

Summary
A key component to the success of M ycobacterium tuberculosis as a pathogen is the ability to sense and adapt metabolically to the diverse range of conditions encountered in vivo, such as oxygen tension, environmental pH and nutrient availability. Although nitrogen is an essential nutrient for every organism, little is known about the genes and pathways responsible for nitrogen assimilation in M . tuberculosis. In this study we have used transcriptomics and chromatin immunoprecipitation and high‐throughput sequencing to address this. In response to nitrogen starvation, a total of 185 genes were significantly differentially expressed (96 up‐regulated and 89 down regulated; 5% genome) highlighting several significant areas of metabolic change during nitrogen limitation such as nitrate/nitrite metabolism, aspartate metabolism and changes in cell wall biosynthesis. We identify GlnR as a regulator involved in the nitrogen response, controlling the expression of at least 33 genes in response to nitrogen limitation. We identify a consensus GlnR binding site and relate its location to known transcriptional start sites. We also show that the GlnR response regulator plays a very different role in M . tuberculosis to that in non‐pathogenic mycobacteria, controlling genes involved in nitric oxide detoxification and intracellular survival instead of genes involved in nitrogen scavenging.
Introduction
Nitrogen is a key component of all biological macromolecules, found in information‐encoding polymers such as DNA and RNA, and structural molecules such as proteins and components of the cell envelope. As such nitrogen assimilation is essential for life, and organisms such as bacteria have evolved a variety of mechanisms to obtain nitrogen from their surroundings and subsequently assimilate it into cellular macromolecules. Historically, the bulk of work published on nitrogen metabolism in bacteria has concentrated on model organisms such as Escherichia coli (see Reitzer, 2003; Leigh and Dodsworth, 2007 for reviews), but recently there have also been a number of reports on nitrogen metabolism in Actinomycetes, including the saprophyte Mycobacterium smegmatis (Amon et al., 2008; Khan et al., 2008; Harper et al., 2010; Behrends et al., 2012; Jenkins et al., 2012; 2013; Williams et al., 2013), and the obligate human pathogen Mycobacterium tuberculosis (Read et al., 2007; Carroll et al., 2008; Malm et al., 2009; Tan et al., 2010; Akhtar et al., 2013; Gouzy et al., 2013; Williams et al., 2013). However, there has been no work published on the global M. tuberculosis nitrogen stress response and its regulation, a gap that is addressed here.
Ammonium is the preferred nitrogen source for many bacteria, but there is recent evidence that asparagine (converted to ammonium for assimilation) may be the preferred nitrogen source in M. tuberculosis (Gouzy et al., 2014). M. tuberculosis can also use alternative nitrogen sources for growth such as urea (Lin et al., 2012) and aspartate (Gouzy et al., 2013). Once inside the cell (either directly or indirectly), ammonium is assimilated into L‐glutamate and L‐glutamine, the two major biosynthetic nitrogen donors. This is either via the low ammonium affinity glutamate dehydrogenase (GDH) enzyme, when nitrogen is plentiful, or by the energy‐requiring, high‐affinity glutamine synthetase/glutamate synthase (glutamine:2‐oxoglutarate aminotransferase; GS/GOGAT) enzymes when nitrogen is scarce (for reviews see Harper et al., 2008; Amon et al., 2010). M. tuberculosis only appears to encode the high affinity GS/GOGAT system, unlike M. smegmatis, which encodes both (Amon et al., 2009). Key nitrogen control enzymes undergo post‐translational modifications in response to nitrogen limitation. The GlnK (PII) signalling protein is adenylylated on a conserved tyrosine residue by GlnD, in response to nitrogen limitation (Williams et al., 2013). This modification is associated with the dissociation of the PII protein from the AmtB porin channel (Javelle and Merrick, 2005; Radchenko et al., 2010), permitting increased ammonium influx (Gruswitz et al., 2007). Glutamine synthetase undergoes a post‐translational de‐adenylylation by GlnE during nitrogen limitation, making it fully active (Carroll et al., 2008) and ensuring maximal glutamine and glutamate synthesis during nitrogen austerity. However, many gaps remain in our knowledge of nitrogen metabolism and regulation in mycobacteria, for example, the signal indicating nitrogen cellular status is unknown. We have shown that in M. smegmatis, the intracellular ratio of 2‐oxoglutarate : glutamine increases during nitrogen limitation and then decreases when nitrogen is available, indicating that this may be the intracellular signal (Behrends et al., 2012). However, how this signal is detected and then manifested in transcriptional and post‐translational responses remains unclear. What, if any, role PII proteins play in the control of the nitrogen response in mycobacteria control is also unknown. In E. coli, PII‐UMP controls the response‐regulator NtrC (Pioszak et al., 2000), but PII‐AMP in mycobacteria does not mediate the transcriptional response to nitrogen limitation (Williams et al., 2013).
The transcriptional response to nitrogen limitation in enteric bacteria is mediated by the two‐component system NtrBC, which activates expression of over 100 genes (Zimmer et al., 2000; Reitzer, 2003), but this system is missing from the Actinomycetes. The equivalent function in Corynebacterium glutamicum is performed by the TetR‐type response‐regulator AmtR, which controls transcription of over 33 genes (Beckers et al., 2005; Burkovski, 2007), while in Streptomyces, the OmpR‐type response‐regulator GlnR controls nitrogen metabolism (Fink et al., 2002), regulating at least 50 nitrogen response genes in Streptomyces coelicolor and at least 44 genes in Streptomyces venezuelae (Tiffert et al., 2008; 2011; Pullan et al., 2011). M. tuberculosis contains a GlnR homolog (Rv0818) with 61% identity to that from S. coelicolor (Tiffert et al., 2011), but the evidence for an AmtR homologue is weak, with Rv3160c only 28% identical to AmtR from C. glutamicum (Harper et al., 2008), and to date, no role has been reported for AmtR in mycobacteria. We have shown that GlnR is the main nitrogen response regulator in M. smegmatis and described the complete GlnR nitrogen‐response regulon. We demonstrated that GlnR regulates the expression of more than 100 genes during nitrogen limitation (Jenkins et al., 2013), many of which are involved in nitrogen metabolism and nitrogen scavenging, and used the Active Modules algorithm, AMBIENT (Bryant et al., 2013), to identify key metabolic reactions and pathways altered in response to nitrogen stress (Williams et al., 2013). We also demonstrated that the aspartate D48 residue is essential for the GlnR‐mediated transcriptional response to nitrogen limitation in M. smegmatis (Jenkins et al., 2012); this residue was recently shown to be critical for stabilization of the GlnR homodimers required for its function (Lin et al., 2014).
The aim of this study was to extend our global nitrogen stress analyses to the obligate human pathogen M. tuberculosis, which has to deal with a more restricted set of environmental variables in terms of nitrogen sources, and to delineate the GlnR regulon. We combined genome‐wide expression profiling, comparing a glnR mutant to the wild‐type strain during nitrogen limited growth, global analysis of GlnR–DNA interactions by chromatin immunoprecipitation and high‐throughput sequencing (ChIP‐seq), and transcriptomics over nitrogen run‐out. We identified key changes in the metabolic network in response to nitrogen limitation, showing that the main nitrogen metabolism‐related response is the production of ammonium both through the reduction of nitrite and from aspartate; however, M. tuberculosis does not appear to scavenge ammonium or other nitrogen sources (e.g. urea) from the environment. We found changes in general metabolism such as an increased methylcitrate–isocitrate lysase cycle and changes in the cell wall phthiocerol dimycoceroserate (PDIM) and peptidoglycan make up. We show that GlnR is a global regulator in mycobacteria, controlling the expression of at least 67 genes in response to nitrogen stress, although intriguingly only the minority of GlnR controlled genes are directly involved in nitrogen metabolism. We identified 35 GlnR‐binding sites, 22 of which controlled the differential gene expression of at least 19 genes in nitrogen limitation. This includes divergently transcribed genes, and both up‐ and down‐regulated genes, showing GlnR functions as an activator and repressor of transcription. Using the 25 GlnR binding sites found in intergenic regions, we identified a consensus DNA‐binding motif. Overall, this work re‐illustrates the complexity of metabolism in mycobacteria and how M. tuberculosis has adapted its nitrogen metabolism for life inside a vertebrate host.
Results
Defining nitrogen stress conditions
Mycobacterium tuberculosis was grown in nitrogen‐free Sauton's medium containing either 1 mM ammonium chloride (nitrogen limiting) or 30 mM ammonium chloride (nitrogen excess) (Fig. 1A). External ammonium levels were monitored and shown to be completely depleted in the nitrogen limiting medium by day 8 (Fig. 1C). To ensure that a transcriptional response was induced in our nitrogen limiting conditions, transcript levels of a gene previously shown to be highly induced in nitrogen limitation in M. smegmatis (nirB) (Jenkins et al., 2012) was monitored. The expression level of nirB was induced in the nitrogen limiting medium at day 8 relative to sigA, correlating with the point of external ammonium depletion (Fig. 1B). Gene expression was unchanged in the nitrogen excess medium over these time points (data not shown).
Figure 1.
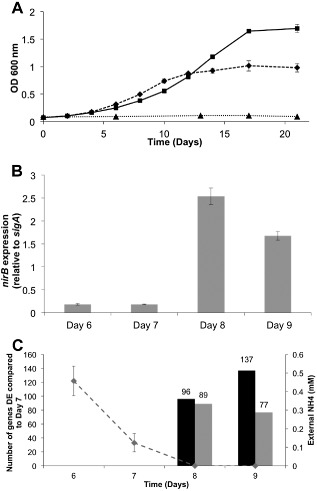
Effect of nitrogen limitation on M . tuberculosis growth and gene expression.
A. M . tuberculosis was grown in nitrogen‐free Sauton's medium (filled triangles), or containing 1 mM ammonium chloride (nitrogen limiting, filled diamonds), or 30 mM ammonium chloride (nitrogen excess, filled squares).
B. The expression of nir B was measured by qRT‐PCR using RNA from three independent cultures, with sig A as internal control. Fold change was calculated as a ratio of the arbitrary expression units, standardised to sig A. Ct values for sig A did not change significantly over nitrogen run‐out.
C. The number of genes showing greater than twofold change in differential expression (DE) over nitrogen run‐out at days 8 and 9 compared with day 7. Black bars show an increase in DE; grey bars a decrease in DE. The concentration of external ammonium concentration (mM) in the growth medium as measured by AquaQuant analysis is also shown (dashed line).
Expression profiling of the nitrogen stress response
RNA samples (three biological replicates, 1 mM ammonium chloride) were taken at three time points (days 7, 8 and 9) and applied to an M. tuberculosis tiling microarray. Fully annotated data have been deposited in BμG@Sbase and ArrayExpress, and can be viewed in File S1. The data were analysed as described to determine which genes were significantly differentially expressed (DE) during nitrogen limitation (days 8 and 9) compared with nitrogen replete (day 7). Genes were considered to be significantly differentially expressed if their expression changed greater than twofold compared with their expression at day 7 with a false discovery rate (FDR) corrected P‐value of < 0.1. A complete list of significantly DE genes identified by these criteria can be viewed in File S2. At nitrogen run‐out, day 8, 185 genes were DE (96 up and 89 down) and at day 9, 214 genes were DE (137 up and 77 down) greater than twofold (Fig. 1C). In total, expression of approximately 5% of the genome was altered significantly upon nitrogen stress.
Metabolic analysis of the nitrogen stress response
To obtain a more holistic view of how changes in gene expression might reflect changes in metabolism, we used AMBIENT (Bryant et al., 2013) and a genome scale metabolic model (Jamshidi and Palsson, 2007) to identify areas of metabolism that were affected in nitrogen limitation, a method we have previously applied successfully to M. smegmatis (Williams et al., 2013). The modules identified ultimately depend on the quality of the genome scale metabolic model, and they do not always fit neatly into traditional pathways. We discovered a large interconnected network of reactions linking 102 enzymes, with both increased (37) and decreased (65) activity (Fig. 2). The genes annotated with each of these metabolic reactions have been overlaid on the same diagram (Fig. S1) to indicate their position in the network. The reactions were grouped into 19 modules according to the AMBIENT analysis (Table 1). Nitrate/nitrite metabolism and nitric oxide detoxification are the major responses to nitrogen stress, and are regulated by GlnR (see discussion later). These potentially produce ammonium for growth as well protecting the cell from toxic effects of NO. A wide range of other biosynthetic activities also respond to nitrogen stress, but most are not obviously linked directly to nitrogen metabolism (Table 1). The methylcitrate/isocitrate lyase in the glyoxylate shunt are up‐regulated, and PDIM and peptidoglycan biosynthesis are increased, indicating changes in energy metabolism and cell envelope biosynthesis, whereas phosphatidyl glycerol shows decreased activity, with links in the network suggesting redistribution of metabolic activity. The M. tuberculosis network identified is more interconnected than that seen in M. smegmatis (Jenkins et al., 2013), but smaller in size, with a number of notable differences. There is no large decrease in modules associated with growth, such as DNA, RNA and protein synthesis, perhaps suggesting M. tuberculosis can maintain its growth rate in the range of habitats encountered in vivo.
Figure 2.
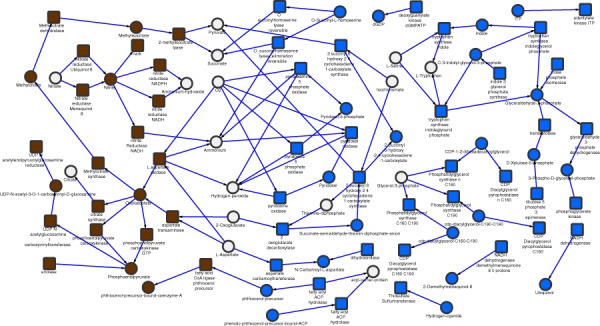
Ambient modules are presented in their metabolic network context, illustrating a redirection of metabolism. Squares represent enzymatic reactions and circles represent metabolites. Grey (blue online) represents negatively regulated, and dark (brown online) positively regulated reactions. No definitive claims can be made about fluxes, so arrows are only indicative and taken from the identity or context of the relevant reaction. For reference, reactions with gene numbers are given in Fig. S1.
Table 1.
AMBIENT modules differentially regulated in response to nitrogen stress
| Module | q‐value | No. of reactions | No. of metabolites | Specific function(s) | Genes |
|---|---|---|---|---|---|
| Up | |||||
| u1 | < 0.001 | 6 | 1 | Nitrite/nitrate reductase activity | Rv0252, Rv0253, Rv0261, Rv0267, Rv1161, Rv1162, Rv1163, Rv1164, Rv1737, Rv2329, Rv2391 |
| u2 | < 0.001 | 11 | 5 | Citrate/oxaloacetate metabolism | Rv0211, Rv0337, Rv0467, Rv0482, Rv0889, Rv0896, Rv1023, Rv1130, Rv1131, Rv1315, Rv1475, Rv1595, Rv1915, Rv3565 |
| u3 | 0.01 | 1 | 1 | Phthiocerol precursor ligase | Rv2930 |
| Down | |||||
| d1 | < 0.001 | 9 | 5 | Rv1408, Rv1436, Rv1437, Rv1438, Rv1449, Rv1611, Rv1612, Rv1613 | |
| d2 | 0.037 | 2 | 1 | Phosphatidylglycerol synthase | Rv1822, Rv2289, Rv2746 |
| d3 | 0.037 | 2 | 1 | Pyridoxine 5 phosphate oxidase | Rv2607 |
| d4 | 0.037 | 2 | 1 | Pyridoxal oxidase | Rv2607 |
| d5 | 0.037 | 2 | 1 | Phosphatidylglycerol synthase | Rv1822, Rv2289, Rv2746 |
| d6 | 0.037 | 2 | 1 | Phosphatidylglycerol synthase | Rv1822, Rv2289, Rv2746 |
| d7 | 0.037 | 2 | 1 | Aspartate carbamoyltransferase | Rv1380, Rv1381 |
| d8 | 0.037 | 2 | 1 | O‐succinylhomoserine lyase | Rv0391, Rv1079 |
| d9 | 0.039 | 1 | 1 | Fatty acyl ACP hydrolase | Rv2928 |
| d10 | 0.041 | 1 | 1 | NADH dehydrogenase demethylmenaquinone 8 | Rv0082, Rv3145, Rv3146, Rv3147, Rv3148, Rv3149, Rv3150, Rv3152, Rv3153, Rv3154, Rv3155, Rv3156, Rv3157, Rv3158 |
| d11 | 0.041 | 1 | 1 | NADH dehydrogenase (ubiquinol) | Rv0082, Rv3145, Rv3146, Rv3147, Rv3148, Rv3149, Rv3150, Rv3152, Rv3153, Rv3154, Rv3155, Rv3156, Rv3157, Rv3158 |
| d12 | 0.041 | 1 | 1 | Thiosulfate sulfurtransferase | Rv2291 |
| d13 | 0.041 | 1 | 1 | Fatty acyl ACP hydrolase | Rv2928 |
| d14 | 0.045 | 1 | 1 | Adenylate kinase ITP | Rv0733 |
| d15 | 0.045 | 3 | 2 | 2‐Oxoglutarate decarboxylase | Rv0555 |
| d16 | 0.045 | 1 | 1 | Deoxyguanylate kinase dGMPATP | Rv1389 |
Up‐ and down‐regulated modules found using AMBIENT in nitrogen stress, indicating the number of reactions and metabolites present in each. Scores are calculated by AMBIENT, taking into account the logarithm of the fold change of the genes associated with each reaction and the connectivity in the metabolic network of each metabolite in the module. The number of genes associated with these reactions according to the M. tuberculosis model (Jamshidi and Palsson, 2007) is also shown. Functional assignments are based on manual inspection of the member reactions of the modules.
Determination of GlnR controlled genes in nitrogen stress
In order to determine the genes regulated by GlnR, we constructed a GlnR null mutant. Substitution of the glnR gene by the hygromycin cassette was confirmed by polymerase chain reaction (PCR) (Fig. S2) and western analysis using a GlnR specific antibody (Fig. S2). The M. tuberculosis glnR KO mutant did not grow with nitrate as the sole nitrogen source, or show any phenotype under nitrogen limitation, and did not up‐regulate nirB expression upon nitrogen limitation (data not shown). There was also no difference in the response of the mutant to environmental stresses including pH 3, hydrogen peroxide or a nitric oxide donor (data not shown). The expression profiles of M. tuberculosis wild type and glnR deletion mutant grown in nitrogen limiting conditions were then obtained. Cells were harvested 1 day after nitrogen run‐out, total RNA was extracted and cDNA hybridised to the M. tuberculosis microarray as described. Data were normalized and genes were considered significantly differentially expressed if they exhibited a difference in gene expression of greater than twofold with an FDR corrected P‐value of < 0.1. Fully annotated microarray data have been deposited in BμG@Sbase and ArrayExpress. A total of 52 genes were significantly up‐regulated and 15 significantly down regulated (File S3). This indicates that GlnR mediates (directly or indirectly) the expression of at least 67 genes.
Identification of GlnR binding sites during nitrogen stress
In order to identify directly regulated GlnR genes, we used ChIP‐seq to identify the location of GlnR binding sites in the genome during nitrogen limitation. Cells were grown in 1 mM (limiting) or 30 mM (excess) ammonium chloride, and DNA–protein complexes were cross‐linked 1 day after ammonium depletion; nitrogen excess samples were cross‐linked at the same time point, cells were then lysed and the DNA sheared by sonication. GlnR‐bound DNA fragments were immunoprecipitated using affinity‐purified anti‐GlnR polyclonal antibody. Prior to Illumina library construction, we performed quantitative PCR on the nitrite reductase (nirB) promoter region to confirm the enrichment of known GlnR binding regions in nitrogen limited cells compared with nitrogen replete cells; a gene thought not to be GlnR regulated (Rv1360) was included as a negative control (Fig. S3). The immunoprecipitated DNA was then prepared for next generation sequencing using the Illumina ChIP‐seq library kit. Sequencing of the DNA libraries generated approximately 144 million reads per sample which were mapped to the M. tuberculosis genome. All ChIP‐seq data files have been deposited into ArrayExpress (accession number E‐MTAB‐2492). GlnR binding regions were identified using the peak‐calling algorithm SISSRs (Narlikar and Jothi, 2012), with peaks defined as significant if they showed greater than fivefold enrichment in the immunoprecipitated sample compared with the input control DNA with a P value of < 0.005. This identified 36 putative GlnR binding sites in nitrogen limitation (Table 2). Of the 36 GlnR binding sites in nitrogen limitation, one (peak 12) was incorrectly identified by SISSRs as a GlnR binding site due to an excess of ribosomal RNA in this region; this was excluded from further analyses. Of the remaining 35 GlnR binding sites identified in nitrogen limitation, 26 were located in intergenic regions and 9 were located within genes. Although identification of the nirB promoter region (peak 1) as a GlnR binding site corroborated our ChIP‐seq data (Table 2, Fig. S3), further validation of four novel GlnR binding sites identified in this study by electromobility shift assays (EMSA) were performed. Two hundred base pair DNA regions, representing the binding regions from peaks 13, 17, 18 and 20, were incubated with increasing concentrations of recombinant GlnR protein. Fig. 3 shows that GlnR binds specifically to these promoter regions with a protein‐concentration dependent shift; the negative control Rv1360 DNA displayed no GlnR binding (Fig. S4). In addition, DNA enrichment of another four novel GlnR binding sites identified in this study (peaks 2, 10, 11 and 23) was verified by rate‐limiting qPCR. Enrichment of these four genomic regions by GlnR IP was observed in nitrogen limitation and not in nitrogen excess (Fig. 4); negative control region showed no enrichment (Fig. S3).
Table 2.
GlnR binding regions identified by ChIP‐seq and corresponding gene expression fold change (wild type vs gln R deletion strain) in M . tuberculosis during nitrogen limitation
| Peak no.a | Coordinatesb | Peak Intensityc | Adjacent gene(s)d | Fold change DEe (wt v glnR ko) | Gene name | Msmeg GlnR regulong | Lsr2‐GlnR overlaph | TSSi | TSS (Exp or Strv) j |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 302811–302851 | 54.53 | Rv0252* | 36.77 | nirB | Y | Y | 302853 | Both |
| 2 | 312651–312691 | 25.91 | Rv0260c* | 44.63 | nnaR | Y | Y | 312659 | Exp |
| 3 | 314191–314231 | 11.84 | Rv0261c | 10.65 | narK3 | Y | None | ||
| 4 | 559811–559851 | 11.21 | Rv0469 | Not Sig | umaA | 559862 | Both | ||
| 5 | 560371–560411 | 7.77 | Within Rv0469 | Not Sig | umaA | y | N/A | ||
| 6 | 1077911–1077951 | 5.4 | Rv0966c* | 0.74 | Rv0966c | 1077836 | |||
| Rv0967 | 0.86 | csoR | 1077954 | Both | |||||
| 7 | 1163531–1163571 | 50.83 | Rv1040c* | 4.75 | PE8 | Y | None | ||
| 8 | 1214391–1214431 | 5.08 | Rv1088 | Not Sig | PE9 | Y | None | ||
| 9 | 1287091–1287131 | 60.55 | Rv1161* | 1.76 | narG | 1287124 | Both | ||
| 10 | 1288291–1288331 | 7.14 | Within Rv1161 | 1.76 | narG | N/A | |||
| 11 | 1304571–1304611 | 34.76 | Within Rv1173 | Not Sig | fbiC | N/A | |||
| 12 | 1471651–1471691 | 5.4 | Ribosomal RNA f | N/A | N/A | N/A | |||
| 13 | 1561351–1561391 | 12.06 | Rv1386* | 0.67 | PE15 | Y | 1561403 | Strv | |
| 14 | 1728391–1728431 | 10.62 | Rv1527c* | 1.33 | pks5 | Y | 1728877 | Strv | |
| 15 | 1728911–1728951 | 12.52 | Rv1527c* | 1.33 | pks5 | 1728877 | Strv | ||
| 16 | 1735631–1735671 | 6.45 | Rv1535 | 0.44 | Rv1535 | 1735507/8 | Both | ||
| 17 | 1744831–1744871 | 30.01 | Rv1542c* | 99.66 | glbN | Y | 1744836 | Strv | |
| 18 | 1753431–1753471 | 30.04 | Rv1548c* | 1.14 | PPE22 | Y | None | ||
| Rv1549 | 0.56 | fadD11 | 1753563 | ||||||
| 19 | 2029771–2029811 | 5.38 | Rv1791 | 1.33 | PE19 | Y | 2029771 | Both | |
| 20 | 2487551–2487591 | 9.56 | Rv2219A* | Not Sig | Rv2219Ac | 2487478 | Both | ||
| Rv2220 | 1.35 | glnA1 | Y | 2487544 | Exp | ||||
| 21 | 2493731–2493771 | 41.58 | Rv2222c | 1.23 | glnA2 | Y | 2493745 | Both | |
| 22 | 2553111–2553151 | 47.26 | Rv2281 | 1.30 | pitB | 2553192 | |||
| 23 | 2563071–2563111 | 9.15 | Rv2291 | 0.63 | sseB | 2562953 | Both | ||
| 24 | 2603491–2603531 | 42.33 | Rv2329c | 2.33 | narK1 | 2603499 | Both | ||
| 25 | 2752931–2752971 | 12.79 | Within Rv2452c | Not Sig | Rv2452c | Y | N/A | ||
| Within Rv2453c | Not Sig | mobA | Y | N/A | |||||
| 26 | 3079111–3079151 | 6.27 | Rv2769c* | 5.11 | PE27 | Y | 3079137 | ||
| 27 | 3477871–3477911 | 6.29 | Within Rv3109 | 0.62 | moaA1 | Y | N/A | ||
| 28 | 3478231–3478271 | 5.47 | Within Rv3109 | 0.62 | moaA1 | Y | N/A | ||
| 29 | 3595491–3595531 | 6.07 | Rv3219 | Not Sig | whiB1 | Y | Y | 3595603 | Both |
| 30 | 3784831–3784871 | 14.79 | Rv3370c | Not Sig | dnaE2 | 3484777 | |||
| Rv3371 | 24.60 | Rv3371 | 3784902 | Strv | |||||
| 31 | 3799991–3800031 | 15.45 | Rv3385c* | Not Sig | vapB46 | 3800093 | Both | ||
| Rv3386 | Not Sig | Rv3386 | 3799988 | Both | |||||
| 32 | 3834631–3834671 | 26.47 | Rv3415c | Not Sig | Rv3415c | Y | None | ||
| Rv3416 | 3.44 | whiB3 | Y | Y | 3834791 | Both | |||
| 33 | 3964871–3964911 | 5.15 | Within Rv3528c | 0.85 | Rv3528c | Y | N/A | ||
| 34 | 3971191–3971231 | 8.71 | Within Rv3533c | 0.91 | PPE62 | Y | N/A | ||
| 35 | 4060611–4060651 | 8.02 | Rv3620c* | Not Sig | esxW | Y | 4060630 | ||
| 36 | 4062351–4062391 | 48.07 | Rv3622c* | 0.81 | PE32 | Y | None | ||
| Rv3623 | Not Sig | lpqG | Y | 4062342 | Both |
Assigned peak number.
Peak coordinates on the M. tuberculosis genome.
Fold enrichment of each peak compared with the input control calculated using SISSRs.
Adjacent gene(s) to peak (italics denotes intragenic).
Fold change in gene expression (wild type vs glnR KO) with FDR < 0.1, Not Sig means FDR > 0.1.
rRNA peak, excluded from further analysis.
Whether or not part of the GlnR regulon in M. smegmatis.
Overlap of GlnR and Lsr2 binding sites (lower case, italics denotes intragenic).
Transcriptional start site (TSS) from (Cortes et al., 2013).
TSS recorded under starvation or exponential growth (Cortes et al., 2013). * Genes in operons.
Figure 3.
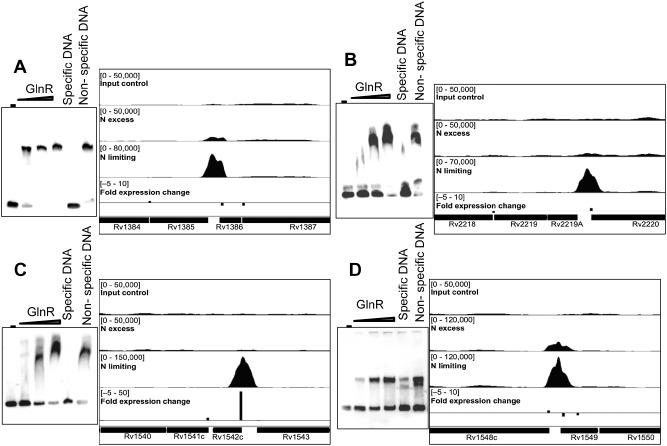
Novel GlnR binding sites identified upstream of differentially expressed genes, with corresponding EMSA to confirm specific GlnR binding.
EMSA were performed by incubating increasing amounts of His‐GlnR recombinant protein with labelled DNA corresponding to the promoter regions of the genes downstream of the GlnR binding site. GlnR binding was visualised in IGV. The top track represents input control DNA, the second track represents GlnR binding in nitrogen excess and the third track represents GlnR binding in nitrogen limiting conditions. Bar height is representative of fold change in gene expression in the wild type compared with the glnR KO mutant in nitrogen limitation. Levels of gene expression are indicated in the bottom track. (A) Peak 13, Rv1386 (PE15); (B) peak 20, Rv2219A; (C) peak 17, Rv1542c (glbN); (D) peak 18, Rv1548c (PPE22). The negative control Rv1360 DNA showed no GlnR binding (Fig. S4).
Figure 4.
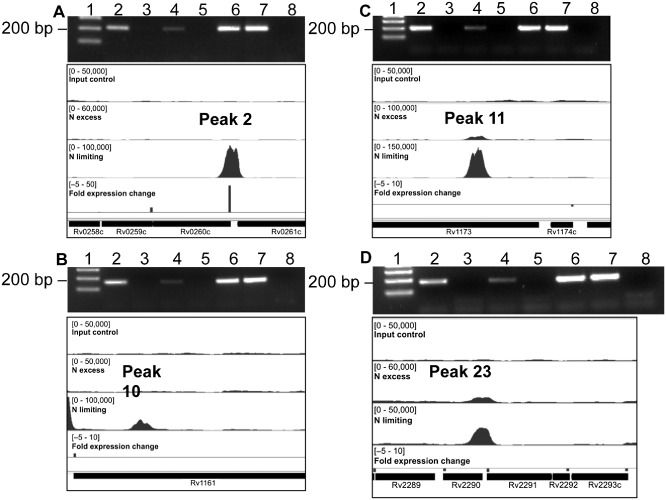
Novel GlnR binding sites identified upstream of differentially expressed genes, with corresponding DNA enrichment verified by qPCR. DNA enrichment for an additional four novel GlnR‐binding regions was observed in nitrogen limitation, but not nitrogen excess using rate‐limiting qPCR. (A) Peak 2, Rv0260c (nna R); (B) peak 10, Rv1161 (narG); (C) peak 11, Rv1173 (fbi C); (D) peak 23, Rv2291 (sse B). Lane 1, size marker, 200 bp arrowed. Lane 2, ChIP nitrogen excess. Lane 3, nitrogen excess no antibody control. Lane 4, ChIP nitrogen limiting. Lane 5, nitrogen limiting no antibody control. Lane 6, input nitrogen excess. Lane 7, input nitrogen limiting. Lane 8, no DNA control. A negative control region showed no enrichment (Fig. S3).
Determination of the GlnR regulon during nitrogen stress
In order to identify the genes directly controlled by GlnR, and thus determine the GlnR regulon, we compared the 35 GlnR binding sites with the profile of transcripts regulated by GlnR during nitrogen limitation (Table 2). Twenty‐one GlnR binding sites corresponded to the significant (twofold cut‐off, FDR corrected P value of ≤ 0.05) differential expression of 10 genes, 9 genes up regulated and 1 down regulated (Table 2), indicating that GlnR functions as both an activator and repressor of transcription. GlnR binding also controlled the expression of one set of divergent genes (peak 18), although DE fall below the twofold cut‐off in both cases. Genes adjacent to 32 predicted GlnR binding sites did not show any differential expression above the twofold cut‐off during nitrogen limitation suggesting that GlnR binds silently to the genome and/or requires additional transcription factors to control expression of these genes. All GlnR peaks and associated DE genes are provided as supplementary data file (File S4).
Analysis of the GlnR DNA binding motif
Initially, all 35 GlnR binding site sequences (200 bp) identified in this study were submitted to MEME to determine if there was a motif that was responsible for GlnR genomic binding. A consensus motif was initially identified, but this motif was only present in 18 of the 35 binding sites (data not shown). Therefore, the MEME search was restricted to just GlnR binding sites in intergenic regions. With the additional constraint, a significant consensus motif was identified which was present in all GlnR regulon binding sites (Fig. 5).
Figure 5.

M ycobacterium tuberculosis GlnR consensus binding motif derived from 20 GlnR binding regions identified during nitrogen limitation. MEME generated GlnR motif from 200 bp DNA sequences surrounding the 20 peaks associated with differential gene expression.
GlnR‐binding and transcriptional start sites
Of the 35 peaks identified by GlnR ChIP‐seq, 27 mapped to potential promoter regions of a total of 33 genes (including divergently transcribed) and the remaining 8 were within coding regions (italics in Table 2). Localisation of transcription factor binding sites within intragenic regions has been described in the case of EspR and linked to a potential additional role as a nucleoid associated protein (Galagan et al., 2013). It is interesting to note in this context that 6 of the 8 intragenic GlnR sites, and 15 out of 27 intergenic GlnR binding sites overlapped with sites recognised by the nucleoid associated protein Lsr2 (Gordon et al., 2010). One of the genes with an internal GlnR site (moaA) showed increased abundance in the GlnR deletion mutant, while narG, which has an additional promoter‐associated GlnR site, had decreased abundance. Localisation of the potential promoter‐associated GlnR sites in the context of a whole genome map of transcription start sites (TSSs) in M. tuberculosis (Cortes et al., 2013) identified adjacent TSSs for 27 of the 33 genes, including seven TSSs below the stringent cut‐off used by Cortes et al. that were detected by visual inspection of the original data using the Artemis genome browser. Four of the TSSs were recorded only under starvation conditions, 2 only during exponential growth, and 15 during both (Table 2). Five of the six genes for which we were unable to identify a relevant TSS also overlapped with Lsr2 sites; the remaining gene (narK3) was strongly down‐regulated in the GlnR knockout (Table 2). Rv1535 is flanked by two riboswitches: an Mbox upstream of Rv1535 and a T‐box upstream of Rv1536 (ileS) (Arnvig et al., 2011), but there are no transcripts evident from the intergenic probes on the tiling microarray flanking Rv1535.
Discussion
Despite more than a century of TB research, including the publication of the genome sequence of M. tuberculosis in 1998, fundamental questions still remain about the biology of the organism, such as the nature of the nutrients available to the pathogen and how it obtains these from its host environment. This lack of basic knowledge about the physiology of the organism has impacted negatively on our ability to identify targets for the urgently needed new drugs. Despite this lack of success, an attractive area for TB drug development remains the discovery of drugs with novel modes of action that inhibit essential metabolic processes. In order to identify such targets, we need a better understanding of host environments and how the bacteria metabolically adapt to them. The ability to sense and respond to the environment is fundamental for bacterial survival, such responses often occur through coordinated and complex programmes of metabolite signalling molecules, signal transduction proteins, metabolic enzymes and gene expression controlled by transcriptional regulators. Although several global studies of the response of M. tuberculosis to various models of in vivo stresses have been performed (for example Voskuil, 2003; Voskuil et al., 2004; Voskuil et al., 2011; Cunningham‐Bussel and Zhang, 2013), the specific metabolic adaptation to nitrogen has not been studied, and the genes/pathways responsible for nitrogen uptake and assimilation are unknown.
The mycobacterial genome (Cole et al., 1998) provides insight into nitrogen metabolic pathways and nitrogen sources the tubercle bacillus can potentially use, but does not indicate which pathways are active and under what conditions. M. tuberculosis contains fewer genes predicted to be involved in nitrogen metabolism than the non‐pathogenic M. smegmatis (see Amon et al., 2009 for a comparison), presumably due to the diverse range of environmental nitrogen sources available in the soil compared with those within a vertebrate host. M. tuberculosis encodes genes for ammonium uptake and assimilation through GS/GOGAT pathways, urea and nitrate assimilation, and amino acid permeases, indicating that it may encounter these compounds in vivo. However, many of these nitrogen sources are toxic to host cells at high levels, limiting their availability in vitro. Analysis of how bacteria respond when nitrogen becomes limiting may provide more general clues about how bacteria survive in vivo starvation.
The identification of glnR in transposon screens in vitro and in vivo is rather variable. However, we would not necessarily expect to see differential expression of glnR – since, as described here, glnR is not hugely up‐regulated under nitrogen stress. Instead GlnR phosphorylation and dimer formation (signal for either unknown) modify its function rather than increased levels of GlnR. A survey of five published studies, and unpublished data from the TBdb resource, identifies two studies where there is no differential expression of glnR under a variety of conditions (THP‐1 infection, starvation, heat shock, Bone Marrow Macrophages (BMM), surfactants/lipids) (Fontan et al., 2008; Schwab et al., 2009). One study (Voskuil et al., 2004) showed a decrease in expression concomitant with the stationary phase and non‐replication persistence. Three studies (Voskuil et al., 2011) (Boshoff et al., 2004) and Unpublished Carbon Sources on TBdb from Liu and Schoolnik (http://www.tbdb.org/expressionHistory.shtml?gn=Rv0818) observed differential expression of glnR on exposure to DETA/NO, H2O2, oleic acid and palmitate.
In previous studies, we examined the transcriptomic response to nitrogen stress of the soil‐dwelling saprophyte M. smegmatis (Williams et al., 2013) and demonstrated that GlnR is the global nitrogen response regulator, controlling the expression of over 100 genes, including those involved in the assimilation and utilisation of nitrogen (Jenkins et al., 2013). Scavenging of nitrogen from a variety of environmental sources was a key area of metabolism up‐regulated in M. smegmatis, and AMBIENT analysis of the transcriptional response identified modules associated with this as well as carbon scavenging and energy generation. In this study, we used a similar approach to investigate how the obligate human pathogen M. tuberculosis alters its metabolism in response to nitrogen stress. The transcriptional data in Fig. S2 highlight the fact that M. tuberculosis does not seem to be adapted to scavenge a diverse range of nitrogen sources, with only the genes associated with asparagine and ornithine transport/metabolism upregulated greater than twofold on nitrogen run‐out at day 8.
Inspection of the AMBIENT network (Fig. 2) and comparison with the GlnR‐binding data (Table 2) show that GlnR regulation of nitrate–nitrite metabolism and nitric oxide detoxification is the major response to nitrogen stress, with both nitrate (narGHJI) and nitrite (nirBD) reductase operons activated by GlnR. The nirBD operon transcription is strongly activated by the intergenic GlnR‐binding site, whereas the narGHJI operon contains two GlnR sites, one promoter‐associated site upstream of narG and one within the gene (Fig. 3, peak 10), and shows only modest activation. Other genes involved in NO detoxification (narK1, narK3, nnaR, glnbN, whiB3) also have GlnR‐binding sites. It may be that production of nitrite by NarGHJI is limited by substrate availability, and not enzyme levels (as reflected by only modest changes in gene expression), but that as ammonium levels fall and metabolism of nitrate increases, a much larger increase in NirBD activity is needed to remove potentially toxic nitrite. GlnR does not regulate narGHJI in M. smegmatis, so the up‐regulation in response to nitrogen stress must be controlled by another mechanism, which is more in keeping with its ability to metabolise a much wider range of nitrogen sources than are evident here. The importance of nitrate–nitrite metabolism to the survival of M. tuberculosis inside human macrophages has recently been reported (Cunningham‐Bussel and Zhang, 2013), with bacterial adaptation to nitrite utilisation likely a key adaptation during infection, regardless of oxygen status. During adaptation to hypoxia, nitrate has also been shown to modulate the tricarboxylic acid cycle in mycobacteria (Eoh and Rhee, 2013), regulating both metabolism and respiratory activity, and the methylcitrate cycle/isocitrate lyase are up‐regulated here.
Mycobacterium tuberculosis also does not show the large hydrogen peroxide response seen in M. smegmatis (Williams et al., 2013), but ahpC, ahpD and katG are all up‐regulated in response to nitrogen stress. A similar response has been reported on exposure of mycobacteria to nitric oxide (Voskuil et al., 2011), leading the authors to propose that mycobacteria are continually primed for oxidative stress defence.
The reduction in bacterial growth rate after nitrogen run‐out is accompanied by a decrease in ribosomal proteins, and the AMBIENT shows how alterations (up and down) in metabolism are linked. For example, Rv2928 (fatty acyl ACP hydrolase) and Rv2930 (fatty acyl CoA ligase) both act on phthiocerol and are down‐ and up‐regulated respectively. Aspartate metabolism, which has been proposed to be an important source of nitrogen in vivo (Gouzy et al., 2013), is altered with less going to pyrimidine biosynthesis, making more aspartate available to be converted into ammonium. Genes down‐regulated in reduced growth rate may not be associated with nitrogen stress per se, but up‐regulated genes are going against the general metabolism down shift so are likely to be part of the nitrogen stress response. RNA was collected when external nitrogen had run out to try and mitigate the effect of the subsequent decrease in growth rate.
Several AMBIENT nodes are marked as ‘none’ in Fig. 2; these are either spontaneous chemical reactions or are indicated as a result of the manual curation of the genome model, suggesting that such a reaction exists, but has not yet been identified.
The GlnR‐regulon in M. tuberculosis is much more restricted in terms of the number of GlnR‐binding sites, with only 26 intergenic ones (Table 1), compared with 53 in M. smegmatis (Jenkins et al., 2013). This fits with the smaller number of nitrogen metabolism genes compared with M. smegmatis. Indeed there are only eight binding site locations shared between the two organisms in terms of the genes controlled. The GlnR‐binding site identified is similar to that found for M. smegmatis with conserved AC residues nine bases apart. As mentioned earlier, nirB is GlnR controlled in both, as are genes associated with nitrogen metabolism such as the nitrate/nitrite transporter narK3 and the transcription factor nnaR. The two glutamine synthetases are GlnR regulated in both organisms, although only glnA1 is differentially expressed in M. tuberculosis. Two other transcription factors, whiB1 and whiB3, feature in both organisms, as does the most highly up‐regulated gene the globin glbN. This encodes a potent mannosylated oxygen‐dependent nitric oxide dioxygenase which protects mycobacteria from toxic NO generated by macrophages (Arya et al., 2013). Its up‐regulation under in vitro conditions may be in response to NO produced as by‐product of nitrate reductase activity. Overall this suggests a central core of genes in both organisms that mediate the bacterial response to nitrogen stress – not only directly through the nitrite reductases and transporters but also indirectly via other transcription factors, which may recruit more general stress response elements, such hydrogen peroxide detoxification (Voskuil et al., 2011) needed for survival. A recent paper (Elharar et al., 2014) shows that survival of M. smegmatis under nutrient limitation, including nitrogen, requires proteasome‐mediated amino acid recycling.
Of the 28 genes for which we identified a TSS, in 18 cases, the midpoint of GlnR‐binding fell within a region between 75 bp upstream and 10 bp downstream of the TSS that is commonly associated with transcription factor regulation. For 11 of these genes, the level of expression in the GlnR knockout was significantly decreased over that in the wild type (Table 2), consistent with a model in which GlnR binding acts by promoting access of RNA polymerase. GlnR homo‐dimer formation is important in its function, and its interaction with other proteins may be governed by post‐translational modifications such as acetylation or small molecule binding (Lin et al., 2014). Any post‐translational modification and interaction with other factors may explain its dual ability to both repress and activate gene expression.
The expression of glutamine synthase genes glnA1 and glnA2, which have previously been identified as components of the GlnR regulon in M. smegmatis, showed little or no change in expression in the knockout. While the overall abundance of glnA1 and glnA2 transcripts also changed only slightly in the starvation model described by Cortes et al., they recorded a switch in the dominant TSS from a distal to a proximal site in the case of glnA1 and from a leaderless to an internal TSS for glnA2. The location of GlnR binding sites at positions −37 (glnA1) and −30 (glnA2) would be consistent with GlnR regulation of transcription from the downstream promoter in each case. The resulting change in 5′ untranslated region may affect the efficiency of translation. A similar switch from distal to proximal TSS occurs in the starvation response of thiosulphate sulphuryltransferase sseB, with an overall increase in transcript abundance in the GlnR knockout. In spite of GlnR binding at −23 with respect to the TSS, there was no change in the level of csoR transcript in the knockout. In this case, the GlnR binding site overlaps that of CsoR itself, which inhibits its own expression under conditions of low copper concentrations (Liu et al., 2007). EsxW, part of the new TB vaccine (Knudsen et al., 2014), shows a similar picture, with a GlnR‐binding site which overlaps with that of Lsr2 (Gordon et al., 2010) and no differential expression. In this case, the lack of altered expression also fits with transcriptional data obtained during mouse infection (Knudsen et al., 2014), used as evidence for its stability and suitability as a vaccine candidate. Transcription may be dually regulated by competition between the two transcription factors in these cases. Similarly for PE15, transcriptional regulation may depend on competition between GlnR and cAMP receptor protein binding to a site at 1561338 (Bai et al., 2007), and for whiB1, there may be competition between CRP (Rickman et al., 2005; Agarwal et al., 2009), Lsr2 (Gordon et al., 2010) and GlnR, suggesting a complex level of regulation.
With the evolution of M. tuberculosis as an obligate vertebrate pathogen, it has lost many of the pathways not required for the assimilation of nutrients readily available in vitro. In keeping with this, the GlnR‐regulon has shrunk overall, with the nitrogen component now seemingly limited to the nitrate and nitrite reductases, which are presumably a main source of nitrogen available in the host. We still do not know how GlnR is activated. There is no obvious pattern linking the GlnR‐binding site, the transcriptional start site and whether GlnR functions as an activator or repressor. Presumably, this depends on post‐translational modification or the interaction with other transcription factors and DNA binding proteins, and indeed the majority of genes that show no differential expression have GlnR‐binding sites that overlap with Lsr2, CRP or both. This suggests that regulation results from a complex interplay between these factors, or that some sites act as depots for DNA‐binding proteins. The GlnR regulated inter conversion of nitrate, nitrite and nitric oxide are increasingly recognised as the key processes in nitrogen assimilation and intracellular survival by M. tuberculosis (Voskuil et al., 2011; Cossu et al., 2013; Cunningham‐Bussel and Zhang, 2013; Eoh and Rhee, 2013), making them important areas for further research.
Experimental procedures
Bacterial strains and growth conditions
Mycobacterium tuberculosis H37Rv wild type (ATCC 27294) and M. tuberculosis H37Rv glnR KO (this study) were used in this study. M. tuberculosis was grown aerobically in Middlebrook 7H9 liquid broth (supplemented with 0.2% glycerol, 0.025% tyloxapol and 10% OADC) at 37°C, 100 rpm. Optimised nitrogen limiting conditions have been described (Behrends et al., 2012; Jenkins et al., 2012). Briefly, a 10 day culture of M. tuberculosis was washed twice in nitrogen‐free Sauton's medium (0.05% (w/v) KH2PO4, 0.05% (w/v) MgSO4, 0.2% (w/v) citric acid, 0.005% (w/v) ferric citrate, 0.2% (v/v) glycerol, 0.0001% (v/v) ZnSO4, 0.015% (v/v) tyloxapol) and inoculated into Sauton's nitrogen‐free medium, supplemented with 1 mM (nitrogen limiting) or 30 mM (nitrogen excess) ammonium chloride (Ultra pure; Sigma) to a starting OD60nm of 0.04. Growth was monitored by OD600nm. Ammonium ions in the culture supernatant were quantified using an AquaQuant Ammonium detection kit (Merck).
Construction of GlnR mutant
The M. tuberculosis glnR KO mutant was constructed by a recombineering approach as previously described (van Kessel and Hatfull, 2007), replacing the entire glnR gene with a hygromycin resistance cassette (Jenkins et al., 2012). Primers used to amplify the flanking regions and to confirm mutant construction are given in Table S1. Successful construction of the mutant was confirmed by flanking PCR and Western analysis for the GlnR protein using a GlnR specific antibody previously described (Jenkins et al., 2013). For complementation experiments, vector pSM57, a 1582 bp EcoRV–SmaI fragment containing glnR cloned into the promoterless pMV306 vector (kind gift from Prof Franz‐Christoph Bange) (Malm et al., 2009), was transformed into the glnR mutant.
EMSA
To analyse GlnR binding to gene promoter regions, DNA fragments were PCR amplified from M. tuberculosis genomic DNA and used in EMSA. Primers used to amplify DNA regions are given in Table S1. His‐GlnR protein was purified as described (Jenkins et al., 2013). DNA fragments were labelled using a DIG Oligonucleotide 3′‐End Labelling Kit (Roche). DNA : protein binding reactions contained 0.4 ng of labelled DNA, 0.5 μg poly d(A–T), 0–0.9 μg His‐GlnR, 25 mM HEPES (pH 7.9), 150 mM NaCl, 2.5 mM MgCl2. The reaction mixture was incubated at 37°C for 15 min, before separation on a pre‐run 6% DNA retardation gel (Life Technologies). Bands were visualised using a LAS‐3000 Fuji imager.
Rate‐limiting PCR (qPCR)
To identify enrichment in GlnR‐immunoprecipitated DNA, a rate‐limiting PCR was performed. DNA was immunoprecipitated and purified as described under ChIP. DNA sequences were amplified using primers listed in Table S1. Reaction mixtures consisted of GlnR‐immunoprecipitated DNA (0.3 ng), 1 × BioMix (Bioline), 1 μM of each primer and 5% (v/v) dimethyl sulfoxide (Sigma). PCR was carried out in a thermocycler T3000 (Biometra); 95°C for 5 min, 23 cycles of 95°C 30 s, 55°C 30 s, 72°C 1 min, with final extension 72°C for 8 min. DNA was visualised on a 2% agarose gel.
RNA isolation
Mycobacterium tuberculosis strains were grown in triplicate in nitrogen limiting conditions until external nitrogen was depleted. Total RNA was extracted from exponentially growing cells using the GTC/Trizol method (Ehrt et al., 2003). Extracted RNA was purified using the RNeasy kit (Qiagen) and residual DNA was removed by TURBO DNA‐free (Ambion Life Technologies) treatment. Superase (ABI Life Technologies) was added and RNA was stored at −80°C. Quality and quantity of RNA were determined using a Bio‐analyser (Agilent).
Quantitative real‐time PCR (qRT‐PCR)
cDNA was amplified from 100 ng of RNA using the SuperScript III First‐Strand Synthesis SuperMix (Invitrogen). qRT‐PCR reactions were carried out in a final volume of 10 μl [1 μl of cDNA, 5 μl of TaqMan PCR master mix (Applied Biosystems), 0.5 μl TaqMan probe (Applied Biosystems)]. Amplification was performed on an Applied Biosystems 7500 Real‐Time System (50°C 5 min, 95°C 10 min, and 40 cycles of 95°C 15 s, 60°C 1 min). Linear amplification and amplification efficiencies for each TaqMan primer/probe were determined. Real‐time analysis was performed on RNA from three independent cultures and quantification of sigA expression served as an internal control. Fold change was calculated as a ratio of the arbitrary expression units, standardised to sigA. Primers and Taqman probe sequences for each gene studied are given in Table S2.
Preparation of labelled cDNA from total RNA
Labelled cDNA was prepared from 1 μg of total RNA using Cy3‐dCTP (GE Healthcare) and SuperScript II reverse transcriptase with random hexamer primers (Invitrogen). Labelled cDNA was purified by Qiagen MinElute column, combined with 10 × CGH blocking agent and 2× Hi‐RPM hybridisation buffer (Agilent) and heated (95°C for 5 min) prior to loading onto microarray slides. Slides were incubated overnight in an Agilent rotating oven at 65°C, 20 rpm. After hybridization, slides were washed (5 min at room temperature) with CGH Wash Buffer 1 (Agilent) and 1 min at 37°C with CGH Wash buffer 2 (Agilent). Slides were scanned immediately, using an Agilent High Resolution Microarray Scanner (Agilent Technologies, Stockport, UK), at 2 μm resolution, 100% PMT. Scanned images were quantified using Feature Extraction software v 10.7.3.1. RNA for tiling microarray hybridization was directly labelled using the Kreatech Universal Linkage System (Leica Microsystems, Milton Keynes, UK), which is based on the stable binding of platinum to the N7 position of guanine, according to the manufacturer's instructions.
Microarray design
The microarray for the wild type vs GlnR KO experiments was constructed by determining all unique genes from the 3992 chromosomal predicted coding sequences of M. tuberculosis H37Rv, downloaded from Ensembl Bacteria Release 5 (http://bacteria.ensembl.org/). Multiple optimal hybridisation 60‐mer oligonucleotide sequences were designed (Oxford Gene Technologies), from which a minimal non‐redundant subset of oligonucleotides were selected with target coverage of three 60‐mers per gene. Arrays were manufactured on the Inkjet in situ synthesized platform (Agilent) using the 8 × 60k format. Analysis of gene expression over nitrogen run‐out was performed using a 180k Tiling array, designed as described (Golby et al., 2013).
Statistical analyses of differential gene expression
Statistical analyses of the gene expression data were carried out using the statistical analysis software environment R together with packages available as part of the Bioconductor project (http://www.bioconductor.org). Data generated from the Agilent Feature Extraction software for each sample were imported into R. Replicate probes were mean summarised and quantile normalised using the pre‐process Core R package. The limma R package (Smyth, 2004) was used to compute empirical Bayes moderated t‐statistics to identify DE genes between time points. Generated P‐values were corrected for multiple testing using the Benjamini and Hochberg FDR. An FDR corrected P‐value cut‐off of less than 0.1 was used to determine significant differential expression, which is more stringent than an uncorrected P‐value of 0.05.
ChIP
Mycobacterium tuberculosis was grown as specified and processed for ChIP using purified rabbit anti‐GlnR polyclonal antibody as described (Jenkins et al., 2013). DNA were prepared for next generation sequencing using the Illumina ChIP‐seq DNA sample preparation kit according to the manufacturer's protocol, with the addition of a second gel extraction step after PCR amplification, to remove excess primer dimers. DNA size and purity were confirmed by DNA Bioanalyser (Agilent) and sequencing conducted on an Illumina HiSeq2000 sequencer (MRC Clinical Sciences Centre, Hammersmith). Sequencing reads were mapped to the M. tuberculosis genome using Bowtie (Langmead et al., 2009), and GlnR binding regions were identified using the peak‐calling algorithm SISSRs (Site Identification for Short Sequence Reads) (Narlikar and Jothi, 2012), with peaks defined as significant if they showed greater than fivefold enrichment in the immunoprecipitated sample compared with the input control DNA with a P value of < 0.005. The SISSR peaks and gene expression data were visualised using the Integrative Genome Viewer (IGV) (Robinson et al., 2011; Thorvaldsdóttir et al., 2013) and screenshots were taken of individual peaks.
Supporting information
The Tiling array design is available in BμG@Sbase (Accession No. A‐BUGS‐47; http://bugs.sgul.ac.uk/A‐BUGS‐47) and also ArrayExpress (Accession No. A‐BUGS‐47). All sequencing data have been deposited in ArrayExpress (Accession No. E‐MTAB‐2492). The other data sets supporting the results of this article are included within the article and its additional files.
Supporting information
Supporting information
Acknowledgements
We thank Dr Kate Gould and the team at BμG@S (Bacterial Microarray Group at St George's, University of London) for the M. tuberculosis microarray design and hybridisations. We also acknowledge Dr Laurence Game and the team at the MRC Clinical Sciences Centre Genomics Lab, Imperial College, for performing the high throughput next generation sequencing. We also thank Dr Huihai Wu (University of Surrey) for bioinformatics support and Prof Douglas Young (NIMR) for discussions about the transcriptional start sites. This work was funded by an MRC studentship to VJ and by Grant BB/G020434/1 from the Biotechnology and Biological Sciences Research Council, UK; the funders played no other role in the study. The authors declare no conflicts of interest.
References
- Agarwal, N. , Lamichhane, G. , Gupta, R. , Nolan, S. , and Bishai, W.R. (2009) Cyclic AMP intoxication of macrophages by a Mycobacterium tuberculosis adenylate cyclase. Nature 460: 98–102. [DOI] [PubMed] [Google Scholar]
- Akhtar, S. , Khan, A. , Sohaskey, C.D. , Jagannath, C. , and Sarkar, D. (2013) Nitrite reductase NirBD is induced and plays an important role during in vitro dormancy of Mycobacterium tuberculosis . J Bacteriol 195: 4592–4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amon, J. , Bräu, T. , Grimrath, A. , Hänssler, E. , Hasselt, K. , Höller, M. , et al (2008) Nitrogen control in Mycobacterium smegmatis: Nitrogen‐dependent expression of ammonium transport and assimilation proteins depends on the OmpR‐type regulator GlnR. J Bacteriol 190: 7108–7116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amon, J. , Titgemeyer, F. , and Burkovski, A. (2009) A genomic view on nitrogen metabolism and nitrogen control in mycobacteria. J Mol Microbiol Biotechnol 17: 20–29. [DOI] [PubMed] [Google Scholar]
- Amon, J. , Titgemeyer, F. , and Burkovski, A. (2010) Common patterns – unique features: Nitrogen metabolism and regulation in Gram‐positive bacteria. FEMS Microbiol Rev 34: 588–605. [DOI] [PubMed] [Google Scholar]
- Arnvig, K.B. , Comas, I. , Thomson, N.R. , Houghton, J. , Boshoff, H.I. , Croucher, N.J. , et al (2011) Sequence‐based analysis uncovers an abundance of non‐coding RNA in the total transcriptome of Mycobacterium tuberculosis . PLoS Pathog 7: e1002342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arya, S. , Sethi, D. , Singh, S. , Hade, M.D. , Singh, V. , Raju, P. , et al (2013) Truncated hemoglobin, HbN, is post‐translationally modified in Mycobacterium tuberculosis and modulates host–pathogen interactions during intracellular infection. J Biol Chem 288: 29987–29999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai, G. , Gazdik, M.A. , Schaak, D.D. , and McDonough, K.A. (2007) The Mycobacterium bovis BCG cyclic AMP receptor‐like protein is a functional DNA binding protein in vitro and in vivo, but its activity differs from that of its M. tuberculosis ortholog, Rv3676. Infect Immun 75: 5509–5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckers, G. , Strösser, J. , Hildebrandt, U. , Kalinowski, J. , Farwick, M. , Krämer, R. , and Burkovski, A. (2005) Regulation of AmtR‐controlled gene expression in Corynebacterium glutamicum: Mechanism and characterization of the AmtR regulon. Mol Microbiol 58: 580–595. [DOI] [PubMed] [Google Scholar]
- Behrends, V. , Williams, K.J. , Jenkins, V.A. , Robertson, B.D. , and Bundy, J.G. (2012) Free glucosylglycerate is a novel marker of nitrogen stress in Mycobacterium smegmatis . J Proteome Res 11: 3888–3896. [DOI] [PubMed] [Google Scholar]
- Boshoff, H. , Boshoff, H.I.M. , Myers, T.G. , Myers, T.G. , Copp, B.R. , Copp, B.R. , et al (2004) The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism – Novel insights into drug mechanisms of action. J Biol Chem 279: 40174–40184. [DOI] [PubMed] [Google Scholar]
- Bryant, W.A. , Sternberg, M.J. , and Pinney, J.W. (2013) AMBIENT: Active Modules for Bipartite Networks – using high‐throughput transcriptomic data to dissect metabolic response. BMC Syst Biol 7: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkovski, A. (2007) Nitrogen control in Corynebacterium glutamicum: Proteins, mechanisms, signals. J Microbiol Biotechnol 17: 187–194. [PubMed] [Google Scholar]
- Carroll, P. , Pashley, C.A. , and Parish, T. (2008) Functional analysis of GlnE, an essential adenylyl transferase in Mycobacterium tuberculosis . J Bacteriol 190: 4894–4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole, S.T. , Brosch, R. , Parkhill, J. , Garnier, T. , Churcher, C. , Harris, D. , et al (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393: 537–544. [DOI] [PubMed] [Google Scholar]
- Cortes, T. , Schubert, O.T. , Rose, G. , Arnvig, K.B. , Comas, I. , Aebersold, R. , and Young, D.B. (2013) Genome‐wide mapping of transcriptional start sites defines an extensive leaderless transcriptome in Mycobacterium tuberculosis . Cell Reports 5: 1121–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossu, A. , Sechi, L.A. , Bandino, E. , Zanetti, S. , and Rosu, V. (2013) Expression profiling of Mycobacterium tuberculosis H37Rv and Mycobacterium smegmatis in acid‐nitrosative multi‐stress displays defined regulatory networks. Microb Pathog 65: 89–96. [DOI] [PubMed] [Google Scholar]
- Cunningham‐Bussel, A. , and Zhang, T. (2013) Nitrite produced by Mycobacterium tuberculosis in human macrophages in physiologic oxygen impacts bacterial ATP consumption and gene expression. Proc Natl Acad Sci USA 110: E4256–E4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrt, S. , Liu, Y. , Mangan, J.A. , Monahan, I.M. , Dolganov, G. , Efron, B. , et al (2003) Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: Insights into the phagosomal environment. J Exp Med 198: 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elharar, Y. , Roth, Z. , Hermelin, I. , Moon, A. , Peretz, G. , Shenkerman, Y. , et al (2014) Survival of mycobacteria depends on proteasome‐mediated amino acid recycling under nutrient limitation. EMBO J 33: 1802–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eoh, H. , and Rhee, K.Y. (2013) Multifunctional essentiality of succinate metabolism in adaptation to hypoxia in Mycobacterium tuberculosis . Proc Natl Acad Sci USA 110: 6554–6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink, D. , Weissschuh, N. , Reuther, J. , Wohlleben, W. , and Engels, A. (2002) Two transcriptional regulators GlnR and GlnRII are involved in regulation of nitrogen metabolism in Streptomyces coelicolor A3(2). Mol Microbiol 46: 331–347. [DOI] [PubMed] [Google Scholar]
- Fontan, P. , Aris, V. , Ghanny, S. , Soteropoulos, P. , and Smith, I. (2008) Global transcriptional profile of Mycobacterium tuberculosis during THP‐1 human macrophage infection. Infect Immun 76: 717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galagan, J.E. , Minch, K. , Peterson, M. , Lyubetskaya, A. , Azizi, E. , Sweet, L. , et al (2013) The Mycobacterium tuberculosis regulatory network and hypoxia. Nature 499: 178–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golby, P. , Nunez, J. , Witney, A. , Hinds, J. , Quail, M.A. , Bentley, S. , et al (2013) Genome‐level analyses of Mycobacterium bovis lineages reveal the role of SNPs and antisense transcription in differential gene expression. BMC Genomics 14: 710–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, B.R.G. , Li, Y. , Wang, L. , Sintsova, A. , van Bakel, H. , Tian, S. , et al (2010) Lsr2 is a nucleoid‐associated protein that targets AT‐rich sequences and virulence genes in Mycobacterium tuberculosis . Proc Natl Acad Sci USA 107: 5154–5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouzy, A. , Wu, T.‐D. , Peixoto, A. , Levillain, F. , Lugo‐Villarino, G. , Gerquin‐Kern, J.‐L. , et al (2013) Mycobacterium tuberculosis nitrogen assimilation and host colonization require aspartate. Nat Chem Biol 9: 674–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouzy, A. , Bottai, D. , Levillain, F. , Dumas, A. , Wallach, J.B. , Caire‐Brandli, I. , et al (2014) Mycobacterium tuberculosis exploits asparagine to assimilate nitrogen and resist acid stress during infection. PLoS Pathog 10: e1003928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruswitz, F. , O'Connell, J. , and Stroud, R.M. (2007) Inhibitory complex of the transmembrane ammonia channel, AmtB, and the cytosolic regulatory protein, GlnK, at 1.96 A. Proc Natl Acad Sci USA 104: 42–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper, C. , Hayward, D. , Wiid, I. , and van Helden, P. (2008) Regulation of nitrogen metabolism in Mycobacterium tuberculosis: A comparison with mechanisms in Corynebacterium glutamicum and Streptomyces coelicolor . IUBMB Life 60: 643–650. [DOI] [PubMed] [Google Scholar]
- Harper, C.J. , Hayward, D. , Kidd, M. , Wiid, I. , and van Helden, P. (2010) Glutamate dehydrogenase and glutamine synthetase are regulated in response to nitrogen availability in Mycobacterium smegmatis . BMC Microbiol 10: 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamshidi, N. , and Palsson, B.Ø. (2007) Investigating the metabolic capabilities of Mycobacterium tuberculosis H37Rv using the in silico strain iNJ661 and proposing alternative drug targets. BMC Syst Biol 1: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javelle, A. , and Merrick, M. (2005) Complex formation between AmtB and GlnK: An ancestral role in prokaryotic nitrogen control. Biochem Soc Trans 33: 170–172. [DOI] [PubMed] [Google Scholar]
- Jenkins, V.A. , Robertson, B.D. , and Williams, K.J. (2012) Aspartate D48 is essential for the GlnR‐mediated transcriptional response to nitrogen limitation in Mycobacterium smegmatis . FEMS Microbiol Lett 330: 38–45. [DOI] [PubMed] [Google Scholar]
- Jenkins, V.A. , Barton, G.R. , Robertson, B.D. , and Williams, K.J. (2013) Genome wide analysis of the complete GlnR nitrogen‐response regulon in Mycobacterium smegmatis . BMC Genomics 14: 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kessel, J.C. , and Hatfull, G.F. (2007) Recombineering in Mycobacterium tuberculosis . Nat Meth 4: 147–152. [DOI] [PubMed] [Google Scholar]
- Khan, A. , Akhtar, S. , Ahmad, J.N. , and Sarkar, D. (2008) Presence of a functional nitrate assimilation pathway in Mycobacterium smegmatis . Microb Pathog 44: 71–77. [DOI] [PubMed] [Google Scholar]
- Knudsen, N.P.H. , Nørskov‐Lauritsen, S. , Lindenstrøm, T. , Andersen, P. , Agger, E.M. , and Aagaard, C. (2014) Tuberculosis vaccine with high predicted population coverage and compatibility with modern diagnostics. Proc Natl Acad Sci USA 111: 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. , Trapnell, C. , Pop, M. , and Salzberg, S.L. (2009) Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh, J.A. , and Dodsworth, J.A. (2007) Nitrogen regulation in bacteria and archaea. Annu Rev Microbiol 61: 349–377. [DOI] [PubMed] [Google Scholar]
- Lin, W. , Mathys, V. , Koh, V.H.Q. , Martínez Gómez, J.M. , Zainul Rahim, S.Z. , Tan, M.P. , et al (2012) Urease activity represents an alternative pathway for Mycobacterium tuberculosis nitrogen metabolism. Infect Immun 80: 2771–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, W. , Wang, Y. , Han, X. , Zhang, Z. , Wang, C. , Wang, J. , et al (2014) Atypical OmpR/PhoB subfamily response regulator GlnR of actinomycetes functions as a homodimer, stabilized by the unphosphorylated conserved asp‐focused charge interactions. J Biol Chem 289: 15413–15425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, T. , Ramesh, A. , Ma, Z. , Ward, S.K. , Zhang, L. , George, G.N. , et al (2007) CsoR is a novel Mycobacterium tuberculosis copper‐sensing transcriptional regulator. Nat Chem Biol 3: 60–68. [DOI] [PubMed] [Google Scholar]
- Malm, S. , Tiffert, Y. , Micklinghoff, J. , Schultze, S. , Joost, I. , Weber, I. , et al (2009) The roles of the nitrate reductase NarGHJI, the nitrite reductase NirBD and the response regulator GlnR in nitrate assimilation of Mycobacterium tuberculosis . Microbiology 155: 1332–1339. [DOI] [PubMed] [Google Scholar]
- Narlikar, L. , and Jothi, R. (2012) ChIP‐Seq data analysis: Identification of protein‐DNA binding sites with SISSRs peak‐finder. Methods Mol Biol 802: 305–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pioszak, A.A. , Jiang, P. , and Ninfa, A.J. (2000) The Escherichia coli PII signal transduction protein regulates the activities of the two‐component system transmitter protein NRII by direct interaction with the kinase domain of the transmitter module. Biochemistry 39: 13450–13461. [DOI] [PubMed] [Google Scholar]
- Pullan, S.T. , Chandra, G. , Bibb, M.J. , and Merrick, M. (2011) Genome‐wide analysis of the role of GlnR in Streptomyces venezuelae provides new insights into global nitrogen regulation in actinomycetes. BMC Genomics 12: 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radchenko, M.V. , Thornton, J. , and Merrick, M. (2010) Control of AmtB‐GlnK complex formation by intracellular levels of ATP, ADP, and 2‐oxoglutarate. J Biol Chem 285: 31037–31045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read, R. , Pashley, C.A. , Smith, D. , and Parish, T. (2007) The role of GlnD in ammonia assimilation in Mycobacterium tuberculosis . Tuberculosis (Edinb) 87: 384–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitzer, L. (2003) Nitrogen assimilation and global regulation in Escherichia coli . Annu Rev Microbiol 57: 155–176. [DOI] [PubMed] [Google Scholar]
- Rickman, L. , Scott, C. , Hunt, D.M. , Hutchinson, T. , Menéndez, M.C. , Whalan, R. , et al (2005) A member of the cAMP receptor protein family of transcription regulators in Mycobacterium tuberculosis is required for virulence in mice and controls transcription of the rpfA gene coding for a resuscitation promoting factor. Mol Microbiol 56: 1274–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, J.T. , Thorvaldsdóttir, H. , Winckler, W. , Guttman, M. , Lander, E.S. , Getz, G. , and Mesirov, J.P. (2011) Integrative genomics viewer. Nat Biotechnol 29: 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab, U. , Rohde, K.H. , Wang, Z. , Chess, P.R. , Notter, R.H. , and Russell, D.G. (2009) Transcriptional responses of Mycobacterium tuberculosis to lung surfactant. Microb Pathog 46: 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth, G.K. (2004) Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3: 1–25. [DOI] [PubMed] [Google Scholar]
- Tan, M.P. , Sequeira, P. , Lin, W.W. , Phong, W.Y. , Cliff, P. , Ng, S.H. , et al (2010) Nitrate respiration protects hypoxic Mycobacterium tuberculosis against acid‐ and reactive nitrogen species stresses. PLoS ONE 5: e13356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdóttir, H. , Robinson, J.T. , and Mesirov, J.P. (2013) Integrative Genomics Viewer (IGV): High‐performance genomics data visualization and exploration. Brief Bioinform 14: 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiffert, Y. , Supra, P. , Wurm, R. , Wohlleben, W. , Wagner, R. , and Reuther, J. (2008) The Streptomyces coelicolor GlnR regulon: Identification of new GlnR targets and evidence for a central role of GlnR in nitrogen metabolism in actinomycetes. Mol Microbiol 67: 861–880. [DOI] [PubMed] [Google Scholar]
- Tiffert, Y. , Franz‐Wachtel, M. , Fladerer, C. , Nordheim, A. , Reuther, J. , Wohlleben, W. , and Mast, Y. (2011) Proteomic analysis of the GlnR‐mediated response to nitrogen limitation in Streptomyces coelicolor M145. Appl Microbiol Biotechnol 89: 1149–1159. [DOI] [PubMed] [Google Scholar]
- Voskuil, M.I. , Schnappinger, D. , Visconti, K.C. , Harrell, M.I. , Dolganov, G.M. , Sherman, D.R. , and Schoolnik, G.K. (2003) Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J Exp Med 198: 705–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voskuil, M.I. , Visconti, K.C. , and Schoolnik, G.K. (2004) Mycobacterium tuberculosis gene expression during adaptation to stationary phase and low‐oxygen dormancy. Tuberculosis 84: 218–227. [DOI] [PubMed] [Google Scholar]
- Voskuil, M.I. , Bartek, I.L. , Visconti, K. , and Schoolnik, G.K. (2011) The response of Mycobacterium tuberculosis to reactive oxygen and nitrogen species. Front Microbiol 2: 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, K.J. , Bennett, M.H. , Barton, G.R. , Jenkins, V.A. , and Robertson, B.D. (2013) Adenylylation of mycobacterial Glnk (PII) protein is induced by nitrogen limitation. Tuberculosis 93: 198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, K.J. , Bryant, W.A. , Jenkins, V.A. , Barton, G.R. , Witney, A.A. , Pinney, J.W. , and Robertson, B.D. (2013) Deciphering the response of Mycobacterium smegmatis to nitrogen stress using bipartite active modules. BMC Genomics 14: 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer, D.P. , Soupene, E. , Lee, H.L. , Wendisch, V.F. , Khodursky, A.B. , Peter, B.J. , et al (2000) Nitrogen regulatory protein C‐controlled genes of Escherichia coli: Scavenging as a defense against nitrogen limitation. Proc Natl Acad Sci USA 97: 14674–14679. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information