 9474–9478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlick, B. , Flucher, B.E. & Obermair, G.J. (2010) Voltage‐activated calcium channel expression profiles in mouse brain and cultured hippocampal neurons. Neuroscience, 167, 786–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuki, K. & Kimura, S. (1997) Dynamic properties of nitric oxide release from parallel fibres in rat cerebellar slices. J. Physiol., 498(Pt 2), 443–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, S.L. & Otis, T.S. (2003) Persistent changes in spontaneous firing of Purkinje neurons triggered by the nitric oxide signaling cascade. J. Neurosci., 23, 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadmouri, A. , Kiyonaka, S. , Barbado, M. , Rousset, M. , Fablet, K. , Sawamura, S. , Bahembera, E. , Pernet‐Gallay, K. et al (2012) Cacnb4 directly couples electrical activity to gene expression, a process defective in juvenile epilepsy. EMBO J., 31, 3730–3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tank, D.W. , Sugimori, M. , Connor, J.A. & Llinas, R.R. (1988) Spatially resolved calcium dynamics of mammalian Purkinje cells in cerebellar slice. Science, 242, 773–777. [DOI] [PubMed] [Google Scholar]
- Walter, J.T. , Alvina, K. , Womack, M.D. , Chevez, C. & Khodakhah, K. (2006) Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat. Neurosci., 9, 389–397. [DOI] [PubMed] [Google Scholar]
- Wittemann, S. , Mark, M.D. , Rettig, J. & Herlitze, S. (2000) Synaptic localization and presynaptic function of calcium channel beta 4‐subunits in cultured hippocampal neurons. J. Biol. Chem., 275, 37807–37814. [DOI] [PubMed] [Google Scholar]
- Xie, M. , Li, X. , Han, J. , Vogt, D.L. , Wittemann, S. , Mark, M.D. & Herlitze, S. (2007) Facilitation versus depression in cultured hippocampal neurons determined by targeting of Ca2+ channel Cavβ4 versus Cavβ2 subunits to synaptic terminals. J. Cell Biol., 178, 489–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwingman, T.A. , Neumann, P.E. , Noebels, J.L. & Herrup, K. (2001) Rocker is a new variant of the voltage‐dependent calcium channel gene Cacna1a. J. Neurosci., 21, 1169–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Substantially reduced size and modestly reduced density of the distal dendritic arbor in lethargic Purkinje neurons.