

Summary
Urinary tract infections (UTI) are among the most common infections in humans. Uropathogenic E scherichia coli (UPEC) can invade and replicate within bladder epithelial cells, and some UPEC strains can also survive within macrophages. To understand the UPEC transcriptional programme associated with intramacrophage survival, we performed host–pathogen co‐transcriptome analyses using RNA sequencing. Mouse bone marrow‐derived macrophages (BMMs) were challenged over a 24 h time course with two UPEC reference strains that possess contrasting intramacrophage phenotypes: UTI89, which survives in BMMs, and 83972, which is killed by BMMs. Neither of these strains caused significant BMM cell death at the low multiplicity of infection that was used in this study. We developed an effective computational framework that simultaneously separated, annotated and quantified the mammalian and bacterial transcriptomes. Bone marrow‐derived macrophages responded to the two UPEC strains with a broadly similar gene expression programme. In contrast, the transcriptional responses of the UPEC strains diverged markedly from each other. We identified UTI89 genes up‐regulated at 24 h post‐infection, and hypothesized that some may contribute to intramacrophage survival. Indeed, we showed that deletion of one such gene (pspA) significantly reduced UTI89 survival within BMMs. Our study provides a technological framework for simultaneously capturing global changes at the transcriptional level in co‐cultures, and has generated new insights into the mechanisms that UPEC use to persist within the intramacrophage environment.
Introduction
Urinary tract infections (UTIs) represent one of the most significant community‐acquired and healthcare‐associated diseases (Foxman, 2010; Horvath et al., 2012). Uncomplicated UTIs result in more than 14 million medical visits and account for almost $4 billion in medical expenditure each year in the USA alone (Salvatore et al., 2011). Approximately 50% of women will experience a UTI at some point in their life, with almost 25% of patients experiencing a recurrence within the first 6 months following treatment of the initial UTI (Salvatore et al., 2011). An estimated 68% of recurrent UTIs arise from the same bacterial strain that caused the initial infection (Hunstad and Justice, 2010). Uropathogenic Escherichia coli (UPEC) is the most common causative agent of UTIs, being responsible for ∼ 80% of all community‐acquired infections (Foxman, 2010). In the majority of acute, uncomplicated UPEC‐mediated UTIs, single cultured isolates are diagnostic of the infection (Willner et al., 2014).
UPEC employ a range of virulence factors, including adhesins, toxins and iron acquisition systems to colonize the urinary tract and cause disease (Totsika et al., 2012; Ulett et al., 2013). Different UPEC strains display extensive genetic diversity owing to the presence of mobile DNA elements such as ‘pathogenicity islands’, prophages and plasmids (Hacker and Kaper, 2000; Mysorekar and Hultgren, 2006; Wiles et al., 2008; Hunstad and Justice, 2010; Hannan et al., 2012). The UPEC strains UTI89 (Mulvey et al., 2001) and 83972 (Lindberg et al., 1975c; Klemm et al., 2007; Zdziarski et al., 2010) are representative of cystitis and asymptomatic bacteriuria (ABU) isolates respectively.
Acute pyelonephritis and ABU represent the two extremes of UTI. Acute pyelonephritis is a severe, acute systemic infection caused by UPEC strains containing virulence genes clustered on pathogenicity islands (Eden et al., 1976; Funfstuck et al., 1986; Stenqvist et al., 1987; Orskov et al., 1988; Johnson, 1991; Welch et al., 2002). Asymptomatic bacteriuria, on the other hand, is an asymptomatic carrier state that resembles commensalism. A single E. coli strain may be present in ABU patients at levels of more than 105 colony‐forming units (CFU) ml−1 for months or years without provoking a host response. Because the majority of ABU‐associated E. coli strains are non‐haemolytic, non‐adherent and lack hemagglutination ability, early studies suggested that this behavior reflected a lack of virulence genes (Lindberg, 1975; Lindberg et al., 1975a, 1975b, 1975c; Eden et al., 1976; Kaijser and Ahlstedt, 1977). Molecular epidemiology has shown, however, that many ABU strains carry virulence genes despite failing to express the phenotype (Plos et al., 1990; 1995; Mabbett et al., 2009).
As with all infectious agents, UPEC must overcome innate immunity, a biological system compromising both cellular mediators (e.g. neutrophils) and soluble mediators (e.g. complement proteins) that act synergistically. Several studies have investigated the role of neutrophils in UPEC‐mediated pathology (Ingersoll et al., 2008; Sivick and Mobley, 2010; Lau et al., 2012; Tourneur et al., 2013), whereas there is a paucity of information on the interactions between UPEC and macrophages, another key cellular component of innate immunity (Tegner et al., 2006). We previously demonstrated that the ability of UPEC to survive in mouse macrophages differs markedly between different strains; some strains, such as UTI89, are able to survive over a 24 h infection period, whereas others, such as 83972, are rapidly killed (Bokil et al., 2011). This suggests that UPEC strains like UTI89 are able to subvert macrophage antimicrobial pathways, though the mechanisms responsible are still unknown. These findings are in keeping with a larger body of literature documenting intraepithelial cell survival of some UPEC strains, both in vitro and in vivo (Hunstad and Justice, 2010).
Next‐generation sequencing technologies provide a powerful approach for studying co‐transcriptomics during infection (‘t Hoen et al., 2008; Hegedus et al., 2009; Jager et al., 2009; Xiang et al., 2010; Huang et al., 2012; Nie et al., 2012; Wang et al., 2012). These direct sequencing methodologies allow the measurement of millions of RNA transcripts in a sample, thus enabling identification of global differences in gene expression under different growth conditions (Morozova and Marra, 2008; Wang et al., 2009). RNA sequencing generates information about absolute transcript levels, avoiding many of the limitations of microarrays (‘t Hoen et al., 2008; Llorens et al., 2011). To further understand the transcriptional programmes that are simultaneously activated during host–pathogen interaction, we developed an approach for isolating total RNA from co‐cultures and analysing the simultaneous changes in expression that take place in the interacting organisms. Previous studies (Hegedus et al., 2009; Xiang et al., 2010; Xiao et al., 2010; Ordas et al., 2011) have focused on either the host or the pathogen, without revealing simultaneous co‐transcriptomic changes that occur during infection. Such methods have also relied on the isolation of species‐specific RNA, which can introduce biases in the analysis.
To gain insights into novel strategies used by UPEC to subvert macrophage antimicrobial responses, we performed a global co‐transcriptomic analysis of UPEC gene expression within murine bone marrow‐derived macrophages (BMMs). We used RNA sequencing to monitor (i) the transcriptional responses of UPEC strains UTI89 and 83972 within the intramacrophage environment across an extended time course, and (ii) differences in macrophage gene expression responses to each strain. Our comprehensive approach has generated new insights into host–pathogen interactions and the possible consequences of these interactions for disease processes.
Results
Analysis of digital gene expression libraries
The UPEC strains UTI89 and 83972 display contrasting intramacrophage survival phenotypes; UTI89 is able to survive in significant numbers, whereas 83972 is rapidly killed (Bokil et al., 2011). To examine the molecular basis for this difference, we investigated the gene expression profiles of UTI89 and 83972 in BMMs in parallel over a 24 h period. For our experimental system, we used a multiplicity of infection (MOI) of 10:1, since this MOI does not have obvious effects on BMM cell viability for either of the strains. As expected, bacterial loads of UTI89 that were recovered from BMM were substantially higher than those of 83972 (Supporting Information Fig. S1). Total RNA was harvested at 2, 4 and 24 h post‐infection (hpi) and global gene expression profiles were analysed using the Illumina Hi‐Seq 2000 Digital Gene Expression Tag Profiling Kit, a tag‐based transcriptome sequencing method. cDNA libraries were prepared from gentamicin‐treated UPEC‐BMM co‐cultures, sequenced and analysed together with bacterial and BMM control samples. The RNA‐Seq files generated for all libraries were pre‐processed by a custom java script (Supporting Information Table S1).
Mapping RNA‐Seq reads
Alignment of sequencing reads to the respective mouse and UPEC reference genomes was performed using TopHat. Sequences were mapped against their respective Mus musculus or E. coli genomes. The resulting BAM files were further used to compute alignment statistics for all libraries employing the samtool flagstat command. These processes are summarized in Supporting Information Table S2. As expected, very few sequence reads were captured from 83972‐BMM co‐cultures at 24 hpi, consistent with the observation that these bacteria were essentially cleared by BMMs at this time point. Hence, subsequent analyses of gene expression in 83972 excluded this specific condition.
BMM transcriptional responses to UPEC
To explore transcriptional relationships between different conditions, we performed dimensionality reduction analysis using minimum curvilinearity embedding (MCE; Cannistraci et al., 2010; 2013). This analysis revealed that the BMM genes whose expression was regulated by infection were not markedly different between the two different UPEC strains at the initial stage, 2 hpi. However, after 4 hpi, a slight difference emerged, which was also maintained at 24 hpi. As expected, however, we observed that the time‐dependent regulation of gene expression (Fig. 1A) was the main pattern that emerged from the data, indicating that the macrophage transcriptional response follows a distinct temporal profile that is common to infection by both strains, and much more distinctive than the difference between the two strains.
Figure 1.
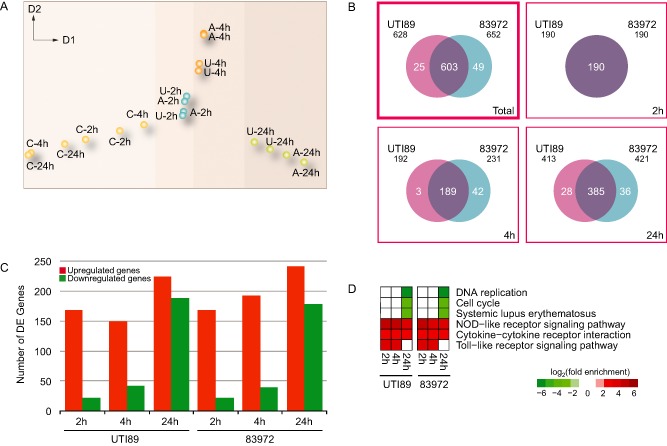
BMM transcriptome analysis.
A. MCE plot of the relationships between conditions for the BMM gene sets. Each sample from two independent biological replicates is represented as a dot in a two‐dimensional space (C, control; U, UTI89; A, 83972).
B. Venn diagrams quantifying the overlap in the response of BMMs to the two UPEC strains for two independent biological replicates. The numbers of DEG are shown for the total response (top left), as well as for each of the three time points (2, 4 and 24 hpi).
C. Histogram of the regulation of BMM DEG showing the numbers of up‐ (red) and down‐regulated (green) genes during the 24 h infection time course.
D. Pathways activated in BMMs during the course of UPEC infection. Each coloured square in the matrix represents a significant fold enrichment (log2) of the respective pathway term at each point. Red, up‐regulated DEG; green, down‐regulated DEG.
To gain further insights into the global transcriptional changes that take place in UPEC‐infected macrophages, we catalogued differentially expressed genes (DEG) in normalized digital gene expression data through pairwise comparisons between controls (uninfected BMM cultures) and treatments (co‐cultures of UPEC‐infected BMMs) using a previously described method (Trapnell et al., 2012) with a threshold of a false‐discovery rate (FDR)‐adjusted P‐value < 0.01 in at least one of the pairwise comparisons. Using this approach, we identified 628 and 652 BMM genes that were differentially regulated following infection with UTI89 or 83972, respectively, over a 24‐h infection time course. Further analyses of the BMM transcriptome identified 603 genes that were commonly regulated by UTI89 and 83972, as well as 25 and 49 BMM genes that were differentially regulated after infection with UTI89 or 83972 respectively (Fig. 1B). The greatest divergence in responses to each strain occurred at the latest time point post‐infection.
To investigate the regulatory patterns of divergently expressed (DE) genes, we clustered genes as either up or down‐regulated. This analysis revealed a rapid response for up‐regulated genes that was maintained throughout the infection time course. In contrast, the number of down‐regulated genes was much lower at 2 and 4 hpi, but increased markedly by 24 hpi (Fig. 1C). The substantial overlap in DEG, as well as their conserved pattern of regulation, suggests that the macrophage transcriptional response to UTI89 and 83972 is broadly conserved.
Functional annotation of DE macrophage genes
The consequences of gene expression changes associated with UPEC infection were characterized by gene ontology (GO) and pathway (KEGG) enrichment analyses of DEG using the DAVID program (da Huang et al., 2009a, 2009b). As shown in Supporting Information Fig. S2A, common highly enriched GO categories for up‐regulated genes included activation of inflammatory responses; regulation of chemokine, cytokine and interleukin‐6 and ‐12 production; T‐cell activation; and regulation of transcription factor (TF) activity. Common biological processes associated with down‐regulated genes included DNA replication initiation, DNA packaging, nucleosome assembly and organization, and cytokinesis. These pathways are consistent with the known activation of innate immune responses by bacterial challenge (Rosenberger and Finlay, 2003; Mogensen, 2009; Portt et al., 2011). As expected, the signalling pathways that were inferred, on the basis of gene expression changes, to be regulated in macrophages upon UPEC infection showed common characteristics between both UPEC strains. Signalling pathways associated with up‐regulated genes included cytokine–cytokine receptor interaction, NOD‐like receptor signalling and Toll‐like receptor (TLR) signalling pathways (Fig. 1D).
We next independently validated both conservation and divergence in macrophage responses to UTI89 versus 83972. Many of the well‐validated TLR target genes such as cytokines (Il1a, Il1b, Il16) and chemokines (Cxcl1, Ccl8) were similarly inducible by both UTI89 and 83972 in BMM (data not shown). Although previous studies have demonstrated pathological roles for extracellular histones in mouse models of sepsis (Xu et al., 2009; 2011), there is little known about the regulation of histone gene expression downstream of TLR signalling. This may reflect the fact that canonical histone mRNAs are not poly‐adenylated, and so their regulated expression may not be captured by traditional microarray approaches. Interestingly, we found that a large suite of histone genes were down‐regulated in response to infection with either UPEC strain (Fig. 2A), consistent with the inhibitory effect of TLR signalling on macrophage proliferation. This observation was validated for several individual histone genes using quantitative reverse transcription‐polymerase chain reaction (RT‐qPCR; Fig. 2B). Our attempts to validate the small number of macrophage genes differentially regulated by UTI89 and 83972 (Fig. 1B) using qPCR were less successful; however, we did confirm differential regulation of the cystine/glutamate exchanger Slc7a11. Whereas Slc7a11 mRNA expression was similarly up‐regulated by UTI89 and 83972 at 4 hpi in infected BMM, its expression remained elevated at 24 hpi in UTI89‐infected BMMs but was significantly reduced at this time point in 83972‐infected BMMs (Fig. 2C).
Figure 2.
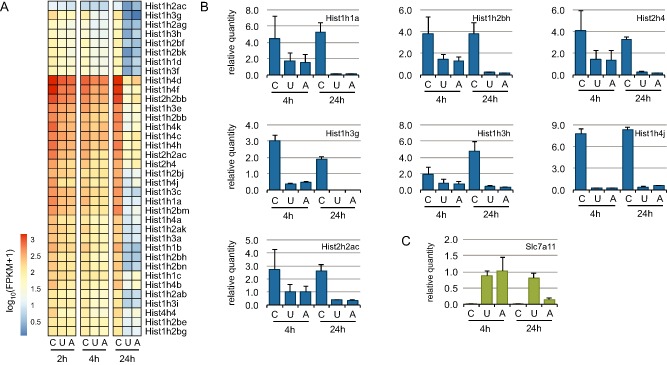
Gene regulation in BMM.
A. Heat map summarizing the expression profiles of histone genes from the RNA‐Seq libraries. The values are log‐transformed FPKM counts.
B. Bar plots showing the relative mRNA levels of selected histone candidate genes determined by RT‐qPCR. qPCR data represent means relative expression ± range (n = 2 independent experiments).
C. Bar plot showing the mean relative levels of the mRNA for Slc7a11, as determined by RT‐qPCR. Error bars denote the range of the two biological replicates (C, control; U, UTI89; A, 83972).
Clustering analysis separated DE macrophage genes into two major clusters (Fig. 3A). Cluster 1 (Fig. 3B), which contained 460 genes, was positively correlated with the profiles of the TFs Arnt (aryl hydrocarbon receptor nuclear translocator), Myc and Pparg (peroxisome proliferator‐activated receptor gamma); and negatively correlated with the profiles of Hif1a (hypoxia‐inducible factor 1‐alpha) and Stat3 (signal transducer and activator of transcription 3); and enriched for pathways associated with DNA replication, cell cycle and systemic lupus erythematosus. Cluster 2 (Fig. 3B), which contained 217 genes, was positively correlated with the profiles of the TFs Hif1a, Nfkb2 (nuclear factor of kappa light polypeptide gene enhancer in B cells 2), Nr3c1 (nuclear receptor subfamily 3, group C, member 1) and Stat3; negatively correlated with the profile of Myc; and enriched for pathways associated with cytokine–cytokine receptor interaction and NOD‐like receptor, TLR, chemokine and Jak‐STAT signalling. These results are in keeping with expectations; TLR signalling activates NF‐κB and HIF (Rossol et al., 2011) and inactivates CSF‐1 signalling. Various studies link the biology of CSF‐1, Myc and Pparg (Dey et al., 2000; Bonfield et al., 2008; Pello et al., 2012), and as expected, down‐regulated genes have an association with Myc and Pparg. That is, TLR signalling switches off signalling via CSF‐1, which itself can signal, in part, by Myc; hence, TLR signalling down‐regulates Myc responses. Apart from the expected patterns, our analysis suggests a potential association between inactivation of CSF‐1 signalling and Arnt (partner for Hif) responses, which has not been reported previously.
Figure 3.
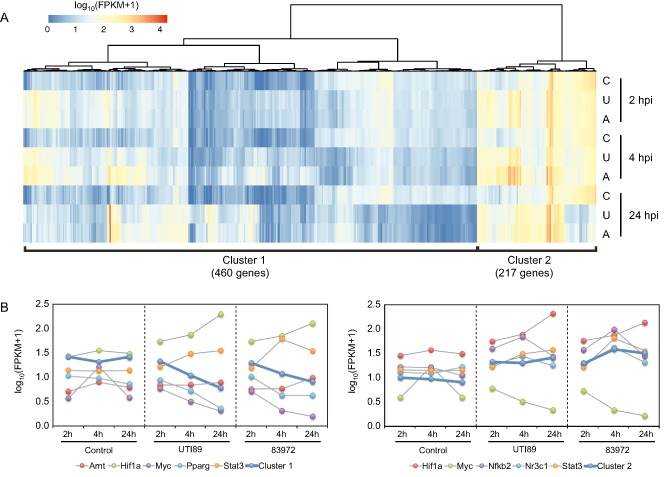
Transcription factors associated with DE BMM genes.
A. Heat map showing results of k‐means clustering of BMM DEG. The values are log‐transformed FPKM counts for all DEG across all RNA‐Seq libraries in the dataset.
B. Expression profiles of TFs associated with binding motifs from the TFBS analysis that are highly correlated with each cluster. Each TF is represented with a different colour, whereas the cluster mean expression is coloured blue. All values are log‐transformed FPKM counts. C, control; U, UTI89; A, 83972.
UPEC transcriptional responses upon infection of BMMs
As was the case with the macrophage gene expression analysis, we initially investigated transcriptional responses in both UPEC strains by performing dimensionality reduction analysis using MCE (Cannistraci et al., 2010; 2013). We identified specific differences in the regulated expression of UPEC genes within the intramacrophage environment (Fig. 4A), which likely reflects the differential pathogenicity and capacity for intramacrophage survival of both strains (Supporting Information Fig. S1; Bokil et al., 2011).
Figure 4.

UPEC transcriptome analysis.
A. MCE plot of the relationships between conditions for the UPEC gene sets. Each sample from two independent biological replicates is represented as a dot in a two‐dimensional space (C, control; U, UTI89; A, 83972).
B. Venn diagrams quantifying the overlap in the response of the UPEC strains in the intramacrophage environment for two independent biological replicates. The numbers of DEG for the total response (top left), as well as for each of the three time points (2, 4 and 24 hpi) are shown.
C. Histogram of the regulation of UPEC DEG showing the numbers of up‐ (red) and down‐regulated (green) genes during the 24‐h infection time.
D. Pathways activated in UPEC during the course of infection. Each coloured square in the matrix represents significant fold enrichment (log2) of the respective pathway term at each point. Red, up‐regulated DEG; green, down‐regulated DEG.
To gain insights into the global transcriptional changes that occur in UPEC during macrophage infection, we applied the same method described above and performed pairwise comparisons between controls (UPEC cultures alone) and treatments (co‐cultures of UPEC‐infected BMMs). Again, we employed an FDR‐adjusted P‐value < 0.01 in at least one of the pairwise comparisons in our dataset as the threshold for differential expression. In total, we identified 137 UTI89 genes and 33 83972 genes that were differentially regulated in BMMs. Surprisingly, an analysis of the intramacrophage transcriptomic profile identified only 13 regulated genes that were common to both strains, whereas 124 genes were uniquely regulated in UTI89, and 20 genes were uniquely regulated in 83972 (Fig. 4B). The UPEC transcriptional profiles also revealed a number of strain‐specific responses; a substantial number of UTI89 genes were up‐regulated at 2 and 4 hpi, whereas a much smaller number of 83972 genes were up‐regulated at these time points (Fig. 4C). This is consistent with the initiation of a UTI89 transcriptional programme that permits intramacrophage survival. In contrast to the transcriptional responses observed for macrophage genes, few UPEC genes were down‐regulated over the infection time course.
Functional annotation of DE UPEC genes
The consequences of gene expression changes associated with UPEC infection were characterized by GO and pathway (KEGG) enrichment analyses of DEG using the DAVID program (da Huang et al., 2009a, 2009b). This again provided clear evidence of strain‐specific biological responses within the intramacrophage environment (Supporting Information Fig. S2B). Highly enriched GO categories associated with the up‐regulated transcriptional programme included genes associated with chemotaxis and motility in UTI89 and genes involved in protein folding in 83972. We did not identify any significantly enriched GO categories associated with down‐regulated genes in common for the two strains. In keeping with the above findings, an analysis of the pathways activated during UTI89 infection revealed enrichment for pathway terms associated with bacterial chemotaxis and flagellar biosynthesis (Fig. 4D). We did not observe significant enrichment for any pathway term in the case of 83972.
UPEC genes associated with intramacrophage survival
We hypothesized that genes selectively up‐regulated by UTI89 may contribute to intramacrophage survival. Such genes included those encoding flagella and those associated with protection against oxidative stress. Notably, several genes encoding flagella‐related proteins were uniquely regulated in UTI89; these DE flagella genes included flgA‐F, flgK, flgL, fliC‐E, fliQ, motA and motB. Figure 5A shows the expression patterns of all flagella‐related genes in UTI89 versus 83972, revealing strong up‐regulation at 2 h with a subsequent gradual decrease over time.
Figure 5.
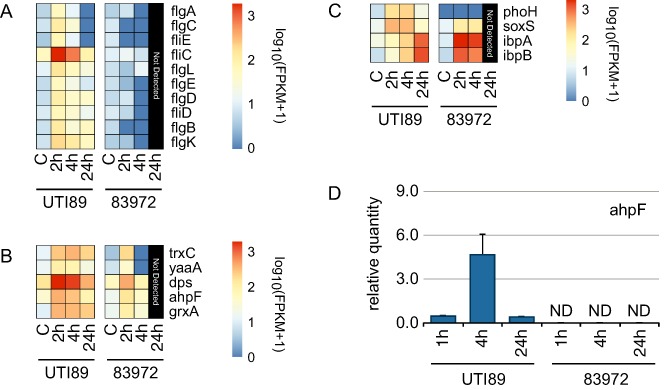
Regulation of UPEC flagella, OxyR regulon and hydrogen peroxide‐induced genes in the intramacrophage environment.
A–C. Heat maps summarizing the RNA‐Seq‐derived expression profiles of flagellar genes (A), OxyR regulon genes (B) and hydrogen peroxide‐induced genes (C). All values are log‐transformed FPKM counts.
D. Bar plot showing the relative mRNA levels of ahpF, as determined by RT‐qPCR. qPCR data represent mean relative expression ± range (n = 2) of two biological replicates; ND, not detected.
A common mechanism used by bacterial pathogens to avoid host innate immune pathways is to employ defence mechanisms against oxidative stress (Imlay, 2013). We therefore clustered the expression patterns of all the OxyR regulon genes (Fig. 5B) and the most strongly hydrogen peroxide‐induced genes in both UPEC strains (Fig. 5C). Interestingly, the OxyR regulon, which included alkyl hydroperoxide reductase subunit F (ahpF), dps (DNA starvation/stationary phase protection protein), grxA (glutaredoxin 1), trxC (thioredoxin 2) and yaaA, was strongly up‐regulated in UTI89, with the effect being apparent at 2 hpi and peaking at 4 hpi. Hydrogen peroxide‐inducible genes, which included ahpF, dps, grxA, heat shock proteins/chaperones (ibpA, ibpB), phoH, soxS (DNA‐binding transcriptional dual regulator), trxC and yaaA showed a similar pattern, with genes being significantly up‐regulated in UTI89 and less so in 83972. Differential regulation of ahpF in UTI89 versus 83972 within macrophages was validated by RT‐qPCR (Fig. 5D).
Although UTI89 persists at 24 h within macrophages, only a relatively small percentage of bacteria (< 5%) survive within BMM at 24 hpi compared with the bacterial loads at 2 hpi (Supporting Information Fig. S1). We therefore reasoned that changes in UPEC gene expression at 24 hpi might be linked to intramacrophage survival. We identified 22 genes that were highly up‐regulated (> 3‐fold) by UTI89 at 24 hpi (Fig. 6A). The most highly expressed of these included those encoding ibpB (encoding a small heat shock protein); pspACDE (encoding the phage‐shock protein system); rpoE and rpoH (encoding sigma factors); smpA (encoding outer membrane lipoprotein); yadR (encoding the iron‐sulphur cluster insertion protein ErpA); yceP (encoding the biofilm formation regulatory protein BssS); yebG (encoding DNA damage‐inducible protein); and UTI89_C2624 and UTI89_C5162‐3 (encoding proteins of unknown function).
Figure 6.
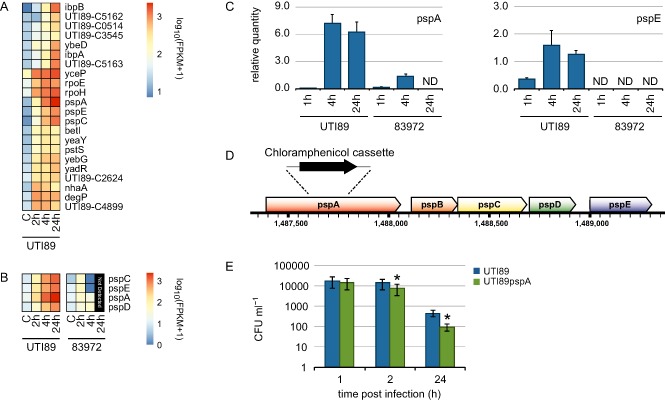
UPEC genes associated with intramacrophage survival.
A and B. Heat maps summarizing the RNA‐Seq‐derived expression profiles of UTI89 genes elevated at 24 hpi (A) and UPEC Psp genes (B).
C. Bar plots showing the relative quantity of pspA and pspE mRNA, as determined by RT‐qPCR. qPCR data represent mean relative expression ± range (n = 2) of two biological replicates; ND, not detected.
D. Insertion site for creation of UTI89 pspA mutant.
E. Intramacrophage survival of UTI89 and UTI89pspA. BMMs were infected at an MOI of 10 and intracellular bacterial survival was assessed at 1, 2 and 24 h of infection. Data are compiled from three independent experiments, and show mean ± standard deviation (*P < 0.05).
Phage‐shock‐protein (Psp)‐related genes, which are required for bacterial survival during extracytoplasmic stress responses and changes in pH (Darwin, 2013), were significantly up‐regulated in UTI89 compared with 83972 (Fig. 6B). Reverse transcription‐polymerase chain reaction confirmed the elevated expression of pspA and pspE in UTI89 compared with 83972 at 24 h post‐infection (Fig. 6C). Finally, we validated the impact of pspA for intramacrophage survival of UTI89 by constructing a UTI89pspA mutant (Fig. 6D), and testing it for intramacrophage survival during a 24 h infection time course in BMM. In this assay, the UTI89pspA mutant was significantly reduced for intracellular survival compared with the wild‐type strain (Fig. 6E).
Discussion
In this study, we determined the co‐transcriptomic programme of UPEC‐infected primary mouse macrophages during an infection time course. The use of two UPEC strains that differed in their ability to survive in these cells enabled the identification of both common and UPEC strain‐specific responses. To our knowledge, this is one of the first RNA‐Seq studies that have simultaneously measured the transcriptomes of both the host and pathogen during an infection (Humphrys et al., 2013). Using only open‐source tools, we developed a computational framework that was capable of successfully separating, annotating and quantifying the mammalian and bacterial transcriptomes. Whereas previous studies (Mysorekar et al., 2002; Bower et al., 2009; Hagan et al., 2010; Duell et al., 2012) have been limited to microarrays, the RNA‐Seq analysis reported here provides a more sensitive, comprehensive, and unbiased coverage of the entire transcriptome. We achieved an average sequencing depth of approximately 24 million tags per library and identified 677 BMM and 157 UPEC genes that were differentially expressed following UPEC infection. By mapping our RNA‐Seq tag data onto transcript databases and genomic sequences, we were able to identify genes that were regulated upon UPEC challenge.
Minimum curvilinearity embedding confirmed the distinct temporal cascade of BMM responses to UPEC as well as differences in transcriptional programmes of the two UPEC strains. The sets of DEG and regulatory processes and pathways identified in both BMMs and UPEC strains suggest coordinated expression and mutual influences. Our study detected 603 DEG in BMMs that were common to infection with UTI89 and 83972, accounting for the vast majority of changes in BMM gene expression. This was not unexpected, given that both UTI89 and 83972 present pathogen‐associated molecular patterns, such as lipopolysaccharide that is recognized by TLR4. Macrophage‐expressed genes that showed differences in regulation in response to these two strains were a very minor component of the total signature and their significance remains to be determined. Despite the general conservation of the BMM response against UPEC infection, we nonetheless expected to detect some selective BMM responses at 24 hpi for UTI89 compared with 83972, since the former condition reflects macrophages that are continuing to cope with intracellular UPEC, whereas the latter corresponds to macrophages that have cleared the infection. Consistent with this supposition, we found that at 24 hpi, BMM responses showed a greater divergence between UTI89 and 83972 (47.8% overlap) compared with earlier time points where this difference was not as evident. Of interest in this regard, was our validation of differential regulation of the cationic amino acid cysteine/glutamate antiporter Slc7a11, which is required for glutamate uptake and glutathione synthesis (Bannai, 1986; Hayes and McLellan, 1999; Pompella et al., 2003; Shih et al., 2006). This gene showed a sustained up‐regulation at 24 hpi following infection with UTI89, whereas it was only transiently up‐regulated after infection with 83972. Given that macrophage antimicrobial responses typically involve subjecting pathogens to oxidative stress, the sustained expression of Slc7a11 upon infection with UTI89 may be required to maintain glutathione levels and cytoprotection during stress responses.
Our data is consistent with previous reports showing inducible cytokine and chemokine expression, as well as activation of pro‐survival pathways (Sester et al., 1999; 2006), during bacterial infection and/or TLR stimulation of macrophages. Moreover, the down‐regulation of DNA replication and cell cycle genes (Fig. 1D) is consistent with the known inhibitory effects of TLR4 signalling on proliferation of cycling macrophages (Sester et al., 1999; 2006). We also found that the genes encoding histones H1, H2, and H4 were dramatically down‐regulated at 24 hpi, and we further confirmed the regulated expression of several of these using RT‐qPCR. A member of the H2A histone family, Hist2h2aa1, was also significantly down‐regulated at 24 hpi in vivo in mouse bladder colonized with UPEC CFT073 (Tan et al., 2012), which is consistent with these findings. Interestingly, previous studies have reported a pathological role for extracellular histones during LPS‐induced septic shock (Xu et al., 2009; Li et al., 2011). Xu et al. (2011) revealed that antibodies against extracellular histones rescued animals from LPS‐mediated death (Xu et al., 2011), and a previous study showed that extracellular histones mediate endothelial dysfunction, organ failure and death during sepsis (Semeraro et al., 2011). Our findings could thus either reflect a host attempt to reduce inflammatory responses upon cell death and histone release, or the consequence of growth‐inhibitory effects of TLR agonists on proliferating macrophages, which would also be expected to lead to down‐regulated histone expression.
A transcription factor‐binding site (TFBS) analysis of the promoter sequences of the two BMM DE gene clusters further showed significant enrichment of seven motifs associated with TFs that have key roles in macrophage functions. The cluster containing inducible inflammation‐related genes correlated positively with the expression of Hif1a, a key pro‐inflammatory transcription factor that drives macrophage inflammatory responses and is up‐regulated in UPEC‐infected mouse bladder at 2 hpi and 24 hpi (Duell et al., 2012; Tan et al., 2012); Nfkb2, which is activated by a wide variety of stimuli such as cytokines, oxidant‐free radicals and bacterial or viral products; the glucocorticoid receptor Nr3c1, which up‐regulates the expression of anti‐inflammatory genes and/or represses the expression of pro‐inflammatory genes; and Stat3, a transcriptional activator stimulated in response to cytokines and growth factors. In vivo, both Nfkb2 and Stat3 are immediately up‐regulated in the bladder following UPEC infection (Duell et al., 2012). On the other hand, the cluster containing down‐regulated cell cycle‐related genes correlated positively with the expression of Arnt, which is involved in the induction of several enzymes that participate in xenobiotic metabolism; Myc, which is required for cell proliferation in response to mitogenic stimuli such as CSF‐1; and Pparg, which regulates fatty acid storage and glucose metabolism. Myc, Pparg and Csf1 are all differentially expressed in bladders of mice infected with UPEC (Duell et al., 2012) supporting the in vivo relevance of these findings.
In contrast to the general conserved pattern of BMM responses to both UPEC strains, the individual transcriptional responses of the two strains in BMMs differed markedly. This is consistent with their contrasting survival patterns in macrophages. Minimum curvilinearity embedding confirmed the different transcriptional programmes of the two UPEC strains, revealing 124 UTI89‐specific DEG, 20 83972‐specific DEG and 13 DEG that were commonly regulated in both pathogens upon infection of BMMs. Given the different survival patterns of both UPEC strains in macrophages, we expected to detect increased expression of genes specific to UTI89 within macrophages at 24 hpi.
Previous studies have shown that flagella contribute to the virulence of a number of pathogenic species (Tomich et al., 2002). Some bacteria, for example, Salmonella enterica Typhimurium and Yersinia enterocolitica, use flagella for invasion of epithelial cells (McNally et al., 2007; Ibarra et al., 2010). Uropathogenic Escherichia coli also employ flagella for the invasion of renal collecting duct cells (Pichon et al., 2009), but flagella‐enhanced uptake of UPEC by macrophages has not been investigated. Our co‐transcriptomic analysis revealed the up‐regulation of multiple genes associated with flagella biosynthesis. This could reflect inducible gene expression in the intramacrophage environment or selective uptake by macrophages of a sub‐population of flagella‐expressing UPEC, which would generate a similar gene expression profile. The acute up‐regulation at 2 hpi and subsequent down‐regulation at 24 hpi would suggest the latter may be the case.
Murine macrophages employ both rapidly produced reactive oxygen species (ROS) and reactive nitrogen species (RNS), produced in a delayed fashion, as bacterial clearance strategies (Flannagan et al., 2009). The observation that UPEC strain UTI89 survives in macrophages suggests that it can overcome these pathways. Although the genes that respond to ROS have been well characterized in E. coli K‐12, their expression during UPEC infection and their role in intramacrophage survival have not yet been studied. Our transcriptomic approach identified UPEC genes that are likely involved in defence against ROS and RNS. Disturbances in the normal redox state of cells can cause toxic effects through the production of peroxides and free radicals that damage all components of the cell, including proteins, lipids and DNA. An earlier study confirmed that the peroxide response regulator OxyR activates most of the genes that are highly induced by hydrogen peroxide (Zheng et al., 2001). Our results show that the response of UTI89 to oxidative stress (up‐regulated expression of dps, grxA, ahpF, soxS, trxC, ibpA and ibpB) is more robust than that of 83972. These differences imply that part of the UTI89 intramacrophage survival strategy includes robust protection against oxidative stress.
Several UTI89 genes were up‐regulated in BMM at 24 hpi, and we selected genes encoding the Psp system for further functional analysis. The Psp system responds to extracytoplasmic stress and contributes to the virulence of several pathogens, including Y. enterocolitica and S. Typhimurium (Karlinsey et al., 2010; Yamaguchi and Darwin, 2012). PspA, the master effector of the Psp system, mediates its response via a dual mechanism: (i) binding to the transcriptional regulator PspF and preventing it from activating the transcription of pspACDE in the absence of extracytoplasmic stress, and (ii) binding to the cytoplasmic membrane‐localized proteins PspB and PspC in the presence of extracytoplasmic stress, thus releasing PspF to induce pspACDE transcription (Yamaguchi et al., 2013). We confirmed a role for the Psp system in UPEC by constructing a pspA mutant (UTI89pspA) and demonstrating reduced intramacrophage survival of this mutant. Despite this statistically significant reduction in survival within macrophages, the biological significance of this effect is at this stage unknown. Inactivation of other stress response systems on the pspA mutant background may be required to reveal a more striking phenotype.
In summary, we have demonstrated the capacity to employ co‐transcriptomics to study host–pathogen interactions. Our novel approach revealed new insights into the mechanisms used by UPEC to avoid macrophage responses and persist in the intramacrophage environment, and has identified multiple target genes for further functional studies. Finally, this manuscript is one of the first to successfully evaluate the expression profiles of two organisms from the same sample using RNA sequencing, and will make an excellent resource for other studies that aim to perform similar analyses in different host–pathogen systems.
Experimental procedures
TIER I: in vitro infection assays
Ethics statement and animal experimentation
A University of Queensland institutional animal ethics committee approved all animal experimentations. Female C57BL/6 mice (6–8 weeks old) were purchased from the Animal Resources Center, Australia. Murine BMMs were generated by the in vitro differentiation of bone marrow cells from C57BL/6 mice on bacteriological plastic plates in the presence of 10 000 U ml−1 recombinant human CSF‐1 (a gift from Chiron) for 6 days, after which cells were harvested and replated onto tissue culture plastic in the presence of CSF‐1 for infection on day 7. BMM were maintained at 37°C (5% CO2) in RPMI 1640 supplemented with 2 mM L‐glutamine (GlutaMAX), 10% heat‐inactivated fetal bovine serum and 50 U ml−1 penicillin and 50 μg ml−1 streptomycin (Life Technologies).
Culture of bacterial strains and infection of mouse BMMs
Escherichia coli UTI89 is a well‐characterized cystitis isolate (Mulvey et al., 2001). Escherichia coli 83972, which was carried without symptoms by a young female, was originally isolated from the urine of this individual (Andersson et al., 1991; Roos et al., 2006). For macrophage infection assays, UPEC strains were cultured statically in Luria‐Bertani (LB) broth at 37°C overnight. Type 1 fimbriae expression was assessed by yeast cell agglutination prior to infection as previously described (Schembri et al., 2000). Bacterial cells were centrifuged and washed in phosphate buffered saline (PBS), and then resuspended in antibiotic‐free media at a concentration of 2 × 108 CFU ml−1. Viable CFU counts of bacterial inocula were routinely confirmed in every infection assay by serial dilution and plating on LB agar.
Intramacrophage survival assays were performed essentially as described previously (Bokil et al., 2011). Briefly, following overnight adherence in antibiotic‐free media, BMMs were infected for 1 h with bacteria at an MOI of 10. Extracellular bacteria were killed by washing twice in 200 μg ml−1 gentamicin, followed by 1 h incubation in media containing the same gentamicin concentration. Subsequent exclusion of extracellular bacteria for the duration of the experiment was performed by incubation in 20 μg ml−1 gentamicin. At appropriate time points (1 h, 2 h and 24 h), cells were washed twice with antibiotic‐free media, and then lysed with PBS/0.01%Trition X‐100. Lysates were cultured on LB agar plates overnight at 37°C and colony counts were used to assess intracellular bacterial loads. For standard infection assays, complete exclusion of viable extracellular bacteria was confirmed by performing colony counts on culture supernatants.
TIER II: RNA preparation and sequencing
Macrophage infection and mRNA isolation, enrichment and purification
Control macrophages, control bacteria and infected macrophages were incubated for an additional 1, 3 and 23 h at 37°C in a humidified 5% CO2 atmosphere, after the initial 1 h infection. Cells were then washed twice with antibiotic‐free media, and then lysed on ice for RNA isolation and purification (RNeasy; Qiagen, Germantown, MD, USA). Microbial total RNA in co‐culture samples was enriched (MICROBEnrich, Ambion). rRNA was removed from all purified RNA samples using kits targeting mammalian and gram‐negative bacterial rRNAs (Ribo‐Zero; Epicenter, Madison, WI, USA). Prior to sequencing, all samples were further quantified and examined for protein and reagent contamination using a Nanodrop ND‐1000 spectrophotometer. RNA samples for analysis were selected based on a 28S/18S rRNA band intensity of 2:1, a spectroscopic A260/A280 nm ratio of 1.8–2.0 and an A260/A230 nm ratio > 1.5.
RNA sequencing
Next‐generation sequencing analyses were performed for two biological replicates on an Illumina Cluster Station and the Illumina HiSeq 2000 System using primarily reagents from the Illumina Gene Expression Sample Preparation Kit and the Illumina Sequencing Chip (Flowcell; Illumina, San Diego, CA, USA). Sequence tags were prepared using the Digital Gene Expression Tag Profiling Kit (Illumina), according to the Illumina protocol.
TIER III: data pre‐processing
Image analysis, base calling and quality calibration were performed using the Solexa Automated Pipeline. Quality control of RNA‐Seq reads were pre‐processed by a custom java script. All sequences generated have a length of 101 bases and have been submitted to the BioProject database of NCBI under BioProject ID: PRJNA256028.
TIER IV: alignment and differential gene expression analysis
Alignment of reads
Bowtie indexes were created for the mouse (version 37.1) and the two E. coli strains (UTI89 and 83972) using the bowtie‐build algorithm and reference sequences from the GenBank database. Our protocol began with mapping of the raw RNA‐Seq reads (fastq files) to the reference genomes using TopHat. TopHat uses Bowtie as an alignment engine and breaks up reads that Bowtie cannot align on its own into smaller pieces (Kim and Salzberg, 2011). Using the standard parameters, we mapped both reads of our paired end libraries. All simulations were performed using a 30 core, high‐memory node cluster system; total computation duration was 3 h.
Transcript annotation
After running TopHat, the resulting alignment files were provided to Cufflinks to generate a transcriptome assembly for each condition. During this analysis step, adapter tags; mitochondrial sequences; poly A, poly C and phiX sequences; and remaining ribosomal sequences were filtered out. Estimated normalized expression levels were reported in fragments (i.e. reads) per kilobase of exon per million mapped reads (FPKM). These assemblies were compared with annotation files using the Cuffcompare utility, which is included in the Cufflinks package (Roberts et al., 2011; Trapnell et al., 2012).
Differential expression analysis
The reads and assemblies were imported to Cuffdiff, which calculates expression levels and tests the statistical significance of observed changes. For comparison of DEG across samples, the number of raw clean tags in each library was normalized to FPKM using the Cufflinks package. The minimum number of alignments in a locus needed to test for significance of changes in that locus between samples was set to 50 fragment alignments. If no testing was performed, changes in the locus were deemed insignificant, and the changes observed in the locus did not contribute to corrections for multiple testing. The Cuffdiff output files were then imported to cummeRbund, which plots abundance and differential expression results as commonly used expression plots for quality control (Trapnell et al., 2012).
TIER V: functional analysis
Dimensionality reduction
Dimensionality reduction is necessary for exploring the relationships between conditions in our experiment. The MCE method performs a non‐linear dimension reduction by embedding high‐dimensional data points into a lower dimensional space using the minimum curvilinear kernel in combination with multidimensional scaling (MDS; Cannistraci et al., 2010) or in alternative the singular value decomposition (Cannistraci et al., 2013). The non‐linear data distances for MDS or SVD were computed and stored in the minimum curvilinear kernel as the traversal distances over the minimum spanning tree between the data points (in our study the samples' conditions) in the multidimensional space (in our case, the gene space). The minimum spanning tree was constructed from the Pearson correlation‐based distances between the samples (Cannistraci et al., 2010):
MCE is a parameter‐free projection algorithm that was shown to be particularly effective in discriminating classes in small‐n (samples: here, conditions), large‐m (features: here, gene expressions) datasets using only the first dimension of embedding (Cannistraci et al., 2010). The fact that our datasets have n < < m led us to adopt MCE algorithm for unsupervised analysis of the patterns present between the different sample conditions.
Clustering of genes
Genes with similar expression patterns often serve overlapping functions. Accordingly, the optimal number of clusters in the dataset was determined by performing a cluster analysis of gene expression patterns using the R package NbClust. Selected lists of expression profiles of DE genes were compiled for each hypothesis tested and clustered using the Ward's methodology.
GO and pathway enrichment analyses of DEG
Genes involved in common biological processes or pathways tend to show overlapping expression profiles. In gene expression profiling analyses, significantly enriched GO terms and pathways were identified by mapping all DEG to terms in the GO and KEGG databases by applying two‐sided Fisher's exact and χ2 tests respectively (da Huang et al., 2009a, 2009b). P‐values were corrected by calculating the FDR, and only GO and pathway terms with a FDR < 0.01 were chosen.
Identification of key BMM TFs
Promoter sequences of all DEG were retrieved using the RSAT and were further input into the RSAT matrix‐scan tool along with mouse‐related JASPAR matrices for TFBS prediction (Thomas‐Chollier et al., 2008; 2011; Turatsinze et al., 2008). The RSAT output was filtered using an adjusted P‐value < 0.05 as a cut‐off, and lists of the most significant TFBSs and their known corresponding TFs were compiled. The expression profiles of the mouse DEG were clustered and each cluster was correlated with TFs profiles using Pearson correlation in R. Finally, the clusters were annotated using GO to fully elucidate the molecular processes in which each TF was involved.
Screening for strain‐specific UPEC gene expression patterns
Flagella‐related gene lists were compiled based on Macnab's review on bacterial flagellar assembly (Macnab, 2003). A list of 35 genes related to the flagellar apparatus was used as background for screening our UPEC datasets. For OxyR regulon and hydrogen peroxide‐induced genes, gene lists were similarly compiled using previously published data on the response of bacteria to hydrogen peroxide (Zheng et al., 2001). Finally, lists of genes involved in Psp regulation were compiled, and our datasets were screened for their expression patterns. The expression patterns of the screened genes were further clustered as described above, and all results were visualized using pheatmap package in R.
Identification of UPEC genes associated with intramacrophage survival
Putative bacterial genes associated with intramacrophage survival were considered as those that remained highly up‐regulated at 24 hpi. Bacterial genes up‐regulated at this time point were compiled and filtered based on their significance of expression. All DEG were filtered using an FDR‐adjusted P‐value of the test statistic < 0.01 and a log2 fold change > 3. The filtered lists of DEG enabled us to screen for survival genes, cluster their expression and further annotate them using the GO database to better understand the biological processes they regulated.
TIER VI: gene validation
cDNA synthesis, primer design and RT‐qPCR
cDNA was synthesized using a SuperScript III First‐Strand Synthesis kit (Invitrogen). Quantitative polymerase chain reaction was then performed on a 7900HT Fast Real‐Time PCR system (Applied Biosystems) using RNA samples from two independent biological replicates, similar to those employed for the RNA‐Seq experiments. TaqMan Fast Universal PCR Master Mix 2X (Applied Biosystems) was used for BMM gene validation, and SYBR Green Master Mix (Applied Biosystems) was used for UPEC gene validation. Each cDNA was analysed in triplicate, after which the average threshold cycle (Ct) per sample was calculated. Raw data were processed with qBase Plus software (Biogazelle), which performs downstream processing of qPCR data. The geNorm algorithm, integrated in the qBase Plus package, was used for determining the optimal number and identity of reference genes needed to normalize the data in both BMM (Actb and Polr2a) and UPEC (gapA and purC) qPCR libraries. Relative expression levels were calculated with the 2‐ΔΔCt method (ΔCt is the difference in Ct between the reference genes and the target gene products); the average Ct value for all genes was used to correct for differences in cDNA input. Other statistical procedures were performed with the R program. All steps, from the experimental design to bioinformatic analysis and gene validation, are summarized in Supporting Information Fig. S3.
Construction of UTI89pspA deletion mutant
Chromosomal DNA purification, PCR and DNA sequencing of PCR products was performed as previously described (Allsopp et al., 2010). The pspA gene was mutated in UTI89 using the λ‐red‐mediated homologous recombination method with some modifications (Datsenko and Wanner, 2000; Allsopp et al., 2012). Briefly, a three‐step PCR procedure was employed to generate a DNA fragment comprising the chloramphenicol cassette from plasmid pKD3 and two 500‐bp fragments homologous to the flanking regions of the pspA gene. The following primers were used: 5376_UTI89pspA FwUP (5′‐gccgtagcgagttcatca) and 5377_UTI89pspA RvUP (5′‐ggaataggaactaaggaggaagcgttgatgttggcatt), 5378_UTI89pspA Fwdn (5′‐cctacacaatcgctcaagacgccgaactgaaagccgat) and 5379_UTI89pspA Rvdn (5′‐taaacagcgccagaccga) to generate the 500bp homology arms; 3746‐Cm.3a (5′‐tcctccttagttcctattcc) and 3747‐Cm.4a (5′‐gtcttgagcgattgtgtagg) to generate the chloramphenicol resistance gene fragment. This DNA fusion product was electroporated into UTI89 harbouring plasmid pKD46, and chloramphenicol resistant mutants were selected and confirmed by PCR (using primers 5375_UTI89pspA FwSc: 5′‐tcgtcgcgcataccaacc and 5380_UTI89pspA Rvsc: 5′‐acttcatccagcaattcgc). The UTI89pspA mutant was confirmed by sequencing.
Supporting information
Fig. S1. Intramacrophage survival of UTI89 versus 83972. Bacterial loads of UTI89 and 83972 within BMM at 2, 4 and 24 hpi in gentamicin exclusion assays were assessed by colony counting. These samples were used for RNA‐Seq analyses. Data represent average CFU ml−1 ± range (n = 2 independent experiments).
Fig. S2. Enriched mouse and UPEC GO terms during the course of infection. Gene ontology terms enriched in BMMs (A) and UPEC (B) during the 24 h course of infection. Each coloured square in the matrix represents a significant fold‐enrichment (log2) of the respective GO term at each point. Red, up‐regulated DEG; green, down‐regulated DEG.
Fig. S3. Bioinformatic analysis pipeline. Summary of the steps followed for the generation and analysis of the RNA‐Seq data produced by next‐generation sequencing. Steps are grouped into six tiers, and details are provided on the algorithms, databases and software used for each of the analyses.
Table S1. Quality control of RNA‐Seq libraries.
Table S2. Alignment statistics of RNA‐Seq reads.
Acknowledgements
This work was supported by grants from the National Health and Medical Research Council (NHMRC) of Australia (APP1005315 and APP1068593). MJS is supported by an Australian Research Council (ARC) Future Fellowship (FT100100657), as well as an honorary NHMRC Senior Research Fellowship (APP1003470). MAS and GCU are supported by ARC Future Fellowships (FT100100662 and FT110101048, respectively), MT is supported by an ARC Discovery Early Career Researcher Award (DE130101169), and SAB is supported by an NHMRC Career Development Fellowship (APP1090456).
References
- Allsopp, L.P. , Totsika, M. , Tree, J.J. , Ulett, G.C. , Mabbett, A.N. , Wells, T.J. , et al (2010) UpaH is a newly identified autotransporter protein that contributes to biofilm formation and bladder colonization by uropathogenic Escherichia coli CFT073. Infect Immun 78: 1659–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allsopp, L.P. , Beloin, C. , Ulett, G.C. , Valle, J. , Totsika, M. , Sherlock, O. , et al (2012) Molecular characterization of UpaB and UpaC, two new autotransporter proteins of uropathogenic Escherichia coli CFT073. Infect Immun 80: 321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson, P. , Engberg, I. , Lidin‐Janson, G. , Lincoln, K. , Hull, R. , Hull, S. , and Svanborg, C. (1991) Persistence of Escherichia coli bacteriuria is not determined by bacterial adherence. Infect Immun 59: 2915–2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannai, S. (1986) Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J Biol Chem 261: 2256–2263. [PubMed] [Google Scholar]
- Bokil, N.J. , Totsika, M. , Carey, A.J. , Stacey, K.J. , Hancock, V. , Saunders, B.M. , et al (2011) Intramacrophage survival of uropathogenic Escherichia coli: differences between diverse clinical isolates and between mouse and human macrophages. Immunobiology 216: 1164–1171. [DOI] [PubMed] [Google Scholar]
- Bonfield, T.L. , Thomassen, M.J. , Farver, C.F. , Abraham, S. , Koloze, M.T. , Zhang, X. , et al (2008) Peroxisome proliferator‐activated receptor‐gamma regulates the expression of alveolar macrophage macrophage colony‐stimulating factor. J Immunol 181: 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bower, J.M. , Gordon‐Raagas, H.B. , and Mulvey, M.A. (2009) Conditioning of uropathogenic Escherichia coli for enhanced colonization of host. Infect Immun 77: 2104–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannistraci, C.V. , Ravasi, T. , Montevecchi, F.M. , Ideker, T. , and Alessio, M. (2010) Nonlinear dimension reduction and clustering by minimum curvilinearity unfold neuropathic pain and tissue embryological classes. Bioinformatics 26: i531–i539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannistraci, C.V. , Alanis‐Lobato, G. , and Ravasi, T. (2013) Minimum curvilinearity to enhance topological prediction of protein interactions by network embedding. Bioinformatics 29: i199–i209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darwin, A.J. (2013) Stress relief during host infection: the phage shock protein response supports bacterial virulence in various ways. PLoS Pathog 9: e1003388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko, K.A. , and Wanner, B.L. (2000) One‐step inactivation of chromosomal genes in Escherichia coli K‐12 using PCR products. Proc Natl Acad Sci U S A 97: 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey, A. , She, H. , Kim, L. , Boruch, A. , Guris, D.L. , Carlberg, K. , et al (2000) Colony‐stimulating factor‐1 receptor utilizes multiple signaling pathways to induce cyclin D2 expression. Mol Biol Cell 11: 3835–3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duell, B.L. , Carey, A.J. , Tan, C.K. , Cui, X. , Webb, R.I. , Totsika, M. , et al (2012) Innate transcriptional networks activated in bladder in response to uropathogenic Escherichia coli drive diverse biological pathways and rapid synthesis of IL‐10 for defense against bacterial urinary tract infection. J Immunol 188: 781–792. [DOI] [PubMed] [Google Scholar]
- Eden, C.S. , Hanson, L.A. , Jodal, U. , Lindberg, U. , and Akerlund, A.S. (1976) Variable adherence to normal human urinary‐tract epithelial cells of Escherichia coli strains associated with various forms of urinary‐tract infection. Lancet 1: 490–492. [PubMed] [Google Scholar]
- Flannagan, R.S. , Cosio, G. , and Grinstein, S. (2009) Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol 7: 355–366. [DOI] [PubMed] [Google Scholar]
- Foxman, B. (2010) The epidemiology of urinary tract infection. Nat Rev Urol 7: 653–660. [DOI] [PubMed] [Google Scholar]
- Funfstuck, R. , Tschape, H. , Stein, G. , Kunath, H. , Bergner, M. , and Wessel, G. (1986) Virulence properties of Escherichia coli strains in patients with chronic pyelonephritis. Infection 14: 145–150. [DOI] [PubMed] [Google Scholar]
- Hacker, J. , and Kaper, J.B. (2000) Pathogenicity islands and the evolution of microbes. Annu Rev Microbiol 54: 641–679. [DOI] [PubMed] [Google Scholar]
- Hagan, E.C. , Lloyd, A.L. , Rasko, D.A. , Faerber, G.J. , and Mobley, H.L. (2010) Escherichia coli global gene expression in urine from women with urinary tract infection. PLoS Pathog 6: e1001187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannan, T.J. , Totsika, M. , Mansfield, K.J. , Moore, K.H. , Schembri, M.A. , and Hultgren, S.J. (2012) Host‐pathogen checkpoints and population bottlenecks in persistent and intracellular uropathogenic Escherichia coli bladder infection. FEMS Microbiol Rev 36: 616–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes, J.D. , and McLellan, L.I. (1999) Glutathione and glutathione‐dependent enzymes represent a co‐ordinately regulated defence against oxidative stress. Free Radic Res 31: 273–300. [DOI] [PubMed] [Google Scholar]
- Hegedus, Z. , Zakrzewska, A. , Agoston, V.C. , Ordas, A. , Racz, P. , Mink, M. , et al (2009) Deep sequencing of the zebrafish transcriptome response to mycobacterium infection. Mol Immunol 46: 2918–2930. [DOI] [PubMed] [Google Scholar]
- ‘t Hoen, P.A. , Ariyurek, Y. , Thygesen, H.H. , Vreugdenhil, E. , Vossen, R.H. , de Menezes, R.X. , et al (2008) Deep sequencing‐based expression analysis shows major advances in robustness, resolution and inter‐lab portability over five microarray platforms. Nucleic Acids Res 36: e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath, D.J., Jr , Dabdoub, S.M. , Li, B. , Vanderbrink, B.A. , and Justice, S.S. (2012) New paradigms of urinary tract infections: implications for patient management. Indian J Urol 28: 154–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Q. , Dong, S. , Fang, C. , Wu, X. , Ye, T. , and Lin, Y. (2012) Deep sequencing‐based transcriptome profiling analysis of Oryzias melastigma exposed to PFOS. Aquat Toxicol 120–121: 54–58. [DOI] [PubMed] [Google Scholar]
- da Huang, W. , Sherman, B.T. , and Lempicki, R.A. (2009a) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Huang, W. , Sherman, B.T. , and Lempicki, R.A. (2009b) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. [DOI] [PubMed] [Google Scholar]
- Humphrys, M.S. , Creasy, T. , Sun, Y. , Shetty, A.C. , Chibucos, M.C. , Drabek, E.F. , et al (2013) Simultaneous transcriptional profiling of bacteria and their host cells. PLoS ONE 8: e80597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunstad, D.A. , and Justice, S.S. (2010) Intracellular lifestyles and immune evasion strategies of uropathogenic Escherichia coli . Annu Rev Microbiol 64: 203–221. [DOI] [PubMed] [Google Scholar]
- Ibarra, J.A. , Knodler, L.A. , Sturdevant, D.E. , Virtaneva, K. , Carmody, A.B. , Fischer, E.R. , et al (2010) Induction of Salmonella pathogenicity island 1 under different growth conditions can affect Salmonella‐host cell interactions in vitro . Microbiology 156: 1120–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlay, J.A. (2013) The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat Rev Microbiol 11: 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingersoll, M.A. , Kline, K.A. , Nielsen, H.V. , and Hultgren, S.J. (2008) G‐CSF induction early in uropathogenic Escherichia coli infection of the urinary tract modulates host immunity. Cell Microbiol 10: 2568–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager, D. , Sharma, C.M. , Thomsen, J. , Ehlers, C. , Vogel, J. , and Schmitz, R.A. (2009) Deep sequencing analysis of the Methanosarcina mazei Go1 transcriptome in response to nitrogen availability. Proc Natl Acad Sci U S A 106: 21878–21882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, J.R. (1991) Virulence factors in Escherichia coli urinary tract infection. Clin Microbiol Rev 4: 80–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaijser, B. , and Ahlstedt, S. (1977) Protective capacity of antibodies against Escherichia coli and K antigens. Infect Immun 17: 286–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlinsey, J.E. , Maguire, M.E. , Becker, L.A. , Crouch, M.L. , and Fang, F.C. (2010) The phage shock protein PspA facilitates divalent metal transport and is required for virulence of Salmonella enterica sv. Typhimurium. Mol Microbiol 78: 669–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. , and Salzberg, S.L. (2011) TopHat‐Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol 12: R72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm, P. , Hancock, V. , and Schembri, M.A. (2007) Mellowing out: adaptation to commensalism by Escherichia coli asymptomatic bacteriuria strain 83972. Infect Immun 75: 3688–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau, M.E. , Loughman, J.A. , and Hunstad, D.A. (2012) YbcL of uropathogenic Escherichia coli suppresses transepithelial neutrophil migration. Infect Immun 80: 4123–4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Liu, B. , Fukudome, E.Y. , Lu, J. , Chong, W. , Jin, G. , et al (2011) Identification of citrullinated histone H3 as a potential serum protein biomarker in a lethal model of lipopolysaccharide‐induced shock. Surgery 150: 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg, U. (1975) Asymptomatic bacteriuria in school girls. V. The clinical course and response to treatment. Acta Paediatr Scand 64: 718–724. [DOI] [PubMed] [Google Scholar]
- Lindberg, U. , Bjure, J. , Haugstvedt, S. , and Jodal, U. (1975a) Asymptomatic bacteriuria in schoolgirls. III. Relation between residual urine volume and recurrence. Acta Paediatr Scand 64: 437–440. [DOI] [PubMed] [Google Scholar]
- Lindberg, U. , Claesson, I. , Hanson, L.A. , and Jodal, U. (1975b) Asymptomatic bacteriuria in schoolgirls. I. Clinical and laboratory findings. Acta Paediatr Scand 64: 425–431. [DOI] [PubMed] [Google Scholar]
- Lindberg, U. , Hanson, L.A. , Jodal, U. , Lidin‐Janson, G. , Lincoln, K. , and Olling, S. (1975c) Asymptomatic bacteriuria in schoolgirls. II. Differences in Escherichia coli causing asymptomatic bacteriuria. Acta Paediatr Scand 64: 432–436. [DOI] [PubMed] [Google Scholar]
- Llorens, F. , Hummel, M. , Pastor, X. , Ferrer, A. , Pluvinet, R. , Vivancos, A. , et al (2011) Multiple platform assessment of the EGF dependent transcriptome by microarray and deep tag sequencing analysis. BMC Genomics 12: 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabbett, A.N. , Ulett, G.C. , Watts, R.E. , Tree, J.J. , Totsika, M. , Ong, C.L. , et al (2009) Virulence properties of asymptomatic bacteriuria Escherichia coli . Int J Med Microbiol 299: 53–63. [DOI] [PubMed] [Google Scholar]
- Macnab, R.M. (2003) How bacteria assemble flagella. Annu Rev Microbiol 57: 77–100. [DOI] [PubMed] [Google Scholar]
- McNally, A. , La Ragione, R.M. , Best, A. , Manning, G. , and Newell, D.G. (2007) An aflagellate mutant Yersinia enterocolitica biotype 1A strain displays altered invasion of epithelial cells, persistence in macrophages, and cytokine secretion profiles in vitro . Microbiology 153: 1339–1349. [DOI] [PubMed] [Google Scholar]
- Mogensen, T.H. (2009) Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 22: 240–273, Table of Contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozova, O. , and Marra, M.A. (2008) Applications of next‐generation sequencing technologies in functional genomics. Genomics 92: 255–264. [DOI] [PubMed] [Google Scholar]
- Mulvey, M.A. , Schilling, J.D. , and Hultgren, S.J. (2001) Establishment of a persistent Escherichia coli reservoir during the acute phase of a bladder infection. Infect Immun 69: 4572–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysorekar, I.U. , and Hultgren, S.J. (2006) Mechanisms of uropathogenic Escherichia coli persistence and eradication from the urinary tract. Proc Natl Acad Sci U S A 103: 14170–14175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysorekar, I.U. , Mulvey, M.A. , Hultgren, S.J. , and Gordon, J.I. (2002) Molecular regulation of urothelial renewal and host defenses during infection with uropathogenic Escherichia coli . J Biol Chem 277: 7412–7419. [DOI] [PubMed] [Google Scholar]
- Nie, Q. , Sandford, E.E. , Zhang, X. , Nolan, L.K. , and Lamont, S.J. (2012) Deep sequencing‐based transcriptome analysis of chicken spleen in response to avian pathogenic Escherichia coli (APEC) infection. PLoS ONE 7: e41645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordas, A. , Hegedus, Z. , Henkel, C.V. , Stockhammer, O.W. , Butler, D. , Jansen, H.J. , et al (2011) Deep sequencing of the innate immune transcriptomic response of zebrafish embryos to Salmonella infection. Fish Shellfish Immun 31: 716–724. [DOI] [PubMed] [Google Scholar]
- Orskov, I. , Svanborg Eden, C. , and Orskov, F. (1988) Aerobactin production of serotyped Escherichia coli from urinary tract infections. Med Microbiol Immunol (Berl) 177: 9–14. [DOI] [PubMed] [Google Scholar]
- Pello, O.M. , De Pizzol, M. , Mirolo, M. , Soucek, L. , Zammataro, L. , Amabile, A. , et al (2012) Role of c‐MYC in alternative activation of human macrophages and tumor‐associated macrophage biology. Blood 119: 411–421. [DOI] [PubMed] [Google Scholar]
- Pichon, C. , Hechard, C. , du Merle, L. , Chaudray, C. , Bonne, I. , Guadagnini, S. , et al (2009) Uropathogenic Escherichia coli AL511 requires flagellum to enter renal collecting duct cells. Cell Microbiol 11: 616–628. [DOI] [PubMed] [Google Scholar]
- Plos, K. , Carter, T. , Hull, S. , Hull, R. , and Svanborg Eden, C. (1990) Frequency and organization of pap homologous DNA in relation to clinical origin of uropathogenic Escherichia coli . J Infect Dis 161: 518–524. [DOI] [PubMed] [Google Scholar]
- Plos, K. , Connell, H. , Jodal, U. , Marklund, B.I. , Marild, S. , Wettergren, B. , and Svanborg, C. (1995) Intestinal carriage of P fimbriated Escherichia coli and the susceptibility to urinary tract infection in young children. J Infect Dis 171: 625–631. [DOI] [PubMed] [Google Scholar]
- Pompella, A. , Visvikis, A. , Paolicchi, A. , De Tata, V. , and Casini, A.F. (2003) The changing faces of glutathione, a cellular protagonist. Biochem Pharmacol 66: 1499–1503. [DOI] [PubMed] [Google Scholar]
- Portt, L. , Norman, G. , Clapp, C. , Greenwood, M. , and Greenwood, M.T. (2011) Anti‐apoptosis and cell survival: a review. Biochim Biophys Acta 1813: 238–259. [DOI] [PubMed] [Google Scholar]
- Roberts, A. , Trapnell, C. , Donaghey, J. , Rinn, J.L. , and Pachter, L. (2011) Improving RNA‐Seq expression estimates by correcting for fragment bias. Genome Biol 12: R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos, V. , Nielsen, E.M. , and Klemm, P. (2006) Asymptomatic bacteriuria Escherichia coli strains: adhesins, growth and competition. FEMS Microbiol Lett 262: 22–30. [DOI] [PubMed] [Google Scholar]
- Rosenberger, C.M. , and Finlay, B.B. (2003) Phagocyte sabotage: disruption of macrophage signalling by bacterial pathogens. Nat Rev Mol Cell Biol 4: 385–396. [DOI] [PubMed] [Google Scholar]
- Rossol, M. , Heine, H. , Meusch, U. , Quandt, D. , Klein, C. , Sweet, M.J. , and Hauschildt, S. (2011) LPS‐induced cytokine production in human monocytes and macrophages. Crit Rev Immunol 31: 379–446. [DOI] [PubMed] [Google Scholar]
- Salvatore, S. , Salvatore, S. , Cattoni, E. , Siesto, G. , Serati, M. , Sorice, P. , and Torella, M. (2011) Urinary tract infections in women. Eur J Obstet Gynecol Reprod Biol 156: 131–136. [DOI] [PubMed] [Google Scholar]
- Schembri, M.A. , Sokurenko, E.V. , and Klemm, P. (2000) Functional flexibility of the FimH adhesin: insights from a random mutant library. Infect Immun 68: 2638–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semeraro, F. , Ammollo, C.T. , Morrissey, J.H. , Dale, G.L. , Friese, P. , Esmon, N.L. , and Esmon, C.T. (2011) Extracellular histones promote thrombin generation through platelet‐dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood 118: 1952–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sester, D.P. , Beasley, S.J. , Sweet, M.J. , Fowles, L.F. , Cronau, S.L. , Stacey, K.J. , and Hume, D.A. (1999) Bacterial/CpG DNA down‐modulates colony stimulating factor‐1 receptor surface expression on murine bone marrow‐derived macrophages with concomitant growth arrest and factor‐independent survival. J Immunol 163: 6541–6550. [PubMed] [Google Scholar]
- Sester, D.P. , Brion, K. , Trieu, A. , Goodridge, H.S. , Roberts, T.L. , Dunn, J. , et al (2006) CpG DNA activates survival in murine macrophages through TLR9 and the phosphatidylinositol 3‐kinase‐Akt pathway. J Immunol 177: 4473–4480. [DOI] [PubMed] [Google Scholar]
- Shih, A.Y. , Erb, H. , Sun, X. , Toda, S. , Kalivas, P.W. , and Murphy, T.H. (2006) Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J Neurosci 26: 10514–10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivick, K.E. , and Mobley, H.L. (2010) Waging war against uropathogenic Escherichia coli: winning back the urinary tract. Infect Immun 78: 568–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenqvist, K. , Sandberg, T. , Lidin‐Janson, G. , Orskov, F. , Orskov, I. , and Svanborg‐Eden, C. (1987) Virulence factors of Escherichia coli in urinary isolates from pregnant women. J Infect Dis 156: 870–877. [DOI] [PubMed] [Google Scholar]
- Tan, C.K. , Carey, A.J. , Cui, X. , Webb, R.I. , Ipe, D. , Crowley, M. , et al (2012) Genome‐wide mapping of cystitis due to Streptococcus agalactiae and Escherichia coli in mice identifies a unique bladder transcriptome that signifies pathogen‐specific antimicrobial defense against urinary tract infection. Infect Immun 80: 3145–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegner, J. , Nilsson, R. , Bajic, V.B. , Bjorkegren, J. , and Ravasi, T. (2006) Systems biology of innate immunity. Cell Immunol 244: 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas‐Chollier, M. , Sand, O. , Turatsinze, J.V. , Janky, R. , Defrance, M. , Vervisch, E. , et al (2008) RSAT: regulatory sequence analysis tools. Nucleic Acids Res 36: W119–W127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas‐Chollier, M. , Defrance, M. , Medina‐Rivera, A. , Sand, O. , Herrmann, C. , Thieffry, D. , and van Helden, J. (2011) RSAT 2011: regulatory sequence analysis tools. Nucleic Acids Res 39: W86–W91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomich, M. , Herfst, C.A. , Golden, J.W. , and Mohr, C.D. (2002) Role of flagella in host cell invasion by Burkholderia cepacia . Infect Immun 70: 1799–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totsika, M. , Moriel, D.G. , Idris, A. , Rogers, B.A. , Wurpel, D.J. , Phan, M.D. , et al (2012) Uropathogenic Escherichia coli mediated urinary tract infection. Curr Drug Targets 13: 1386–1399. [DOI] [PubMed] [Google Scholar]
- Tourneur, E. , Ben Mkaddem, S. , Chassin, C. , Bens, M. , Goujon, J.M. , Charles, N. , et al (2013) Cyclosporine A impairs nucleotide binding oligomerization domain (Nod1)‐mediated innate antibacterial renal defenses in mice and human transplant recipients. PLoS Pathog 9: e1003152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell, C. , Roberts, A. , Goff, L. , Pertea, G. , Kim, D. , Kelley, D.R. , et al (2012) Differential gene and transcript expression analysis of RNA‐seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turatsinze, J.V. , Thomas‐Chollier, M. , Defrance, M. , and van Helden, J. (2008) Using RSAT to scan genome sequences for transcription factor binding sites and cis‐regulatory modules. Nat Protoc 3: 1578–1588. [DOI] [PubMed] [Google Scholar]
- Ulett, G.C. , Totsika, M. , Schaale, K. , Carey, A.J. , Sweet, M.J. , and Schembri, M.A. (2013) Uropathogenic Escherichia coli virulence and innate immune responses during urinary tract infection. Curr Opin Microbiol 16: 100–107. [DOI] [PubMed] [Google Scholar]
- Wang, F. , Hu, S. , Liu, W. , Qiao, Z. , Gao, Y. , and Bu, Z. (2012) Deep‐sequencing analysis of the mouse transcriptome response to infection with Brucella melitensis strains of differing virulence. PLoS ONE 6: e28485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z. , Gerstein, M. , and Snyder, M. (2009) RNA‐Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10: 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch, R.A. , Burland, V. , Plunkett, G., 3rd , Redford, P. , Roesch, P. , Rasko, D. , et al (2002) Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli . Proc Natl Acad Sci U S A 99: 17020–17024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiles, T.J. , Kulesus, R.R. , and Mulvey, M.A. (2008) Origins and virulence mechanisms of uropathogenic Escherichia coli . Exp Mol Pathol 85: 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willner, D. , Low, S. , Steen, J.A. , George, N. , Nimmo, G.R. , Schembri, M.A. , and Hugenholtz, P. (2014) Single clinical isolates from acute uncomplicated urinary tract infections are representative of dominant in situ populations. mBio 5: e1064–e1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang, L.X. , He, D. , Dong, W.R. , Zhang, Y.W. , and Shao, J.Z. (2010) Deep sequencing‐based transcriptome profiling analysis of bacteria‐challenged Lateolabrax japonicus reveals insight into the immune‐relevant genes in marine fish. BMC Genomics 11: 472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, S. , Jia, J. , Mo, D. , Wang, Q. , Qin, L. , He, Z. , et al (2010) Understanding PRRSV infection in porcine lung based on genome‐wide transcriptome response identified by deep sequencing. PLoS ONE 5: e11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, J. , Zhang, X. , Pelayo, R. , Monestier, M. , Ammollo, C.T. , Semeraro, F. , et al (2009) Extracellular histones are major mediators of death in sepsis. Nat Med 15: 1318–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, J. , Zhang, X. , Monestier, M. , Esmon, N.L. , and Esmon, C.T. (2011) Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J Immunol 187: 2626–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi, S. , and Darwin, A.J. (2012) Recent findings about the Yersinia enterocolitica phage shock protein response. J Microbiol 50: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi, S. , Reid, D.A. , Rothenberg, E. , and Darwin, A.J. (2013) Changes in Psp protein binding partners, localization and behaviour upon activation of the Yersinia enterocolitica phage shock protein response. Mol Microbiol 87: 656–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdziarski, J. , Brzuszkiewicz, E. , Wullt, B. , Liesegang, H. , Biran, D. , Voigt, B. , et al (2010) Host imprints on bacterial genomes – rapid, divergent evolution in individual patients. PLoS Pathog 6: e1001078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, M. , Wang, X. , Templeton, L.J. , Smulski, D.R. , LaRossa, R.A. , and Storz, G. (2001) DNA microarray‐mediated transcriptional profiling of the Escherichia coli response to hydrogen peroxide. J Bacteriol 183: 4562–4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Intramacrophage survival of UTI89 versus 83972. Bacterial loads of UTI89 and 83972 within BMM at 2, 4 and 24 hpi in gentamicin exclusion assays were assessed by colony counting. These samples were used for RNA‐Seq analyses. Data represent average CFU ml−1 ± range (n = 2 independent experiments).
Fig. S2. Enriched mouse and UPEC GO terms during the course of infection. Gene ontology terms enriched in BMMs (A) and UPEC (B) during the 24 h course of infection. Each coloured square in the matrix represents a significant fold‐enrichment (log2) of the respective GO term at each point. Red, up‐regulated DEG; green, down‐regulated DEG.
Fig. S3. Bioinformatic analysis pipeline. Summary of the steps followed for the generation and analysis of the RNA‐Seq data produced by next‐generation sequencing. Steps are grouped into six tiers, and details are provided on the algorithms, databases and software used for each of the analyses.
Table S1. Quality control of RNA‐Seq libraries.
Table S2. Alignment statistics of RNA‐Seq reads.