

Abstract
Biomaterials are continually being designed that enable new methods for interacting dynamically with cell and tissues, in turn unlocking new capabilities in areas ranging from drug delivery to regenerative medicine. In this review, we explore some of the recent advances being made in regards to programming biomaterials for improved drug delivery, with a focus on cancer and infection. We begin by explaining several of the underlying concepts that are being used to design this new wave of drug delivery vehicles, followed by examining recent materials systems that are able to coordinate the temporal delivery of multiple therapeutics, dynamically respond to changing tissue environments, and reprogram their bioactivity over time.
Keywords: Bionanoscience, biomaterials, drug delivery, cancer, bacteria, nanoparticles
Introduction
Diseases such as cancer and infection are many times heterogeneous in nature and exhibit a remarkable ability to dynamically adapt to a variety of microenvironments and chemical cues to promote proliferation and survival.1,2 Cancers often accomplish this through high mutation rates,3 while bacterial infections develop innate resistance due to repeated sub-lethal challenges or enhance their survival through the formation of biofilms.4 Treating these conditions commonly involves the use of systemic delivery of cytotoxic drugs that target rapidly dividing cells, block specific pathways that confer survival benefits, or interfere with bacteria-specific building blocks. While many times this basic systemic strategy can be effective, there are associated side effects such as unintended targeting of healthy dividing cells, for instance those located in the hair follicles or mucous membranes, or killing of beneficial probiotic bacteria. To increase effectiveness while concomitantly reducing unwanted side effects, new strategies are being developed for targeted delivery to the complete collection of heterogeneous disease locations by taking advantage of common biomarkers or features across all sites.5,6 Ideally, these delivery strategies not only concentrate delivery in target locations but also preserve bioactivity of sensitive biologic drugs and enable the ability to program multi-functional therapeutics, which has frequently been shown to improve patient outcomes.7,8
In recent years, several biomaterial-based strategies have been developed to tackle these challenges. In the context of cancer, many approaches take advantage of the enhanced permeation and retention (EPR) effect that leads to accumulation of nanoparticles at the target site due to poorly structured vasculature within tumors.9 However, these strategies still do not address the complete set of aforementioned issues with systemic therapies, target diseases with a static rather than potentially more effective temporally coordinated strategy, and typically cannot be tailored to an individual patient’s specific disease profile.10
To address these limitations, recent research is now looking into developing multi-functional programmable biomaterials that can address many of the shortcomings of the prior generation of biomaterials. These materials generally exhibit both dynamic and temporal tunability, allowing for stimuli-responsive behavior that can actively adapt to both the progression of the disease and any potential changes deemed necessary by the clinician. Furthermore, these platforms are highly customizable and allow for a more personalized medicine approach, whether it be choosing which drugs to deliver for combinatorial therapy or changing the stimuli-specific responses of the biomaterial.11
In this review, we have outlined some of the widely used approaches and highlighted some of the latest advanced therapeutic applications of multi-functional programmable biomaterials. We begin by introducing some of the underlying characteristics and behavior of these enabling technologies, specifically oligonucleotide-based nanotechnologies and click chemistries. Afterwards, we give several examples of using each enabling technology for applications relating to cancer and infectious diseases. Finally, we offer a future outlook and provide potential avenues for new research in this rapidly developing field.
Enabling technologies
Previous efforts to define programmable materials often look at synergistic and sequential drug delivery systems. Synergistic systems require presence of two or more triggers to facilitate drug delivery, while sequential systems rely on multiple stimuli to achieve drug release or spur sequential drug delivery.12 However, from a materials point of view, programmability is not limited to these two approaches and can be extended to any materials capable of changing their properties over time and interacting in a uni- or bi-directional manner with cells. In general, such programmable materials consist of multiple modules that can be assembled prior to in vivo administration or refilled once in vivo. The first part of this review introduces some of the prominent enabling technologies and highlights the advantages and disadvantages of each approach.
Delivery platforms
Two of the most common experimental platforms used to prepare drug delivery systems are nanoparticles and hydrogels. Traditionally, nanoparticles have been used to encapsulate and protect drugs, concentrate their activity, improve targeting to tumors,9 and sites of infection;13 together these features work to reduce negative side effects compared to delivery of a free drug.8 One advantage of using nanoparticles is that they come in a variety of sizes, shapes, chemistries, and stimuli-responsive elements,14 providing a seemingly countless number of possibilities.9,15 This provides a diverse palate for designing nanoparticles; for example, the size of a nanoparticle alone can influence which organs it will preferentially accumulate in Blanco et al.16 Some examples of nanoparticles include liposomes for hydrophobic and hydrophilic drug delivery,17 layer-by-layer-coated particles for sustained delivery of therapeutics,18 dendrimers for multi-functional therapies,19 and gold nanorods for optically tunable drug release.20
On the other hand, hydrogels can be an efficient platform for sustaining the delivery of therapeutics over longer periods of time compared to nanoparticles with similar material composition.21 Their larger size and potential for direct injection via shear thinning22 allow them to be placed at a specific location and retain concentrated function locally, in turn reducing systemic exposure.23 Since hydrogel parameters such as degradation rate and swelling are tuneable, they can be used as short-term delivery vehicles, stimuli-triggered release platforms,24 as well as refillable drug depots.25 Additionally, it is possible to incorporate combinations of therapeutics that are locally activated by various responsive elements,26 such as MMP degradable linkers,27 enabling on-demand, and stimuli-specific delivery of therapeutics. Finally, there are multiple methods to adjust the properties of hydrogels (e.g. pore size,28 stiffness29) as well as ways to incorporate bioactive molecules that recruit target cells into the material30 and modify their behavior.
While we have highlighted nanoparticles and hydrogels, there are a vast array of possibilities not delineated in this review and also other numerous delivery platforms, such as film coatings for implants31 and porous membranes for long-term delivery.32 When considering the appropriate platform from the bevy of options, form follows function and thus the environment, use, and specific context should be carefully understood before designing the appropriate delivery system. One of the challenges in this regard has been development of fabrication protocols that make it possible to reproducibly combine multiple bioactive components on one platform. Commercially available end-functionalized peptides, nucleic acids, and polymers that can participate in highly efficient and biocompatible reactions provide biologically focused laboratories simple means to develop, test, and implement combinatorial approaches both in vitro33 and in vivo.34
Click and stimuli-responsive chemistries
A powerful tool in making materials programmable are click chemistries, a class of reactions that proceed at physiological conditions with high efficiency, rapid reaction kinetics, are bio-orthogonal, and result in no by-products.35 One of the first click reactions developed is the Cu-catalyzed azide-alkyne cycloaddition reaction36 (Figure 1(a)). Unfortunately, difficulty in removing the cytotoxic copper ions has precluded its use in many biological applications. In 2007, the Bertozzi group overcame this limitation by developing a copper-free bio-orthogonal chemistry for in vivo imaging. Their new approach took advantage of the reaction between azides and difluorinated cyclooctynes containing ring strain and electron-withdrawing groups37 to replace the Cu catalyst. The catalogue of click chemistries has been continuously expanding since then, and it is currently possible to perform multiple orthogonal reactions in “one pot” without the need for a catalyst.35 Importantly, the high biocompatibility of copper-free click reactions enables the labeling of not only synthetic materials34 but also cell surfaces.38
Figure 1.

There are a variety of enabling technologies that underlie the ability to program functionality into therapeutic materials. (a) Click chemistry reactions are convenient for their high reaction efficiency, bio-orthogonality, specificity, and low toxicity. The stereotypical click reaction is the Cu-catalyzed alkyne-azide reaction, although the Cu catalyst can present toxicity problem in vivo. Other click reactions such as the one between an azide and dibenzocyclooctyne (DBCO) do not require Cu catalysts, instead being catalyzed by the ring strain present in the cyclooctyne. (b) Nucleic acids provide an additional avenue for programming functionality into materials. Aptamers can be used both for targeting over-expressed proteins on cell surfaces and as a therapeutic. (c) Finally, the breakdown and release of materials are tuneable by using enzyme degradable sequences, such as those that are substrates for MMPs. (A color version of this figure is available in the online journal.)
Stimuli-responsive linkers are also particularly useful for introducing programmability into drug delivery systems. The first generation of stimuli-responsive materials made use of functional groups including hydrazones and disulphides to take advantage of non-specific stimuli such as pH and reducing environments, respectively.39 Recently, an increasingly popular concept is to incorporate groups that are cleaved by specific physical or biological entities such as proteases40 (Figure 1(c)). Depending on the intended application, researchers can optimize the system to be cleaved by enzymes located extracellularly, intracellularly, or both. A key challenge in using biologically degradable sequences is that it should be highly preferential for cleavage to occur by enzymes upregulated specifically within diseased tissues. For example, extracellularly located enzymes such as MMPs or hyaluronidase play an important role in turnover of the extracellular matrix (ECM), enabling researchers to use such a property to activate systems in vivo.27
Oligonucleotides
Over the past 20 years, DNA/RNA nanotechnology has revolutionized diagnostics, drug delivery, and intracellular as well as extracellular imaging due to, amongst other things, the guiding principle of using a simple and well-studied molecule as a building block to develop more complex bioactive and versatile systems to control cellular function. In 1996, Tyagi and Kramer developed the concept of a molecular beacon,41 an approximately 15–30 base-long RNA/DNA sequence consisting of an 18–30 base-long loop stabilized by a 5–7 base-pair stem.42 This secondary structure is stable under ambient or body temperature but can be destabilized by the binding of a complementary sequence during the hybridization process, such as during selective interference with mRNA. Both the 5′ and 3′ ends of the oligonucleotide can be modified with functional groups such as a fluorophore-quencher pair or Förster resonance energy transfer (FRET) pair, which makes it possible to simultaneously report on the binding events.43 For example, by choosing the loop sequence to match mRNA up-regulated in target cells, researchers can down-regulate expression of specific genes while simultaneously monitoring the process.44
Oligonucleotides can also be used as effective drug delivery vehicles.45 Like the aforementioned FRET pairs, drugs can be covalently conjugated to either end of the oligonucleotide as well as to inner bases. Additional drug delivery approaches make use of the unique molecular structure of oligonucleotides. For example, the double-stranded stem region of molecular beacons has standard π-π stacking of aromatic rings that are attractive sites for drugs, such as doxorubicin (Dox), that intercalate the duplex DNA regions46 (Figure 1(b)).
Another advantage of oligonucleotides is their ability to bind selected target receptors, molecules, and cells with high affinity and specificity, which makes them an ideal component in targeted delivery platforms.47 This property arises from the tendency of single-stranded oligonucleotides to fold onto themselves and form thermodynamically favorable secondary structures that can be further stabilized by binding appropriate ligands. Sequences that bind to desired ligands with sufficient affinity are named aptamers and selected using an in vitro directed evolution process called SELEX.48,49 The key parameter to take into consideration is the binding constant (KD), which is a ratio between the off-rate and on-rate constants. This value should ideally lie in at least the nM or pM range to maximize the in vivo efficacy and minimize the required dosage.50
Some of the potential disadvantages of oligonucleotides as a material are their susceptibility to degradation by either exonucleases that cut DNA/RNA at the terminal ends or endonucleases that cut oligonucleotides within the strand. Because of this, oligonucleotides are frequently attached to other molecules, such as polyethylene-glycol (PEG), which blocks exonuclease activity and slows renal filtration by increasing molecular weight above the 30–50 kDa molecular mass cutoff of the renal glomerulus.50 Furthermore, the 2′-OH group on the ribose of RNA nucleotides is frequently replaced by molecules such as a 2′-fluoro or 2′-O-methyl to increase resistance to degradation by endonucleases. Alternatively, more stable DNA analogs, such as locked nucleic acids (LNA)51 and peptide nucleic acid (PNAs)52 can be used, however, the hydrophobicity of PNAs presents unique challenges for use in vivo. Finally, it is critical to ensure there is no unintended priming of the immune system by excessive extracellular RNA or DNA, which in some cases can mimic conditions found during infection.53
Recent in vitro and in vivo applications
In the subsequent sections of this review, we focus on recent developments in programmable nanoparticles and hydrogels as the primary means of delivery. We have specifically highlighted approaches that target cancer and bacterial infections; however, it should be reiterated that many of these approaches are non-specific and can be easily adjusted for other conditions and diseases by simply changing the drug of choice or associated biomarkers.
Multi-functional therapies
The beneficial effect of combinatorial and repeated chemotherapy has been known since the 1960s, when it was first used to treat acute lymphoblastic leukaemia. While highly effective, one of the biggest concerns of combinatorial therapy is the systemic cytotoxicity.54 Initial research in the biomaterials field generally adapted existing materials approaches in order to more efficiently coordinate the delivery of multiple therapeutics. For instance, in 2005 Sengupta and colleagues developed a system termed “nanocells” for sequential drug delivery to improve cancer treatments.55 These nanocells consisted of a pegylated-phospholipid block-copolymer envelope that encapsulated combretastatin and a nuclear nanoparticle made of poly(lactic-co-glycolic) acid (PLGA) conjugated with Dox. Following accumulation in the tumor via the EPR effect, combretastatin was released within 12 h and promoted the collapse of the vasculature. Meanwhile, Dox was conjugated to PLGA, which gave rise to a release profile that extended over 15 days. Since Dox-PLGA is only bioactive as small fragments, this strategy delayed the temporal activation of Dox relative to combretastatin. When comparing similar dosages, in vivo experiments revealed that nanocells resulted in a significant decrease in the size of tumors compared to no treatment, nanocells with just Dox or combretastatin, and simultaneous systemic injections.
More recently, Lee and colleagues demonstrated that inhibition of epidermal growth factor receptor by erlotinib reprograms cells from triple-negative breast cancer and non-small cell lung cancer to be more susceptible to death by DNA damage. Subsequent treatment with Dox 24 h after erlotinib results in dramatically more cell death than simultaneous delivery or delivery staggered by only 1 h.56 These results provide compelling evidence that not only are the drugs being delivered important but also the release times and sequence of therapies. In order to take advantage of this effect, Morton et al.57 designed a liposome-based system that initially released erlotinib with first-order release kinetics (80% released at 50 h), followed by a slower linear release of Dox (35% released at 50 h) (Figure 2). These liposome systems were further functionalized with folate to improve in vivo targeting, since folate receptors are up-regulated in numerous cancers including ovarian carcinomas, endometrial carcinomas, and certain prostate cancers.58 In vivo studies with xenograft-bearing NCr nude mice found increased shrinkage of tumors compared to liposomes loaded with only Dox.
Figure 2.
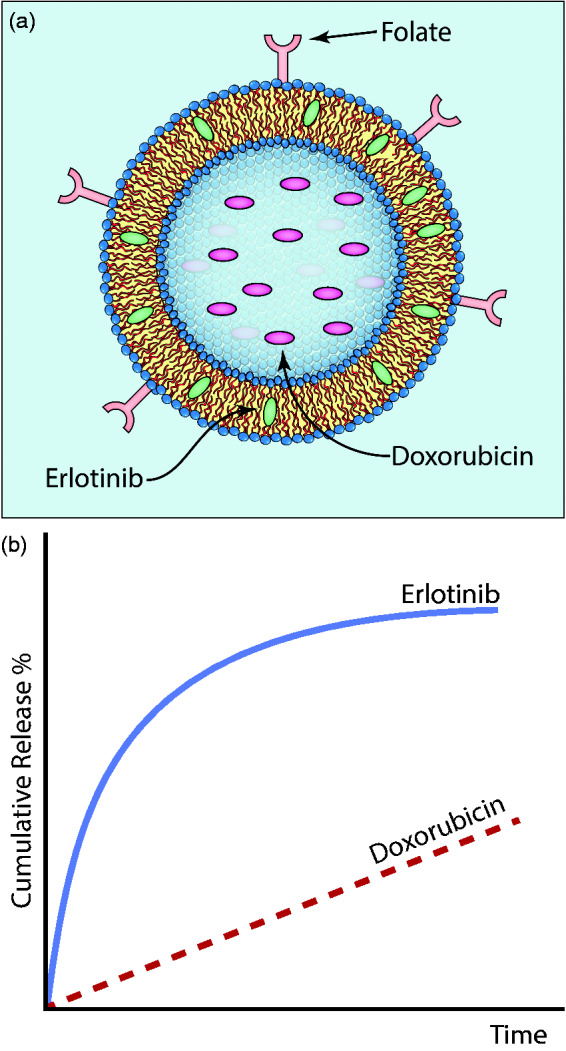
(a) Morton et al.57 have designed a liposome to target cancer cells and stagger the release of encapsulated therapeutics. In this case, folate conjugated to the surface of liposomes promoted enhanced binding to triple-negative breast cancer cells. Erlotinib loads into the core of the lipid bilayer due to its hydrophobic nature, while Dox loads into the aqueous vesicle core. (b) This arrangement of therapeutics allows for the initial release of erlotinib, followed by a slower release of Dox. Using this specific sequence of therapy results in the rewiring of cancer cells to make them more susceptible to DNA damage, dramatically increasing the efficacy of this combination of drugs. (A color version of this figure is available in the online journal.)
Another recent report explored the development of a layer-by-layer nanoparticle to synergistically block MAPK and PI3K,59 two cellular signaling pathways that have significant crosstalk and feedback and can enable signaling associated with drug resistance. In this study, the authors delivered selumetinib (to block Mek1/2) and PX-866 (to inhibit PI3K) as a means to treat a triple-negative breast cancer cell line and a lung cancer cell line possessing a RAS mutation, determining that delivery via nanoparticles increased cancer cell death over treatment with just free drug.
In 2011, von Maltzahn et al.60 reported on an interesting nanoparticle system that recruits complimentary nanoparticles by using the signaling cascade of targeted cells as the means of interaction. They devised a two-component nanoparticle system where the first nanoparticle activates a coagulation cascade locally in a tumor followed by a second nanoparticle that recognizes fibrin and targets the enzymatic activity during coagulation. Activation of the coagulation cascade is achieved either through the use of gold nanorods, which can be heated using near-infrared light, or tumor-targeting human protein tissue factor (tTF-RGD), which induces coagulation by binding to the angiogenic αvβ3 receptor. After confirmation of coagulation, the team delivered Dox-loaded liposomes with fibrin-binding peptides and coagulation transglutaminase FXIII to ensure homing towards the coagulation regions.
Another method of enhancing multi-functionality of nanoparticles is by adding coatings that interact with cancer cell-specific membrane receptors. Hyaluronic acid is especially suitable in this regard as it is a natural ligand of CD44,61 which is up-regulated in a variety of cancers, rapidly degradable, and easily modified to form hydrogels that respond to changes in environment.62 In one approach, Jiang et al.63 developed a system consisting of a TRAIL-loaded hyaluronic acid-based outer shell and Dox-loaded liposome-based inner shell, which was further modified with the cell penetrating peptide R8H3. Upon arrival at the tumor, the nanoparticle’s outer shell rapidly degrades due to hyaluronidase, releasing TRAIL and thus triggering the caspace-3 signaling pathway involved in programmed cell death. Following this, the inner liposome breaks down to release Dox. This system of sequential delivery showed synergistic effects when tested on MDA-MB-231 cells in vitro and MDA-MB-231 xenograft mouse models in vivo.
Nucleotide-based approaches
Many groups have explored responsive nucleotide approaches for treating cancer that aim to ensure the drug will be released via a stimulus inside the target cell. Sun et al.64 have developed a self-degradable DNA and anticancer drug delivery system for Dox. The nanoparticle, termed a nanoclew, is composed of folic acid conjugated to DNA with electrostatically bound Dox and positively charged DNase nanocapsules. The nanocapsules consist of a polymeric shell with acid-sensitive cross linkers that prevent DNase from being released at physiological pH. However, when the particles are taken up by cancer cells and enter the endolysosome, the polymer shell degrades due to the low acidity, releasing the DNase. The DNase then breaks down the DNA backbone of the nanoclew particle, in turn releasing the electrostatically bound Dox.
Another approach has been developed by the Artzi group and uses RNA nanoparticles that target-specific mRNA sequences.65,66 In one example, the Artzi group attached antisense DNA hairpins to gold nanoparticles.44 The hairpins were complimentary to the mRNA sequence of multidrug resistance protein 1 (MRP1) and contained intercalated 5-fluorouracil (5-FU) within the double helix (Figure 3). The functionalized nanoparticles were placed inside an injectable hydrogel to facilitate localized delivery and retention of the therapeutic nanoparticles to specific tumor locations in an orthotopic breast cancer mouse model using MDA-MB-231 triple negative breast cancer cells. Upon delivery, cells took up the functionalized gold nanoparticles, and if MRP1 mRNA was present, the hairpins hybridized with the mRNA to silence it and promote the release of 5-FU. Results from the in vivo testing showed a decrease in tumor size of approximately 90%, along with significant silencing of MRP1.
Figure 3.

Nanoparticles have been developed that contain DNA antisense oligonucleotide hairpins conjugated to gold nanoparticles. 5-FU can then be loaded within the double helix of the hairpins. Following entry into cancer cells, the DNA hairpins hybridize with and silence the mRNA associated with 5-FU drug resistance while simultaneously releasing the bound 5-FU. This strategy has been shown to significantly increase effectiveness of 5-FU on 5-FU resistant cancer cells.44 (A color version of this figure is available in the online journal.)
Other groups have also explored using nucleic acids as targeting vectors. Aptamer-siRNA chimeras have shown great promise in targeted delivery of siRNA into cells for a variety of diseases including cancer67–69 and HIV.70–72 These systems have two levels of specificity, the aptamer antigen and the siRNA target. Thus, this bimodal targeting strategy requires cells to possess both the ligand for the aptamer and the target mRNA for siRNA interference. In the context of prostate cancer, chimeras have been developed that bind to prostate-specific membrane antigen (PSMA).73 In vitro studies have shown increased cellular uptake and decreased cell viability for PSMA expressing cell lines, while in vivo studies with nude mice have shown decreases in tumor size for only tumors that express PSMA. Other groups have investigated DNA-protein chimeras, which work in a similar manner albeit with the minor change of delivering a protein rather than siRNA.74,75 Ultimately, these chimera models can be adapted for conjugation onto nanoparticles or hydrogels for more specific and localized delivery and potentially can be used to deliver other non-coding RNA such as miRNA, circRNA, or lncRNA.
Along with using nucleic acids to enhance or reduce protein expression, they can also be used as binding sites in order to “refill” depots with a drug of interest. Employing hydrogels as drug depots coupled with aptamers as drug binding sites offers a promising delivery approach. Brudno et al.76 recently pioneered the use of refillable hydrogels containing either specific nucleotide sequences or click chemistry groups25 that can bind drugs modified with the complementary binding partner (Figure 4). These hydrogel depots concentrate free drug to a specific area, in turn achieving high local concentration while mitigating potential off-target effects endemic to systemic delivery. Furthermore, the ability to reload these systems minimizes the need for numerous medical interventions, which in many cases may necessitate surgery. This platform, while initially studied as a drug delivery system for tumors, has vast appeal in a wide variety of contexts including bacterial infections, drug eluting stents, and osteoarthritis where either concentrated, but localized release of drugs is critical or accessibility to the target location is problematic.77
Figure 4.

Click chemistry has been used to concentrate systemic drugs at the site of interest,25,95 which can be accomplished in several ways. (Top) Hydrogels functionalized with a molecule such as DBCO can be injected into a localized tissue site. Subsequent injection with azide-tagged therapeutics enables concentration at the hydrogel site via the click reaction. (Bottom) Cells can also be labeled with azide groups to directly target the cells themselves. Subsequent injection with therapeutics labeled with the complimentary click group, in this case DBCO, results in concentration at the membranes of the cells expressing azides followed by subsequent uptake. (A color version of this figure is available in the online journal.)
From the point of programmable nucleotide platforms, treatment of infectious diseases and cancer shares many similarities. With the recent rise in antibiotic resistance, localized targeting, combinatorial therapies, and consistent delivery will help to extend the useful life of different antibiotics and prevent the outbreak of antibiotic-resistant bacteria such as MRSA, PRSP, and MDR Pseudomonas aeruginosa.78,79 In this respect, functional nucleotides have the ability to combine multiple strategies and approaches in order to effectively eliminate bacteria. Although selectively targeting bacteria necessitates a different set of objectives and targets than targeting cancer cells, recent research highlighting the importance of minimizing the impact antibiotics have on the natural microbiome justifies the development of more effective and precise therapies.80,81
Similar to the previously delineated drug depot approach, nucleotide-based approaches have shown great promise in both delivering localized therapies and controlling bacteria-specific drug release. Zhang et al.82 have developed oligonucleotide-functionalized hydrogels that can act as both loading and release sites for antibiotics. Using tetracycline and two separate nucleotide sequences, they measured a large increase in antibiotic uptake with only a slight increase in release time, which they attribute to using oligonucleotides with low affinities. Compared to hydrogels with no oligonucleotides, they delayed the formation of Escherichia coli colonies by 24 h. Importantly, the refilled hydrogels exhibited comparable release profiles to new hydrogels, allowing for localized uptake and release of antibiotics over multiple iterations.
Using a similar nucleotide approach, Kavruk et al.83 have developed aptamer-gated mesoporous silica nanoparticles loaded with vancomysin, which they have termed NanoKeepers.84 The aptamers blocked the pores and only opened when they bound the target molecule on the surface of the target bacteria, in this case Staphylococcus aureus. Compared to Staphylococcus epidermidis, which does not express the same aptamer antigens, S. aureus showed a 15-fold decrease in minimum inhibitory concentration. As expected, there was a consistent decrease in colony-forming units (CFU) for 24 h from 103/mL to slightly below 100/mL. This is compared to a final CFU count of 109/mL after 24 h of unchecked growth. While this approach is not explicitly stated as refillable, these nanoparticles can be easily localized and reloaded using additional aptamers or click chemistries without losing their efficacy or potency.
From a broader perspective, the literature is still scarce in this field and to our knowledge only one other group has developed an aptamer-nanoparticle approach for antibiotic release.85 Considering the wide range of known aptamers that target bacteria86–88 and recent developments in targeting cancer cells (previously described), there is the potential to develop new treatment options for a variety of pathogens that have been stubborn or immune to more traditional therapies.
Click chemistry
As was highlighted earlier, drug depots can be used with either aptamers or click sites as targeted binding sites for drug reloading. Similarly, many of the examples previously outlined can also be modified for use with click chemistry. Historically, click chemistries have been employed for synthesizing cancer therapies and nanoparticles themselves;89 however, only recently have they been used as a mechanism by which therapies can be made to responsively adapt dynamically to changes in the cancer microenvironment.
While the advent of copper-free click chemistry enabled in vivo experiments, the ability to generate azides in vivo through metabolic labeling has opened a promising avenue to further explore new therapies. The Bertozzi group has developed synthetic azidosugars that can be used to modify cell surfaces in vivo.90–92 One sugar in particular, tetraacetylated N-azidoacetyl-d-mannosamine (Ac4ManNAz), has been used to localize azides on cell surfaces in vivo via an interaction with sialic acids on the cell membrane.93 A drug with cyclooctyne moieties is then injected to react with the azides on the cell surface in vivo with rapid kinetics, ensuring the click reactions occur before metabolic clearance will flush out the drug (Figure 4).
Using this approach, Kim and colleagues have been able to develop a therapy targeting lung cancer cells.94,95 Initially, chitosan nanoparticles loaded with Ac4ManNAz were injected into tumor-bearing mice and taken up at the site of the tumor via the EPR effect.95 Once in the cytosol, the Ac4ManNAz generated azides on the cell surface as described previously. A second chitosan nanoparticle, this time surface functionalized with bicyclo[6.1.0]nonyne (BCN) and loaded with photosensitizer chlorine e6, was then injected intravenously. BCN then underwent a click reaction with the azides on the surface, leading to specific uptake and enabling subsequent phototherapy. This strategy offers a particular advantage over antigen-based targeting since click reactions are not receptor limited and a significant amount of azides can be generated on the cell membrane.
In the context of bacterial infections, click chemistry approaches for polymer coatings have gained headway as promising strategies for contact killing and/or fouling resistance.96 Shakiba et al.97 have developed a dual orthogonal click reaction approach that allows for highly customizable surface modifications for subsequent small molecule and peptide immobilization. They initially designed a set of “adsorbate” structures that bind to gold surfaces with an azide group on the terminal end. They then synthesized a middle molecule, a maleimide-terminated alkyne composed of two click groups. The alkyne reacts with the azide on the surface while the maleimide group performs another click addition with thiols via thiol-Michael addition, in this case cysteine-terminated poly(L-lysine). Due to the wide variety of click reactions, this strategy of using a dual click functionalized “intermediary” molecule allows for adaptable surfaces that can offer the advantages of orthogonal reactions without having to change the surface chemistry of the biomaterial in vivo, often a much harder issue to overcome, while still having flexibility with the antimicrobial strategy.
Biomaterials for cell recruitment
Taking a different approach, groups have also looked at using programmable biomaterials to deliver specific stimuli to modulate the behavior of cells in vivo. While many of the examples previously expounded can fall in this category, the focus here is to delineate strategies where the cell will become the therapeutic agent rather than a small molecule drug, a nucleic acid, or nanoparticle.
The pioneering work by the Mooney Lab on programmable vaccines to fight cancer was one of the first systems to employ a cell recruitment strategy.98,99 These approaches look at modulating dendritic cells by (1) recruiting cells in an engineering biomaterial using cytokines, (2) presenting the antigen of interest to the cells, and (3) releasing them back into the environment to target the tumor. More recently, they demonstrated that injectable hydrogels loaded with mesoporous silica rods spontaneously self-assemble into a 3D microenvironment that allows for cells to migrate through the pores between rods.30 It was found that the high aspect ratio of these rods results in many CD11c+ dendritic cells being recruited to the site of injection. The hydrogel was also loaded with granulocyte-macrophage colony-stimulating factor, unmethylated cytosine-phosphate-guanine oligonucleotide sequence, and ovalbumin. This combination treatment recruits and stimulates dendritic cell growth, programs them to respond to a specific Toll-like receptor (TLR9), and then modulates downstream effects in draining lymph nodes, specifically by promoting the maturation of B cells.
Other strategies have sought to program cell behaviour using light as a physical stimulus. Lee et al.100 have designed a light-triggered system to selectively activate caged arginylglycylaspartic acid (RGD) in hydrogels to modulate inflammation. While light-triggered RGD release had previously been studied,101 this new approach has demonstrated great spatial and temporal control, thus allowing the group to finely control inflammation and vascularization following implantation of a non-fouling hydrogel. Upon activation of RGD via UV light, numerous neutrophils (NIMP-R14+) and macrophages (CD68+) were seen present near the hydrogel. Interestingly, this study was able to directly compare the temporal effect of RGD on a single platform, showing that delayed release of RGD leads to less fibrosis and vascularization.
Conclusion and outlook
In this review, several examples of programmable biomaterials that exhibit both dynamic and temporal modalities have been highlighted as the next generation of strategies for treating complex diseases. The ability to evolve concomitantly with the disease and offer multi-pronged treatment options provides researchers, clinicians, and patients with more effective therapeutics and new capabilities.
Certain outstanding challenges must still be addressed, both from an engineering and biological perspective. From the perspective of delivery platforms, nanoparticle-based approaches still predominantly rely on the EPR effect.102 While this has distinct advantages, it is a largely passive process that has fundamental limits of efficacy, including reliance on vascularization, the relatively rapid rate of clearance by the renal system, and potential immune response from long-term treatments. Hydrogels, on the other hand, are able to circumvent some of these issues due to their ability to be placed near the site of interest and locally deliver drugs over a prolonged period of time.103,104 Further research is necessary to both determine what biological markers are appropriate for developing responsive elements and the effectiveness of that response. We envision that such materials may identify sudden changes to tumor or bacterial environments and deliver the appropriate drug without the need for clinical intervention.
Recent years have shown an ever expanding toolbox of technologies at researchers’ and clinicians’ disposal.105,106 At the same time, further work is still necessary to continue this positive trend. For example, oligonucleotide approaches are still largely one dimensional in their uses (e.g. react to a stimulus to release a drug). Due to the simplicity of oligonucleotides and their ability to be readily incorporated onto virtually any platform, future systems may strive towards increasing multi-functionality (e.g. theranostic capabilities coupled with readouts of efficacy). While researchers have begun to use biological moieties, we believe that there is still great potential to further expand this approach. For example, the approaches outlined for cell recruitment98 and using biological cascades to achieve better drug delivery60 are innovative and potentially promising strategies.
Ultimately, the success and justification of these therapies will hinge on their ability to not only surpass systemic therapies but also the advantages of previous generations of nanomedicine. In the end, it is the ability to engineer interactions between materials and cells to be a bi-directional feedback system, one that is able to actively adapt and transform in response to cellular changes, that will unlock new potential for treating the most stubborn of diseases.
Acknowledgements
MTK acknowledges the Whitaker International Program for fellowship support. AS acknowledges the Department of Bioengineering at Imperial College London for PhD studentship support.
Authors’ contributions
The first two authors contributed equally to this paper. All authors contributed to the conception, research, and writing of this review article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144: 646–74. [DOI] [PubMed] [Google Scholar]
- 2.Costerton JW. Bacterial biofilms: a common cause of persistent infections. Science 1999; 284: 1318–22. [DOI] [PubMed] [Google Scholar]
- 3.Loeb LA, Loeb KR, Anderson JP. Multiple mutations and cancer. Proc Natl Acad Sci U S A 2003; 100: 776–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stewart PS, Costerton JW. Antibiotic resistance of bacteria in biofilms. Lancet 2001; 358: 135–8. [DOI] [PubMed] [Google Scholar]
- 5.Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, Buechler P, Isaacs WB, Semenza GL, Simons JW. Overexpression of hypoxia-inducible factor 1α in common human cancers and their metastases. Canc Res 1999; 59: 5830–5. [PubMed] [Google Scholar]
- 6.Schuetz P, Christ-Crain M, Müller B. Biomarkers to improve diagnostic and prognostic accuracy in systemic infections. Curr Opin Crit Care 2007; 13: 578–85. [DOI] [PubMed] [Google Scholar]
- 7.Sanvicens N, Marco MP. Multifunctional nanoparticles – properties and prospects for their use in human medicine. Trends Biotechnol 2008; 26: 425–33. [DOI] [PubMed] [Google Scholar]
- 8.Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov 2008; 7: 771–82. [DOI] [PubMed] [Google Scholar]
- 9.Wang AZ, Langer R, Farokhzad OC. Nanoparticle delivery of cancer drugs. Annu Rev Med 2012; 63: 185–98. [DOI] [PubMed] [Google Scholar]
- 10.Al-Lazikani B, Banerji U, Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nat Biotechnol 2012; 30: 679–92. [DOI] [PubMed] [Google Scholar]
- 11.Oliva N, Unterman S, Zhang Y, Conde J, Song HS, Artzi N. Personalizing biomaterials for precision nanomedicine considering the local tissue microenvironment. Adv Healthc Mater 2015; 4: 1584–99. [DOI] [PubMed] [Google Scholar]
- 12.Pacardo DB, Ligler FS, Gu Z. Programmable nanomedicine: synergistic and sequential drug delivery systems. Nanoscale 2015; 7: 3381–91. [DOI] [PubMed] [Google Scholar]
- 13.Zazo H, Colino CI, Lanao JM. Current applications of nanoparticles in infectious diseases. J Contr Release 2016; 224: 86–102. [DOI] [PubMed] [Google Scholar]
- 14.Wohl BM, Engbersen JFJ. Responsive layer-by-layer materials for drug delivery. J Contr Release 2012; 158: 2–14. [DOI] [PubMed] [Google Scholar]
- 15.De M, Ghosh PS, Rotello VM. Applications of nanoparticles in biology. Adv Mater 2008; 20: 4225–41. [Google Scholar]
- 16.Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol 2015; 33: 941–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev 2013; 65: 36–48. [DOI] [PubMed] [Google Scholar]
- 18.Costa RR, Alatorre-Meda M, Mano JF. Drug nano-reservoirs synthesized using layer-by-layer technologies. Biotechnol Adv 2015; 33: 1310–26. [DOI] [PubMed] [Google Scholar]
- 19.Kesharwani P, Jain K, Jain NK. Dendrimer as nanocarrier for drug delivery. Prog Polym Sci 2014; 39: 268–307. [Google Scholar]
- 20.Alkilany AM, Thompson LB, Boulos SP, Sisco PN, Murphy CJ. Gold nanorods: their potential for photothermal therapeutics and drug delivery, tempered by the complexity of their biological interactions. Adv Drug Deliv Rev 2012; 64: 190–9. [DOI] [PubMed] [Google Scholar]
- 21.Hoffman AS. Hydrogels for biomedical applications. Adv Drug Deliv Rev 2012; 64: 18–23. [DOI] [PubMed] [Google Scholar]
- 22.Mealy JE, Rodell CB, Burdick JA. Sustained small molecule delivery from injectable hyaluronic acid hydrogels through host-guest mediated retention. J Mater Chem B Mater Biol Med 2015; 3: 8010–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoare TR, Kohane DS. Hydrogels in drug delivery: progress and challenges. Polymer 2008; 49: 1993–2007. [Google Scholar]
- 24.Strong LE, Dahotre SN, West JL. Hydrogel-nanoparticle composites for optically modulated cancer therapeutic delivery. J Contr Release 2014; 178: 63–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brudno Y, Desai RM, Kwee BJ, Joshi NS, Aizenberg M, Mooney DJ. In vivo targeting through click chemistry. ChemMedChem 2015; 10: 617–20. [DOI] [PubMed] [Google Scholar]
- 26.Murphy NP, Lampe KJ. Mimicking biological phenomena in hydrogel-based biomaterials to promote dynamic cellular responses. J Mater Chem B 2015; 3: 7867–80. [DOI] [PubMed] [Google Scholar]
- 27.Patterson J, Hubbell JA. Enhanced proteolytic degradation of molecularly engineered PEG hydrogels in response to MMP-1 and MMP-2. Biomaterials 2010; 31: 7836–45. [DOI] [PubMed] [Google Scholar]
- 28.O’Brien FJ, Harley BA, Yannas IV, Gibson LJ. The effect of pore size on cell adhesion in collagen-GAG scaffolds. Biomaterials 2005; 26: 433–41. [DOI] [PubMed] [Google Scholar]
- 29.Rape AD, Zibinsky M, Murthy N, Kumar S. A synthetic hydrogel for the high-throughput study of cell-ECM interactions. Nat Commun 2015; 6: 8129–8129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim J, Li WA, Choi Y, Lewin SA, Verbeke CS, Dranoff G, Mooney DJ. Injectable, spontaneously assembling, inorganic scaffolds modulate immune cells in vivo and increase vaccine efficacy. Nat Biotechnol 2015; 33: 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hetrick EM, Schoenfisch MH. Reducing implant-related infections: active release strategies. Chem Soc Rev 2006; 35: 780–9. [DOI] [PubMed] [Google Scholar]
- 32.Jeon G, Yang SY, Kim JK. Functional nanoporous membranes for drug delivery. J Mater Chem 2012; 22: 14814–34. [Google Scholar]
- 33.Tamura M, Yanagawa F, Sugiura S, Takagi T, Sumaru K, Kanamori T. Click-crosslinkable and photodegradable gelatin hydrogels for cytocompatible optical cell manipulation in natural environment. Sci Rep 2015; 5: 15060–15060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li N, Binder WH. Click-chemistry for nanoparticle-modification. J Mater Chem 2011; 21: 16717–34. [Google Scholar]
- 35.Jewett JC, Bertozzi CR. Cu-free click cycloaddition reactions in chemical biology. Chem Soc Rev 2010; 39: 1272–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meldal M, Tornøe CW. Cu-catalyzed azide-alkyne cycloaddition. Chem Rev 2008; 108: 2952–3015. [DOI] [PubMed] [Google Scholar]
- 37.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Copper-free click chemistry for dynamic in vivo imaging. Proc Natl Acad Sci U S A 2007; 104: 16793–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao W, Schafer S, Choi J, Yamanaka YJ, Lombardi ML, Bose S, Carlson AL, Phillips JA, Teo W, Droujinine IA, Cui CH, Jain RK, Lammerding J, Love JC, Lin CP, Sarkar D, Karnik R, Karp JM. Cell-surface sensors for real-time probing of cellular environments. Nat Nanotechnol 2011; 6: 524–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Böhme D, Beck-Sickinger AG. Drug delivery and release systems for targeted tumor therapy. J Pept Sci 2015; 21: 186–200. [DOI] [PubMed] [Google Scholar]
- 40.Alley SC, Okeley NM, Senter PD. Antibody-drug conjugates: targeted drug delivery for cancer. Curr Opin Chem Biol 2010; 14: 529–37. [DOI] [PubMed] [Google Scholar]
- 41.Tyagi S, Kramer FR. Molecular beacons: probes that fluoresce upon hybridization. Nat Biotechnol 1996; 14: 303–8. [DOI] [PubMed] [Google Scholar]
- 42.Zheng J, Yang R, Shi M, Wu C, Fang X, Li Y, Li J, Tan W. Rationally designed molecular beacons for bioanalytical and biomedical applications. Chem Soc Rev 2015; 44: 3036–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marras SAE. Selection of fluorophore and quencher pairs for fluorescent nucleic acid hybridization probes. Meth Mol Biol 2006; 335: 3–16. [DOI] [PubMed] [Google Scholar]
- 44.Conde J, Oliva N, Artzi N. Implantable hydrogel embedded dark-gold nanoswitch as a theranostic probe to sense and overcome cancer multidrug resistance. Proc Natl Acad Sci U S A 2015; 112: E1278–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu G, Niu G, Chen X. Aptamer-drug conjugates. Bioconjug Chem 2015; 26: 2186–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Z, Duan J-H, Song Y-M, Ma J, Wang F-D, Lu X, Yang X-D. Novel HER2 aptamer selectively delivers cytotoxic drug to HER2-positive breast cancer cells in vitro. J Transl Med 2012; 10: 148–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou J, Rossi JJ. Cell-type-specific, aptamer-functionalized agents for targeted disease therapy. Mol Ther Nucleic Acids 2014; 3: e169–e169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990; 249: 505–10. [DOI] [PubMed] [Google Scholar]
- 49.Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature 1990; 346: 818–22. [DOI] [PubMed] [Google Scholar]
- 50.Keefe AD, Pai S, Ellington A. Aptamers as therapeutics. Nat Rev Drug Discov 2010; 9: 537–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Braasch DA, Corey DR. Locked nucleic acid (LNA): fine-tuning the recognition of DNA and RNA. Chem Biol 2001; 8: 1–7. [DOI] [PubMed] [Google Scholar]
- 52.Ray A, Norden B. Peptide nucleic acid (PNA): its medical and biotechnical applications and promise for the future. FASEB J 2000; 14: 1041–60. [DOI] [PubMed] [Google Scholar]
- 53.Surana S, Shenoy AR, Krishnan Y. Designing DNA nanodevices for compatibility with the immune system of higher organisms. Nat Nanotechnol 2015; 10: 741–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.DeVita VT, Chu E. A history of cancer chemotherapy. Canc Res 2008; 68: 8643–53. [DOI] [PubMed] [Google Scholar]
- 55.Sengupta S, Eavarone D, Capila I, Zhao G, Watson N, Kiziltepe T, Sasisekharan R. Temporal targeting of tumour cells and neovasculature with a nanoscale delivery system. Nature 2005; 436: 568–72. [DOI] [PubMed] [Google Scholar]
- 56.Lee MJ, Ye AS, Gardino AK, Heijink AM, Sorger PK, MacBeath G, Yaffe MB. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell 2012; 149: 780–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Morton SW, Lee MJ, Deng ZJ, Dreaden EC, Siouve E, Shopsowitz KE, Shah NJ, Yaffe MB, Hammond PT. A nanoparticle-based combination chemotherapy delivery system for enhanced tumor killing by dynamic rewiring of signaling pathways. Sci Signal 2014; 7: ra44–ra44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parker N, Turk MJ, Westrick E, Lewis JD, Low PS, Leamon CP. Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Anal Biochem 2005; 338: 284–93. [DOI] [PubMed] [Google Scholar]
- 59.Dreaden EC, Kong YW, Morton SW, Correa S, Choi KY, Shopsowitz KE, Renggli K, Drapkin R, Yaffe MB, Hammond PT. Tumor-targeted synergistic blockade of MAPK and PI3K from a layer-by-layer nanoparticle. Clin Canc Res 2015; 21: 4410–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Von Maltzahn G, Park J-H, Lin KY, Singh N, Schwöppe C, Mesters R, Berdel WE, Ruoslahti E, Sailor MJ, Bhatia SN. Nanoparticles that communicate in vivo to amplify tumour targeting. Nat Mater 2011; 10: 545–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.John CWY, Abatangelo G. Functions of hyaluronan in wound repair. Wound Repair Regen 1999; 7: 79–89. [DOI] [PubMed] [Google Scholar]
- 62.Burdick JA, Prestwich GD. Hyaluronic acid hydrogels for biomedical applications. Adv Mater 2011; 23: H41–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jiang T, Mo R, Bellotti A, Zhou J, Gu Z. Gel-liposome-mediated co-delivery of anticancer membrane-associated proteins and small-molecule drugs for enhanced therapeutic efficacy. Adv Funct Mater 2014; 24: 2295–304. [Google Scholar]
- 64.Sun W, Jiang T, Lu Y, Reiff M, Mo R, Gu Z. Cocoon-like self-degradable DNA nanoclew for anticancer drug delivery. J Am Chem Soc 2014; 136: 14722–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Conde J, Bao C, Tan Y, Cui D, Edelman ER, Azevedo HS, Byrne HJ, Artzi N, Tian F. Dual targeted immunotherapy via in vivo delivery of biohybrid RNAi-peptide nanoparticles to tumor-associated macrophages and cancer cells. Adv Funct Mater 2015; 25: 4183–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Segovia N, Pont M, Oliva N, Ramos V, Borrós S, Artzi N. Hydrogel doped with nanoparticles for local sustained release of siRNA in breast cancer. Adv Healthc Mater 2015; 4: 271–80. [DOI] [PubMed] [Google Scholar]
- 67.Gilboa-Geffen A, Hamar P, Le MTN, Wheeler LA, Trifonova R, Petrocca F, Wittrup A, Lieberman J. Gene knockdown by EpCAM aptamer-siRNA chimeras suppresses epithelial breast cancers and their tumor-initiating cells. Mol Canc Ther 2015; 14: 2279–91. [DOI] [PubMed] [Google Scholar]
- 68.Dassie JP, Liu X-Y, Thomas GS, Whitaker RM, Thiel KW, Stockdale KR, Meyerholz DK, McCaffrey AP, McNamara JO, Giangrande PH. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat Biotechnol 2009; 27: 839–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lai W-Y, Wang W-Y, Chang Y-C, Chang C-J, Yang P-C, Peck K. Synergistic inhibition of lung cancer cell invasion, tumor growth and angiogenesis using aptamer-siRNA chimeras. Biomaterials 2014; 35: 2905–14. [DOI] [PubMed] [Google Scholar]
- 70.Neff CP, Zhou J, Remling L, Kuruvilla J, Zhang J, Li H, Smith DD, Swiderski P, Rossi JJ, Akkina R. An aptamer-siRNA chimera suppresses HIV-1 viral loads and protects from helper CD4(+) T cell decline in humanized mice. Sci Transl Med 2011; 3: 66ra6–66ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhou J, Li H, Li S, Zaia J, Rossi JJ. Novel dual inhibitory function aptamer-siRNA delivery system for HIV-1 therapy. Mol Ther 2008; 16: 1481–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wheeler LA, Trifonova R, Vrbanac V, Basar E, McKernan S, Xu Z, Seung E, Deruaz M, Dudek T, Einarsson JI, Yang L, Allen TM, Luster AD, Tager AM, Dykxhoorn DM, Lieberman J. Inhibition of HIV transmission in human cervicovaginal explants and humanized mice using CD4 aptamer-siRNA chimeras. J Clin Invest 2011; 121: 2401–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McNamara JO, Andrechek ER, Wang Y, Viles KD, Rempel RE, Gilboa E, Sullenger BA, Giangrande PH. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat Biotechnol 2006; 24: 1005–15. [DOI] [PubMed] [Google Scholar]
- 74.Zhang Z, Li S, Chen N, Yang C, Wang Y. Programmable display of DNA-protein chimeras for controlling cell-hydrogel interactions via reversible intermolecular hybridization. Biomacromolecules 2013; 14: 1174–80. [DOI] [PubMed] [Google Scholar]
- 75.Pippig DA, Baumann F, Strackharn M, Aschenbrenner D, Gaub HE. Protein-DNA chimeras for nano assembly. ACS Nano 2014; 8: 6551–5. [DOI] [PubMed] [Google Scholar]
- 76.Brudno Y, Silva EA, Kearney CJ, Lewin SA, Miller A, Martinick KD, Aizenberg M, Mooney DJ. Refilling drug delivery depots through the blood. Proc Natl Acad Sci U S A 2014; 111: 12722–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Couvreur P. Drug delivery: replenishing reservoirs in vivo. Nat Nanotechnol 2014; 9: 874–5. [DOI] [PubMed] [Google Scholar]
- 78.Hawkey PM, Jones AM. The changing epidemiology of resistance. J Antimicrob Chemother 2009; 64: i3–10. [DOI] [PubMed] [Google Scholar]
- 79.Nikaido H. Multidrug resistance in bacteria. Annu Rev Biochem 2009; 78: 119–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jakobsson HE, Jernberg C, Andersson AF, Sjölund-Karlsson M, Jansson JK, Engstrand L. Short-term antibiotic treatment has differing long-term impacts on the human throat and gut microbiome. PLoS One 2010; 5: e9836–e9836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sommer MOA, Dantas G, Church GM. Functional characterization of the antibiotic resistance reservoir in the human microflora. Science 2009; 325: 1128–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang X, Soontornworajit B, Zhang Z, Chen N, Wang Y. Enhanced loading and controlled release of antibiotics using nucleic acids as an antibiotic-binding effector in hydrogels. Biomacromolecules 2012; 13: 2202–10. [DOI] [PubMed] [Google Scholar]
- 83.Kavruk M, Celikbicak O, Ozalp VC, Borsa BA, Hernandez FJ, Bayramoglu G, Salih B, Arica MY. Antibiotic loaded nanocapsules functionalized with aptamer gates for targeted destruction of pathogens. Chem Commun 2015; 51: 8492–5. [DOI] [PubMed] [Google Scholar]
- 84.Hernandez FJ, Hernandez LI, Kavruk M, Arıca YM, Bayramoğlu G, Borsa BA, Öktem HA, Schäfer T, Özalp VC. NanoKeepers: stimuli responsive nanocapsules for programmed specific targeting and drug delivery. Chem Commun 2014; 50: 9489–92. [DOI] [PubMed] [Google Scholar]
- 85.Sundaram P, Wower J, Byrne ME. A nanoscale drug delivery carrier using nucleic acid aptamers for extended release of therapeutic. Nanomedicine 2012; 8: 1143–51. [DOI] [PubMed] [Google Scholar]
- 86.Shum KT, Lui EL, Wong SC, Yeung P, Sam L, Wang Y, Watt RM, Tanner JA. Aptamer-mediated inhibition of Mycobacterium tuberculosis polyphosphate kinase 2. Biochemistry 2011; 50: 3261–71. [DOI] [PubMed] [Google Scholar]
- 87.Blum JH, Dove SL, Hochschild A, Mekalanos JJ. Isolation of peptide aptamers that inhibit intracellular processes. Proc Natl Acad Sci U S A 2000; 97: 2241–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chen F, Zhou J, Luo F, Mohammed A-B, Zhang X-L. Aptamer from whole-bacterium SELEX as new therapeutic reagent against virulent Mycobacterium tuberculosis. Biochem Biophys Res Commun 2007; 357: 743–8. [DOI] [PubMed] [Google Scholar]
- 89.Such GK, Johnston APR, Liang K, Caruso F. Synthesis and functionalization of nanoengineered materials using click chemistry. Prog Polym Sci 2012; 37: 985–1003. [Google Scholar]
- 90.Prescher JA, Dube DH, Bertozzi CR. Chemical remodelling of cell surfaces in living animals. Nature 2004; 430: 873–7. [DOI] [PubMed] [Google Scholar]
- 91.Saxon E, Luchansky SJ, Hang HC, Yu C, Lee SC, Bertozzi CR. Investigating cellular metabolism of synthetic azidosugars with the staudinger ligation. J Am Chem Soc 2002; 124: 14893–902. [DOI] [PubMed] [Google Scholar]
- 92.Saxon E, Bertozzi C. Cell surface engineering by a modified staudinger reaction. Science 2000; 287: 2007–10. [DOI] [PubMed] [Google Scholar]
- 93.Chang PV, Prescher JA, Sletten EM, Baskin JM, Miller IA, Agard NJ, Lo A, Bertozzi CR. Copper-free click chemistry in living animals. Proc Natl Acad Sci U S A 2010; 107: 1821–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Koo H, Lee S, Na JH, Kim SH, Hahn SK, Choi K, Kwon IC, Jeong SY, Kim K. Bioorthogonal copper-free click chemistry in vivo for tumor-targeted delivery of nanoparticles. Angew Chem Int Ed Engl 2012; 51: 11836–40. [DOI] [PubMed] [Google Scholar]
- 95.Lee S, Koo H, Na JH, Han SJ, Min HS, Lee SJ, Kim SH, Yun SH, Jeong SY, Kwon IC, Choi K, Kim K. Chemical tumor-targeting of nanoparticles based on metabolic glycoengineering and click chemistry. ACS Nano 2014; 8: 2048–63. [DOI] [PubMed] [Google Scholar]
- 96.Ganewatta MS, Miller KP, Singleton SP, Mehrpouya-Bahrami P, Chen YP, Yan Y, Nagarkatti M, Nagarkatti P, Decho AW, Tang C. Antibacterial and biofilm-disrupting coatings from resin acid-derived materials. Biomacromolecules 2015; 16: 3336–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shakiba A, Jamison AC, Lee TR. Poly(L-lysine) interfaces via dual click reactions on surface-bound custom-designed dithiol adsorbates. Langmuir 2015; 31: 6154–63. [DOI] [PubMed] [Google Scholar]
- 98.Kim J, Mooney DJ. In vivo modulation of dendritic cells by engineered materials: towards new cancer vaccines. Nano Today 2011; 6: 466–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ali OA, Huebsch N, Cao L, Dranoff G, Mooney DJ. Infection-mimicking materials to program dendritic cells in situ. Nat Mater 2009; 8: 151–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lee TT, García JR, Paez JI, Singh A, Phelps EA, Weis S, Shafiq Z, Shekaran A, Del Campo A, García AJ. Light-triggered in vivo activation of adhesive peptides regulates cell adhesion, inflammation and vascularization of biomaterials. Nat Mater 2015; 14: 352–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wirkner M, Weis S, San Miguel V, Álvarez M, Gropeanu RA, Salierno M, Sartoris A, Unger RE, Kirkpatrick CJ, del Campo A. Photoactivatable caged cyclic RGD peptide for triggering integrin binding and cell adhesion to surfaces. Chembiochem 2011; 12: 2623–9. [DOI] [PubMed] [Google Scholar]
- 102.Maeda H, Nakamura H, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliv Rev 2013; 65: 71–9. [DOI] [PubMed] [Google Scholar]
- 103.Schaal JL, Li X, Mastria E, Bhattacharyya J, Zalutsky MR, Chilkoti A, Liu W. Injectable polypeptide micelles that form radiation crosslinked hydrogels in situ for intratumoral radiotherapy. J Contr Release 2016; 228: 58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fakhari A, Subramony JA. Engineered in-situ depot-forming hydrogels for intratumoral drug delivery. J Contr Release 2015; 220: 465–75. [DOI] [PubMed] [Google Scholar]
- 105.Tang W, Becker ML. ‘Click’ reactions: a versatile toolbox for the synthesis of peptide-conjugates. Chem Soc Rev 2014; 43: 7013–39. [DOI] [PubMed] [Google Scholar]
- 106.Lutolf MP. Integration column: artificial ECM: expanding the cell biology toolbox in 3D. Integr Biol 2009; 1: 235–41. [DOI] [PubMed] [Google Scholar]