

Abstract
Tyrosinemia type 1 is an inherited metabolic disorder attributable to deficiency of fumarylacetoacetate hydrolase enzyme. Here we report an eight month-old male Saudi infant who presented with jaundice, fever, and disturbed level of consciousness accompanied by abdominal distension, hepatomegaly and ascites with features suggestive of rickets. The diagnosis of tyrosinemia typ 1was confirmed based on clinical and biochemical findings.
Keywords: Tyrosinemia type I, Inherited metabolic disorder, Child, Saudi Arabia
Introduction
Tyrosinemia type 1 is an autosomal recessive inherited metabolic disorder attributed to deficiency of fumarylacetoacetate hydrolase (FAH), which is a terminal enzyme in the metabolism of tyrosine. The gene for this enzyme has been mapped to the long arm of chromosome 15 [1]. Its prevalence has been reported as 1: 100.000 [2]. While primarily synthesized in the liver, FAH is also synthesized at moderate amounts in kidneys, adrenal glands, lungs, heart, intestines, stomach, pancreas, lymphocytes and skeletal muscles [1]. The patients with tyrosinemia die in the early years of their lives as a result of hepatic insufficiency. The literature reveals a markedly increased risk of hepatocellular carcinoma among the survivors [2].
Case Report
An eight-month-old Saudi male infant presented with abdominal distension, fever, jaundice, black stool and disturbed level of consciousness for 3 days prior to admission to the Pediatric Intensive Care Unit (PICU) at King Fahd Hospital, Al-Baha. He was the first child of first degree cousin parents. His paternal uncle died at the age of 8 month with unknown cause.
Our patient experienced an upper respiratory tract infection a week before admission. He had no other significant medical history or history of travel abroad. Immunizations were up to date. He was born by normal vaginal delivery after uneventful pregnancy. Birth weight was 3.1kg.
On physical examination (Figure 1), the patient looked sick, pale, jaundiced and drowsy. His weight and length were 7.5kg and 69cm at 10th and 25th percentile for his age, respectively. Head circumference was 43.5cm at 10th percentile. He had wide anterior fontanel (3x4cm), and widened wrists. There was mild lower limb edema. Fine crackles were audible bilaterally on the chest. Abdomen was distended with everted umbilicus. Liver was palpable 5cm below right costal margin with 7cm span. Liver was not tender with an irregular firm surface. Spleen was just palpable below left costal margin. There was positive shifting dullness test for ascites, with scrotal edema and normal male genitalia. Central nervous system examination revealed a drowsy infant. Cranial nerves were intact. Fundal examination was normal. There was increased tone and reflexes in the lower limbs.
Figure 1.

The patient with tyrosinemia type 1. Note the abdominal distension and visible veins.
Complete blood count showed Hb level was 8gm/dl, platelets count was 28 x103/mm3, red blood cell (RBC): 3.3x106/mm3, WBC: 4.8x103/mm3 (neutrophils: 43%, lymphocytes: 55%, monocytes: 2%). Erythrocyte sedimentation rate (ESR) was 12mm/hr, C-reactive protein was 0.8ug/dl and urine analysis showed albumin RBCs.
Biochemical examination showed Na: 143mmol/L, K: 3.7mmol/L, chloride: 105mmol/L, urea 12.5mmol/L, creatinine 27 umol/L, glucose: 2.9mmol/L, Ca: 2.2mmol/L, phosphorus: 0.64mmol/L, Mg: 0.57mmol/L, albumin: 28 μmol/L, total bilirubin: 35μmol/L, direct bilirubin: 17μmol/L, alkaline phosphatase 426 U/L, AST: 65 U/L, gamma GT: 130 U/L, LDH: 566 U/L, and ammonia: 121 μmmol/L.
Coagulation screening showed prothrombin time (PT): 26.8 sec (control: 12.5sec), partial thromboplastin time (PTT): 61.1sec (control: 32.5 sec), fibrinogen: 0.5gm/L (control: 3.3) and fibrin degradation products (FDP): 2ug/mL (normal value: 0); peripheral blood film examination showed no abnormal cells. Blood alpha-fetoprotein was very high as 24804 IU/ml (N: 0-11.3), and arterial blood gases examination showed compensated metabolic acidosis. Blood, urine and eye swab culture and sensitivity (C/S) showed no growth. Tracheal aspirate C/S showed methicillin-resistant Staphylococcus aureus (MRSA, sensitive only to vancomycin). The left wrist X-ray showed evidence of rickets. Abdomen ultrasonography revealed hepatosplenomegaly with moderate ascites. Liver examination showed multiple hyperechoic masses (Figure 2). Abdominal computed tomography (CT) showed hepatomegaly with multiple rounded, hyperdense masses of varying size, splenomegaly and ascites (Figure 3A). Brain CT showed marked bifrontal cerebral atrophy (Figure 3B). Electroencephalography (EEG) revealed normal results.
Figure 2.

Abdominal ultrasonography of the patient showing multiple hepatic hyperechoic masses and ascites.
Figure 3.
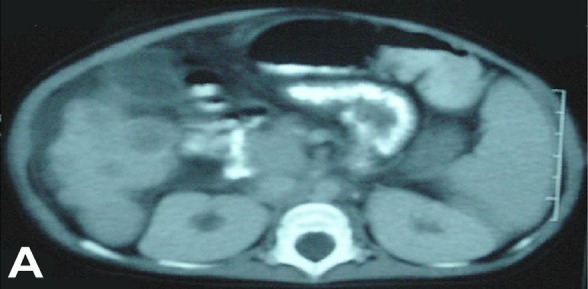

(A) Abdominal CT of the patient revealed multiple hepatic rounded hyperdense masses and splenomegaly. (B) Brain CT showed bifrontal cerebral atrophy.
Blood phenylalanine, tyrosine and methionine levels were 269 μmol/L (N.: 37-129), 897 μmol/L (N: 32-275) and 897 μmol/L (N.: 32-275) respectively. Urine examination for organic acids showed increased levels of succinylacetone, P-hydroxyphenylacetate, and P-hydroxyphenylpyruvate levels. In the light of these findings, the infant was diagnosed to have tyrosinemia type I and was prescribed a phenylalanine and tyrosine restricted diet (special formula milk). Then he was also enrolled in 2-(nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC) treatment program with calcitriol and phosphorus solution for vitamin D resistant rickets. Following the NTBC treatment, there was much improvement regarding neurological status and liver function.
Discussion
Tyrosinemia has three distinctive types. Type I is characterized by progressive liver disease, increased risk of hepatocellular carcinoma, neurological crises and renal tubular dysfunction. It is also characterized by hypophosphatemic rickets. In acute type, hepatic insufficiency develops before six months of age as a result of micro and macronodular cirrhosis. In subacute type however, hepatomegaly, irregular bleeding and rickets are observed after six months. Chronic type manifests itself with hepatomegaly, rickets and growth retardation after one year of age [3]. Tyrosinema type II, which is also known as oculocutaneous tyrosinemia, develops as a result of the deficiency of hepatic tyrosine amino transferase. Clinical findings include mental and motor retardation, corneal ulcerations and hyper keratotic lesions of the digits, palms and soles [4]. In tyrosinemia type III, there is lack of 4- hydroxyphenyl- pyruvate dioxygenase enzyme. All the patients suffer from growth retardation, convulsions, and ataxia. The most distinguishing characteristic of type I tyrosinemia is liver and kidney involvement [4], as seen in our patient.
The increased levels of serum tyrosine and methionine and urine succinyl acetone provided the key for diagnosis. Conventional treatment involves phenylalanine and tyrosine limited diet prescription. Liver transplantation has proven effective in many of the patients. Another treatment modality is the use of (NTBC), which are strong inhibitors of 4-hydroxyphenylpyruvate dehydrogenase involved in the second step of tyrosine metabolism [3,4]. Our patient, whose symptoms started at eight months of age, was reported to have no complaints previously. He presented with bleeding, and hepatomegaly, clinical, laboratory and radiological evidence of rickets, and neurological involvement. Laboratory studies revealed prolonged PT, PTT, low level of fibrinogen and high FDP level which indicate acute liver injury. Through the findings of clinical and laboratory examination, the diagnosis was established as sepsis, disseminated intravascular coagulation (DIC) and rickets. The patient was also further investigated for metabolic diseases. He was put through a 14 - day course of vancomycin due to 100.000 MRSA colonies growth in the tracheal aspirate culture. Fibriogen and FDP levels improved after antibiotic treatment but prolongation of PT and PTT continued. However, the prolonged PT and PTT were also incompatible with the liver function tests and unresponsive to three consecutive days of intramuscular injection of vitamin K and fresh frozen plasma transfusions.
In a study conducted on 32 tyrosinemia type I patients, nephromegaly (47%), hyperechogenicity of kidneys (47%) and nephrocalcinosis (16%), aminoaciduria (82%), hypercalciuria (67%), tubular acidosis (59%), decreased glomerular filtration rate (48%) were found [5]. Our patient had most of these abnormalities including decreased tubular phosphorus reabsorption and aminoaciduria. Another study, conducted on 8 patients, reports nephromegaly, tubulopathy and vitamin D resistant rickets in 50%, 80% and 50% of the patients respectively [6].
Succinylacetone, the actual toxic substance in tyrosinemia type I, is responsible for liver and kidney pathologies. This toxic substance accumulation is prevented by NTBC treatment. According to the literature, risk for hepatocarcinoma risk has been markedly prevented following this treatment [7,8]. Furthermore, the results of a study conducted on 101 patients have shown no hepatocellular carcinoma development for two years [9]. There may also be a 10-15 fold increase in the serum AFP level. An abrupt increase in serum AFP is a red flag for detection of hepatocellular carcinoma [3,5]. Our patient had very high AFP level.
The existence of multiple nodular lesions in the liver in the abdominal CT and the markedly high value of AFP level were suggestive of hepatoma. We also considered other diagnoses such as tyrosinemia, embryonic carcinoma, malignant teratoma and dissiminating malignancy. While it is not possible to conduct a liver biopsy to assist the diagnosis due to the abnormal coagulation tests, the working diagnosis was tyrosinemia type 1 in view of the high serum level of tyrosine, and the increase in urinary excretion of succinylacetone [9]. The definitive confirmatory test for tyrosinemia is demonstration of low enzyme in fresh liver biopsy or by identification of a disease causing mutation on DNA study. Due to logestic issues we were unable to study DNA in our patient.
References
- 1.Bergeron A, D’Astous M, Timm DE, Tanguay MR. Structural and Functional Analysis of Missense Mutations in Fumarylacetoacetate Hydrolase, the Gene Deficient in Hereditary Tyrosinemia type 1. The journal of Biological chemistry 2000; 276 (18): 1525–1531. [DOI] [PubMed] [Google Scholar]
- 2.Croffie JM, Gupta SK, Chong SKF, Fitzgerald JF. Tyrosinemia Type 1 Should Be Suspected in Infants With Severe Coagulapathy Even in the Absence of other signs of Liver failure. Pediatrics 1999; 103 (3): 675–678. [DOI] [PubMed] [Google Scholar]
- 3.Grompe M. The pathophysiology and Treatment of Hereditary Tyrosinemia Type 1. Seminars in Liver Disease 2001; 21 (4): 563–571. [DOI] [PubMed] [Google Scholar]
- 4.Van Spronsen FT, Thomasse Y, Smit GP, Leonard JV, Clayton PT, Fidler V et al. Hereditary tyrosinemia type 1: A New clinical classification with difference in prognosis on dietary treatment. Hepatology 1994; 20 (5): 1187–1190. [PubMed] [Google Scholar]
- 5.Forget S; Patriquin BH; Dulrois J, Lafortune M, Merouani A, Paradis K et al. The kidney in children with tyrosinemia: sonographic, CT and biochemical findings. Pediatr Radiol 1999; 29: 104–108. [DOI] [PubMed] [Google Scholar]
- 6.Laine J, Salo MK, Krogerus L Karkainen J, Walhroos O, Holberg C. Nephropathy of Tyrosinemia and its longterm outlook. J Pediatr Gastroenterology and Nutrition 1997; (24): 113–114. [Google Scholar]
- 7.Grant A Mitchell and Maria A Lambert. Hereditary Tyrosinemia: An over-view, www.meadjohnson.com/metabolics/hereditarytyrosinemia.html.
- 8.Holme E and Lindstedt S. Tyrosinemia type 1 and NTBC (2-nitro-4-trifluoromethylbenzoyl-1, 3-cyclohexanedione) J Inhert Metab Dis 1998; 21 (5): 507–17. [DOI] [PubMed] [Google Scholar]
- 9.Ronald scott C. and Seattle WA. Tyrosinemia: Background; www.meadjolmson.com/metabolics/tyrosinemia.