

Abstract
Walker-Warburg syndrome (WWS) is a rare and lethal autosomal recessive disorder, caused by defective glycosylation of α-dystroglycan that is important for muscle integrity and neuronal migration. Mutations in six genes involved in the glycosylation of α-dystroglycan (POMT1, POMT2, POMGNT1, FCMD, FKRP and LARGE) have been identified in WWS patients, and others remain under study. Prenatal diagnosis may be possible by means of prenatal ultrasonography, or magnetic resonance imaging.
We report a patient demonstrating the typical clinical features of lissencephaly, congenital muscular dystrophy and ocular abnormalities, in addition to other features including hydrocephalus, occipital encephalocele, agenesis of the corpus collosum, microphthalmia, ventricular septal defect, and rocker bottom feet deformity.
Keywords: Walker-Warburg syndrome, Lissencephaly, Congenital muscular dystrophy, Glycosylation of α-dystroglycan
Introduction
Walker-Warburg syndrome (WWS) is a rare form of congenital muscular dystrophy (CMD) associated with brain and eye abnormalities [1, 2]. It has a worldwide distribution, the overall incidence is unknown but a survey in North- eastern Italy has reported an incidence rate of 1.2 per 100,000 live births [1–3]. WWS is the most severe form of CMD, with most of the children dying before the age of three.
Diagnostic features of the syndrome include lissencephaly type II, cerebellar malformation, retinal malformation, and CMD [1–6]. Other uncommon features have been identified, although they are not necessary to establish a diagnosis of WWS.
WWS is caused by defective glycosylation of a α-dystroglycan that is important for muscles integrity and neuronal migration [7–11]. Mutations in six genes involved in the glycosylation of α-dystroglycan (POMT1, POMT2, POMGNT1, FCMD, FKRP and LARGE) have been identified in WWS patients, others remain under study [4, 7, 9, 11, 12]. Laboratory investigations usually show elevated levels of creatinine kinase (CK), myopathic/dystrophic muscle pathology, and altered α -dystroglycan. WWS is an autosomal recessive disorder. In families with one affected child, the risk of having another child with the disease is 25% [7, 9].
Management is mainly supportive and preventive, and the condition is usually lethal within the first few months of life. Survival beyond three years is unusual. Prenatal diagnosis may be useful; it allows appropriate counseling and optimization of obstetrical management. Pre-implantation genetic diagnosis may also be available for families in which the diseases-causing mutation has been identified in an affected baby.
We report this case demonstrating the classical criteria of WWS in addition to other multiple uncommon features.
Case Report
This newborn boy is an outcome of a term pregnancy that ended by an emergency caesarean section, due to cord presentation. The baby cried after resuscitation. Apgar score was 4 at1, 8 at 5, and 9 at 10 minutes. Pregnancy passed uneventfully, mother received folic acid for 2 months preconception, and was on regular antenatal care. A routine antenatal ultrasonographic examination at 20 weeks showed no abnormality. He is the second and only living child to first-degree Palestinian couple, his elder sister born at term by an elective cesarean section due to intrauterine growth retardation (IUGR) and breech presentation. Her birth weight was 1.8Kg (below the 3rd percentile), and she was noted to have multiple congenital anomalies in the form of hypotonia, multiple atrioventricular septal defects (AVSDs), multiple bilateral renal stones, unilateral micropthamia and blindness. A CT scan done at that time was reported to have shown evidence of brain atrophy.
Pulmonary artery banding was performed by the age of 3 months, but despite that her condition did not improve much, as she still experienced recurrent attacks of cyanosis and shortness of breath. She died suddenly at home one month later. At that time no specific diagnosis of her condition was reached.
Clinical examination of our index patient at birth revealed a floppy dysmorphic baby. Weight was 3.0 Kg (at 10th percentile) for age, and head circumference 31cm (below the 3rd percentile). He was hypotonic with absent deep tendon reflexes, poor muscle tone, and sluggish primitive reflexes. He also showed the following dysmorphic features (Figure 1): an occipital encephalocele, rocker bottom feet deformity, bilateral micropthalmia, corneal opacity and scaring, and had a pan systolic murmur at the left lower sternal border.
Figure 1.
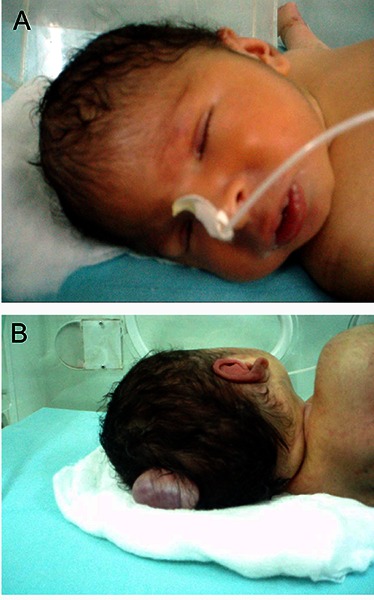
Clinical photograph showing (A) the small head and the nasogastric tube set for feeding due to poor sucking. Right-sided microphthalmia is more evident. (B) Occipital encephalocele.
The baby was admitted to the Neonatal Intensive Care Unit, where he was investigated. Routine biochemical investigations were normal, chest X- ray showed cardiomegaly, an echocardiography done at one week of age revealed a patent foramen ovale and a small membranous ventricular septal defect of 4 mm in diameter. Abdominal and pelvic ultrasound were normal. Serum CK level was high at 404 U/L, when tested at three weeks of age, and raised later to reach 3532U/L after two months. Karyotyping was normal, more detailed genetic studies were not available.
Cranial computed tomography (CT), taken by the age of 10 days (Figure 2), showed irregular gray – white matter junction with simplified gyral pattern, prominent and widely spaced lateral ventricles, dilated third and forth ventricles, agenesis of the corpus callosum, and an occipital encephalocele.
Figure 2.
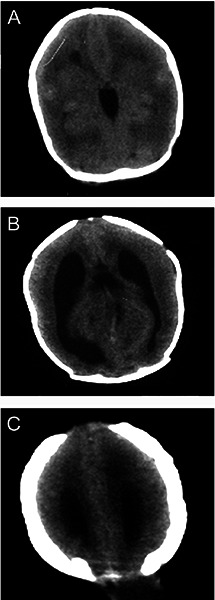
Cranial computed tomography (CT) revealed (A) smooth brain and irrgular gray-white matter junction, (B) ventriculomegaly and absent corpus callosum, and (C) occipital encephalocele.
A ventriculo-peritonial shunt was set due to a rapid increase in the head circumference (31cm-36cm) in two weeks. Repair of the encephalocele was done at the same operation. At 4 weeks of age, he started physiotherapy sessions, and was discharged home 2 weeks later.
At the age of 9 months, he developed generalized epilepsy controlled by sodium valporate. The patient died at the age of 15 months after having seizures complicated by sever aspiration pneumonia.
Discussion
More than 25 neuronal migration disorders resulting in death or improper positioning of the cortical neurons have been described in humans. In cobblestone neocortex, the post-mitotic neurons do not respond to their stop signals, crossing through the neocortex, bypass the glia limtans, and invade the subarachnoid space. The resulting cortex is chaotically structured, consisting of an irregular lissencephalic surface and absence of lamination [1, 5].
Traditionally, cases of lissencephaly were divided into two groups [1–7]:
Type I, or classical lissencephaly, where the normal six –layer cortex seen at histological analysis is replaced by abnormally thick, remodeled, four –layer cortex.
Type II lissencephaly is characterized by a disorganized unlayered cortex, and while in type I, lissencephaly many neurons fail to reach the cortical plate, in type II lissencephaly many neurons move too far into the subpial space [2]. Walker-Warburg syndrome (WWS), muscle –eye-brain (MEB) disease, and Fukuyama congenital muscular dystrophy (FCMD) are the three major entities of this group. Patients are classified into these three entities based on the severity of the phenotype and the presence of syndrome- specific symptoms. WWS being the most severe of the group, especially with regard to the brain phenotype [1, 3].
The definition of CMD as a clinical entity began in 1908 with Howard’s landmark description of a neonate with muscular dystrophic changes [1, 3]. In 1960, WWS was the eponym suggested by Dobyns et al [1] after Walker’s description in 1942 of a child with hydrocephalus, lissencephaly and eye malformation, and Warburg’s report in 1978 of cases with retinal detachment associated with hydrocephalus.
In WWS patients hypoglycosylation resulted from mutations in either the protein O- mannosyltransferase 1 (POMT 1), or one of POMGNT1, and FCMD genes. Glycosylaton of α –dystroglycan, which plays a key role in bridging the cytoskeleton of muscle and CNS cells with extra cellular matrix proteins, is important for muscle integrity and neuronal migration [7, 9, 10]. In 2005, Reeuwijk et al [11] identified POMT2 as the fourth causative gene in WWS. In 2007, LARGE was added as a fifth causative gene in a Saudi family with WWS [12]. These genes account for about only one third of the WWS cases while other several genes remain to be discovered [7].
In reviewing 63 cases of WWS, Dobyns et al [1]established the following diagnostic criteria:
type II lissencephaly, cerebellar malformation, retinal malformation, and CMD. In addition, there are two frequent abnormalities, ventricular dilatation, with or without hydrocephalus, and anterior chamber malformation (Peter’s anomaly) which are helpful, although not necessary to establish a diagnosis [1, 5, 9].
Congenital macrocephaly with hydrocephalus is more common than congenital microcephaly. Dandy -Walker malformation is sometimes associated with posterior cephalocele. Additional abnormalities includes slit like ventricles, microphthalmia, ocular colobomas, congenital cataract and genital anomalies in males [4, 6, 8].
Laboratory investigations usually show elevated CK, myopathic/dystrophic muscle pathology and altered α –dystroglycan [5]. In patients with CMD, CK may reach thousands early in life, reflecting wide spread muscle necrosis in the prenatal period. Later on, these patients have low or normal CK because of lack of muscle mass and limited mobility [5].
Although molecular genetic study was not possible in our case, the diagnosis was confirmed in the index case by the typical clinical picture and the CT findings in association with clinical and biochemical evidence of CMD [8]. Moreover, in view of the clinical similarities between the two siblings plus a definite diagnosis of WWS in the index case, we believe it is reasonable to assume that the second sibling was suffering from the same disease.
Management of WWS is only supportive and preventive; if seizures develop, they usually need to be treated with anticonvulsants. A few children may require a neurosurgical procedure such as shunting of hydrocephalus or encephalocele operation, as in the case of our patient. Physical therapy was reported by some to prevent worsening of contractures although its efficacy has not been established. Feeding needs to be monitored and supplemental nasogastric or gastric tube feeding provided as in our case.
WWS is a lethal disease with most of affected children dying before the age of three [1, 2, 3].
Ultrasounographic findings of ventriculomegaly, posterior fossa abnormalities, encephalocele and ocular abnormalities can be suggestive of WWS prenataly. However, a diagnosis of delayed cortical development should not be considered before 20 weeks of gestation, because in the early second trimester the normal brain is still smooth [5]. In addition, the degree of cerebral involvement in lissencephaly varies across a wide spectrum, and only the severe forms, such as complete agyri are likely to be detectable at prenatal ultrasonography, explaining in part the normal prenatal ultrasound of our patient at 20 weeks gestation [8]. While magnetic resonance imaging (MRI) might be very useful in detecting and confirming abnormal cortical development, and less severe forms of lissencephaly, it is not usually performed unless ultrasound findings are abnormal, and even if MRI was performed, mild forms of lissencephaly might not be detected if the examination is performed too early in the pregnancy [8].
Prenatal molecular diagnosis might be possible only when a mutation has been identified previously [2, 11], and pre-implantation genetic diagnosis may also be available for families in which the diseases-causing mutation has been identified in an affected baby.
References
- Dobyns WB, Pagon RA, Armstrong D, Curry CJ, Greenberg F, Grix A, et al. Diagnostic criteria for Walker –Warburg syndrome. Am J Med Genet.x 1989; 32(2): 195–210. [DOI] [PubMed] [Google Scholar]
- Vajsar J, Schachter H. Review Walker-Warburg syndrome. Orphanet Journal of Rare Diseases. 2006; 1:29 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews KD. Muscular dystrophy overview and diagnosis. Neurol Clin 2003; 21: 795–816. [DOI] [PubMed] [Google Scholar]
- Salih M A M. Muscular Dystrophies and Myopathies in Arab Populations In: Teebi AS, ed Genetic Disorders among Arab Populations (2nd ed). Berlin Heidelberg: Springer-Verlag, 2010: 158–179. [Google Scholar]
- Kanoff RJ, Curless RG, Petito C, Falcone S, Siatkowski RM, Pegoraro E. Walker Warburg syndrome: neurological features and muscle membrane structure. Pediatr Neurol. 1998; 18(1): 76–80. [DOI] [PubMed] [Google Scholar]
- Adachi Y, Poduri A, Kawaguch A, Yoon G, Salih MA, Yamashita F, et al. Congenital microcephaly with a simplified gyral pattern: iassociated findings and their significance. AJNR Am J Neuroradio 2011. . [DOI] [PMC free article] [PubMed]
- Beltrán-Valero de Bernabé D, van Bokhoven H, van Beusekom E, Van den Akker W, Kant S, Dobyns WB, et al. A homozygous nonsense mutation in the Fukutin gene causes a Walker – Warburg syndrome phenotype. Journal of Medical Genetics 2003; 40: 845–848. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandeep Ghai, Fong Katherine W., Ants Toi, David Chitayat, Sophia Pantazi, Susan Blaser. Prenatal US and MR Imaging findings of lissencephaly: Review of fetal cerebral sulcal development. Radio Graphics 2006; 26: 389–405. [DOI] [PubMed] [Google Scholar]
- Cormand B, Pihko H, Bayes M, Valanne L, Santavuori P., B. Talim, R, et al. Clinical and genetic distinction between Walker Warburg syndrome and muscle –eye- brain disease. Nurology 2001; 56: 1059–1069. [DOI] [PubMed] [Google Scholar]
- Marcio M, Vasconcelos Cassia, Guedes R., Marcio Sotero, Monica M. Viera. Walker Warburg syndrome report of two cases. Arquivos de Neuro-Psiquiatria 1999; 57n 3A. [DOI] [PubMed] [Google Scholar]
- van Reeuwijk J, Janssen M, van den Elzen C, Beltran-Valero de Bernabe´ D, Sabatelli P, Merlini L, et al. POMT2 mutations cause α –dystroglycan hypoglycosylation and Walker- Warburg syndrome. J Med Genet 2005; 42(12): 907–912. Epub 2005. May 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Reeuwijk J, Grewal PK, Salih MA, de Bernabe D B-V, Mc Laughlan JM, Michielse CB, et al. Intragenic deletion in LARGE gene causes Walker-Warburg Syndrome. Human Genetics 2007; 121: 685–690. [DOI] [PMC free article] [PubMed] [Google Scholar]