

Abstract
Lung adenocarcinomas with mutant epidermal growth factor receptor (EGFR) respond to EGFR-targeted tyrosine kinase inhibitors (TKIs), but resistance invariably occurs. We found that the Janus kinase (JAK)/signal transduction and activator of transcription 3 (STAT3) signaling pathway was aberrantly increased in TKI-resistant EGFR-mutant non–small cell lung cancer (NSCLC) cells. JAK2 inhibition restored sensitivity to the EGFR inhibitor erlotinib in TKI-resistant cell lines and xenograft models of EGFR-mutant TKI-resistant lung cancer. JAK2 inhibition uncoupled EGFR from its negative regulator, suppressor of cytokine signaling 5 (SOCS5), consequently increasing EGFR abundance and restoring the tumor cells’ dependence on EGFR signaling. Furthermore, JAK2 inhibition led to heterodimerization of mutant and wild-type EGFR subunits, the activity of which was then blocked by TKIs. Our results reveal a mechanism whereby JAK2 inhibition overcomes acquired resistance to EGFR inhibitors and support the use of combination therapy with JAK and EGFR inhibitors for the treatment of EGFR-dependent NSCLC.
INTRODUCTION
Lung cancer is the most frequent cause of cancer death (1), and non–small cell lung cancer (NSCLC) is the most common subtype. Somatic activating mutations of the tyrosine kinase domain of the epidermal growth factor receptor (EGFR) are found in about 26% of all patients with lung adenocarcinoma and confer sensitivity to first-generation EGFR tyrosine kinase inhibitors (TKIs), gefitinib and erlotinib (2, 3). Clinical responses are variable, although most patients exhibit good response rates to these inhibitors. However, acquired resistance to TKIs unfailingly occurs, in most cases (>60%) due to the acquisition of “gatekeeper” mutations (T790M) in the EGFR, which is thought to alter kinase ATP (adenosine 5′-triphosphate) affinity above that of gefitinib or erlotinib (4, 5). Progression-free survival with TKI treatment is only 9 to 12 months, and overall survival is less than 20 months (2, 3). Notably, the acquisition of secondary mutations in EGFR emphasizes a continued dependence on EGFR signaling in these cancers. The need to overcome both innate and acquired resistance has been a major therapeutic challenge.
EGFR is a member of the ERBB/human epidermal growth factor receptor (HER) family of membrane-bound receptor tyrosine kinases (RTKs) (6). Aberrant regulation of EGFR, including gain-of-function mutations and overexpression, is a common feature of many epithelial malignancies, which has led to the development of EGFR TKIs (7). We previously described that signal transduction and activator of transcription 3 (STAT3) is persistently tyrosine-phosphorylated or activated (pSTAT3) in NSCLC (cell lines and primary tumors) due to EGFR-driven up-regulation of interleukin-6 (IL-6) expression, leading to a feed-forward IL-6/Janus kinase (JAK)/STAT3 loop. Furthermore, JAK inhibition abrogates proliferation in NSCLC cell lines, including those that are TKI-resistant (8). JAK1/2 inhibitors have shown promise in preclinical models of NSCLC (9–15). Inhibitors of JAKs were developed for immunologic suppression for organ transplantation and for the treatment of myeloproliferative neoplasms in patients with activating mutations in the JAK2 pathway (16, 17) and are in early-phase clinical trials for lymphomas and solid tumors on the basis of promising preclinical studies (11–15, 18–20).
Our present study investigated the mechanisms by which JAK inhibition represses cell growth in NSCLC cells, alone or in combination with TKIs. Here, we found that JAK2 inhibition overcame acquired resistance to TKIs in EGFR-mutant lung adenocarcinoma in vitro and in vivo.
RESULTS
JAK2 inhibition resensitizes TKI-resistant cells and xenograft models to erlotinib
We previously demonstrated by immunohistochemistry that pSTAT3 is present in 42% of NSCLC cells that have wild-type EGFR and in 88% of NSCLC cells that have mutant EGFR, mediated through increased IL-6/JAK signaling (8). Further examination of this cohort of samples revealed that 31% of EGFR-mutant NSCLC tumors had high expression (immunohistochemistry) of pSTAT3. Here, we sought to determine the relevance of JAK/STAT3 activation in tumors that had developed resistance to TKIs. Patients with EGFR-mutant NSCLC had their tumors rebiopsied upon development of acquired resistance to erlotinib or gefitinib (hereafter referred to collectively as TKI) (5). We examined the abundance of pSTAT3 in 10 TKI-resistant tumors, 4 of which were matched against the untreated primary tumor. We determined that the abundance of pSTAT3 was high (score 2 to 3+) in 68% (4 of 6) of unmatched samples and either similar or increased in all four matched specimens compared to the respective pre-TKI samples (fig. S1A) (21, 22). These results led us to hypothesize that pSTAT3 may be a relevant target in TKI-resistant disease.
We tested this hypothesis by treating TKI-resistant, pSTAT3+ NSCLC cell lines (H1975, PC-9R, and H1650) and xenografts with a JAK inhibitor (JAKi; AZD1480) alone or in combination with a TKI (erlotinib) as a negative control. Treatment with JAKi reduced the abundance of pSTAT3 and inhibited the proliferation of cultured cells, with median inhibitory concentrations in the range of 0.25 to 1.50 μM (Fig. 1, A and B, and fig. S1B) (8, 10, 12, 23). Furthermore, in vivo studies demonstrated a significant inhibitory effect of JAKi as a single agent on the growth of NSCLC xenograft tumors (Fig. 1C). In contrast, TKI (erlotinib) alone did not inhibit the proliferation of TKI-resistant cell lines H1975 and PC-9R and partially inhibited the proliferation of semiresistant H1650 in vitro and in vivo (Fig. 1, A and C), as previously demonstrated (24–26). However, the TKI-resistant cell lines (H1975 and PC-9R) and the TKI-semiresistant cell line (H1650) were rendered sensitive to TKI by the addition of JAKi, evidenced by decreased cell viability and increased apoptosis in cultured cells (Fig. 1A and fig. S1C). The abundance of pSTAT3, phosphorylated EGFR (pEGFR), and the downstream effector phosphorylated extracellular signal–regulated kinase (pERK) in these cultured TKI-resistant cell lines were markedly reduced upon combination treatment with JAKi and TKI, whereas expectedly, TKI alone had no effect (Fig. 1B and fig. S1D). In vivo, dual blockade of JAK and EGFR led to the greatest inhibition of tumor growth when compared to either JAKi or TKI alone in H1975, PC-9R, and H1650 xenografts (Fig. 1C). The enhanced inhibitory effect of combination treatment was accompanied by decreased pSTAT3, pEGFR, and pERK abundance and reduced proliferation (by Ki67 staining) (fig. S1E). These data indicate that combined JAKi/TKI treatment is superior to monotherapy and can overcome resistance to EGFR inhibitors in EGFR-mutant NSCLC.
Fig. 1. Synergistic antiproliferative effects of combined EGFR blockade with JAK inhibition.
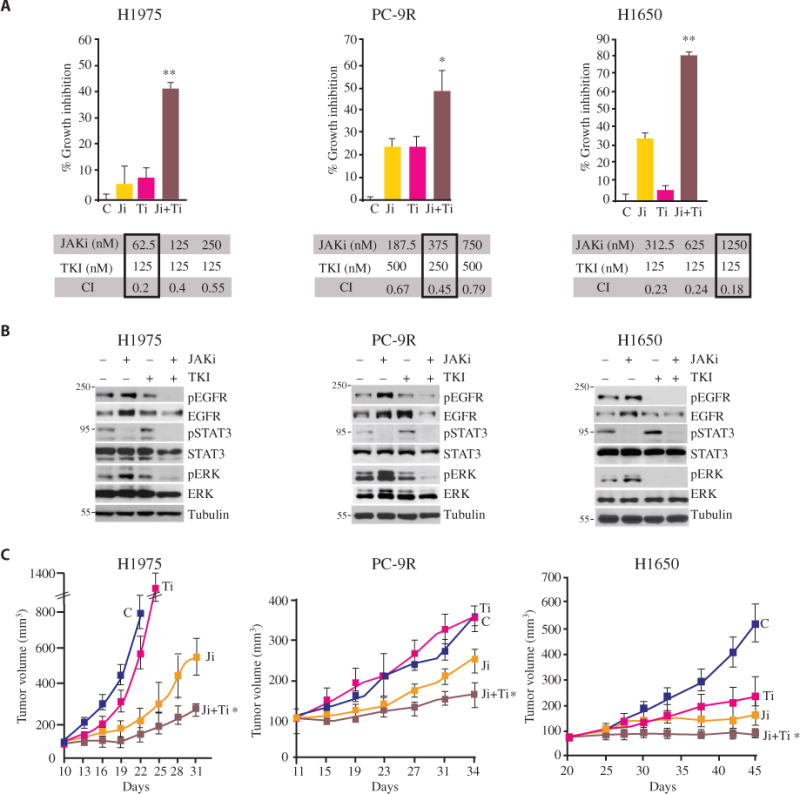
(A) MTT-based proliferation assay in H1975, PC-9R, and H1650 cells treated with the TKI erlotinib (Ti) in combination with the JAKi AZD1480 (Ji). Data are means ± SEM from five replicates in three independent experiments. *P < 0.05; **P < 0.001, versus AZD1480 alone (two-tailed Student’s t test). Below each graph are the representative combination indices (CIs) of erlotinib (TKI) in combination with AZD1480 (JAKi): CI < 0.9 indicates synergy, CI between 0.9 and 1.1 is additive, and CI > 1.1 indicates antagonism. The drug dosage combinations used in the MTT assay are boxed. (B) Western blotting in lysates from H1975, PC-9R, and H1650 cells treated with JAKi (AZD1480, 1 μM), TKI (erlotinib, 0.2 μM), or the combination for 1 hour. Blots are representative of three experiments and are quantified in fig. S1D. (C) Tumor volume tracking in mice bearing xenografts of H1975, PC-9R, or H1650 cells and treated with vehicle (C), AZD1480, erlotinib, or the combination (Ji + Ti) for 12 to 25 days. Doses are provided in Materials and Methods. Data are means ± SEM (n = 5 to 7 mice per group). *P < 0.05, AZD1480 versus the combination (two-tailed Student’s t test).
JAK2 inhibition increases EGFR signaling
Having determined that dual JAK and EGFR inhibition overcame acquired TKI resistance, we next sought to define the mechanisms underlying this phenomenon. Treating cell lines (H1975, PC-9R, and H1650), xenografts (H1975 and PC-9R), and transgenic EGFR-mutant, TKI-resistant NSCLC mouse models [EGFR L858R + T790M (27)] with JAKi reduced the abundance of pSTAT3 in the tumor cells (Figs. 1B and 2, A and B, and figs. S1E and S2, A and B). However, JAK inhibition led to an increase in EGFR signaling. Specifically, the abundance of EGFR, pEGFR, and pERK was increased with no apparent effect on the abundance of phosphorylated AKT or phosphorylated S6 (Fig. 2 and fig. S2, A to E). Additionally, JAK inhibition enhanced the EGFR-mediated RAS activation in cell lines (fig. S3A). Treating cell lines with JAKi resulted in increased pERK abundance after 10 min, which slowly returned to baseline as that of pSTAT3 reappeared over 12 to 24 hours (fig. S3B). The short time scale required for the JAK inhibition–mediated increase in pERK abundance suggested an effect on signaling rather than de novo transcription or translation. A similar phenomenon was observed in vivo. We treated tumor-bearing mice with a single dose of JAKi and observed a rapid increase in pERK abundance with a reciprocal reduction in pSTAT3 abundance 4 to 6 hours after administration of the drug. Twenty-four hours later, as pSTAT3 abundance returned, we observed a concomitant reduction in ERK activation (assessed by pERK staining) (fig. S3C).
Fig. 2. JAK inhibition or depletion enhances EGFR-ERK signaling.
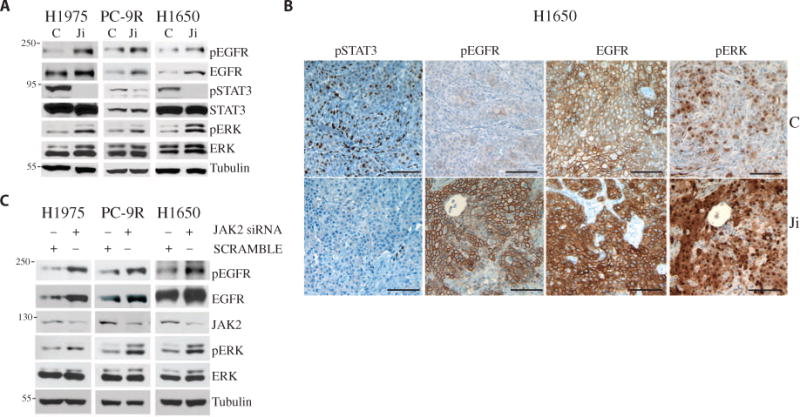
(A) Western blotting as indicated in lysates from H1975, PC-9R, and H1650 cells treated with AZD1480 (1 μM) for 1 hour. Blots are representative of three experiments and are quantified in fig. S2E. (B) Staining for pSTAT3, pEGFR, EGFR, and pERK in representative tumor sections from H1650 xenografts treated with vehicle control (C) or AZD1480 (Ji) (30 mg/kg, twice daily for 3 weeks). Scale bars, 100 μm. (C) Western blotting as indicated in lysates from H1975, PC-9R, and H1650 cells transfected with control (SCRAMBLE) or JAK2 siRNA. Blots are representative of three experiments and are quantified in fig. S3E.
To determine whether the effect of JAK inhibition on ERK phosphorylation was specifically mediated through JAKs, we reduced JAK2 expression in NSCLC lines using small interfering RNAs (siRNAs). Depletion of JAK2 increased the abundance of total EGFR, pEGFR, and pERK (Fig. 2C and fig. S3, D and E). Additionally, we found that several JAK2 inhibitors increased the amount of pEGFR and pERK in NSCLC cell lines, suggesting that the effect was not reagent-specific (fig. S3F). Conversely, we asked whether overexpression of a constitutively active form of JAK2 (JAK2V617F) (28) could decrease pEGFR abundance in NSCLC cell lines. Transient transfection of JAK2V617F into PC-9R cells led to a reduction in pEGFR and pERK abundance (fig. S3G). Together, our data demonstrate that JAK2 inhibition enhances EGFR signaling in NSCLC cell lines, xenografts, and transgenic mice.
JAK2 inhibition increases the surface abundance of EGFR by decreasing the association of EGFR with SOCS5
We hypothesized that altered EGFR turnover could account for the increase in EGFR abundance and signaling. We first examined the effect of JAK inhibition on the levels of membrane-associated EGFR using fluorescently conjugated EGF. JAKi treatment of NSCLC cells led to an increase in the surface staining of EGF-bound EGFR compared to control (Fig. 3A). We obtained similar results using a cell surface EGFR biotinylation assay, which revealed an increase in membrane-associated EGFR in response to JAKi, but no effect on the surface expression of c-MET (another RTK) (Fig. 3B and fig. S4A). These experiments were done in the absence of ligand (after extensive washing of the cells), which suggests that JAK2 inhibition decreased ligand-independent turnover of EGFR, thus resulting in an increase in the steady-state cell surface EGFR/pEGFR abundance.
Fig. 3. JAK2 inhibition increases surface EGFR expression through SOCS5.
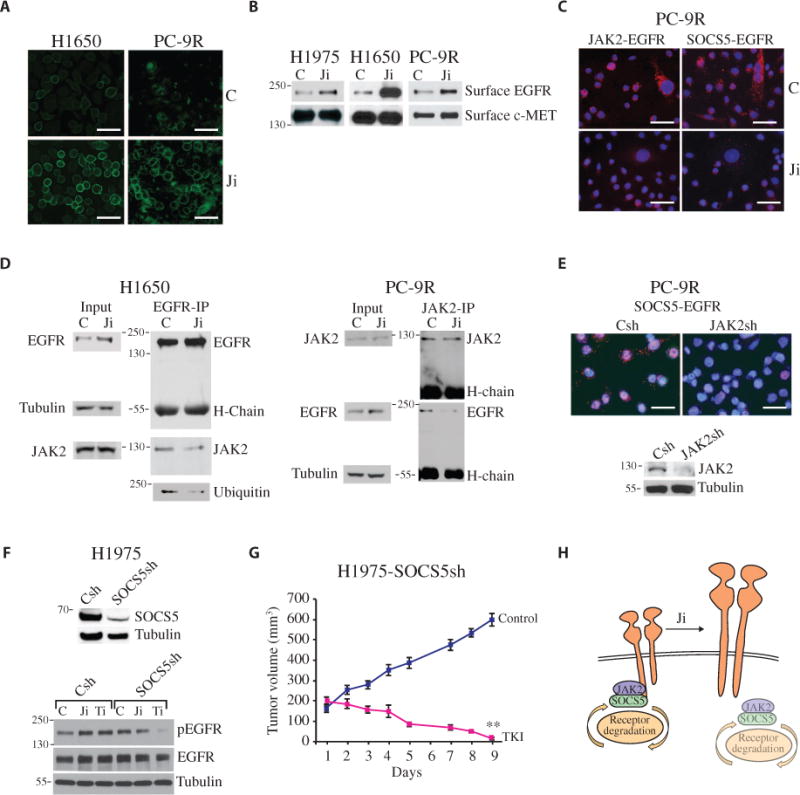
(A) Detection of cell surface–bound EGFR on PC-9R cells using Alexa Fluor–EGF at 4°C after a 1-hour treatment with AZD1480 or control. Scale bars, 50 μm. (B) Serum-starved H1975, H1650, and PC-9R cells were pretreated with AZD1480 or control for 1 hour. Surface proteins were biotinylated, precipitated with avidin resin beads, and analyzed by Western blot for EGFR and c-MET. Blots are representative of three experiments and are quantified in fig. S4A. (C) Detection of JAK2-EGFR and SOCS5-EGFR interactions by Duolink staining in PC-9R cells treated with AZD1480 or control for 1 hour. Scale bars, 50 μm. (D) Cell lysates from H1650 and PC-9R cells treated with control or AZD1480 were immunoprecipitated with an antibody against EGFR or JAK2 and analyzed by Western blot for EGFR, JAK2, and ubiquitin. Loading controls were the heavy-chain (H-chain) immunoglobulin G (IgG) for the co-IP, and tubulin for the input. Blots are representative of three experiments and are quantified in fig. S4D. (E) PC-9R cells expressing JAK2 shRNA (JAK2sh) or vector control (Csh) constructs analyzed for SOCS5 and EGFR interactions by Duolink staining. Scale bars, 50 μm. Cell lysates were analyzed for JAK2 and tubulin. (F) Western blot for SOCS5 and tubulin in lysates from H1975 cells expressing scrambled control or SOCS5 shRNA (SOCS5sh). Control or SOCS5sh cells were treated with control, AZD1480 (1 μM), or erlotinib (0.2 μM) for 1 hour and analyzed for pEGFR, EGFR, and tubulin by Western blot. Representative blots are shown (n = 3). (G) Tumor volumes in mice bearing H1975-SOCS5Sh xenografts and treated with vehicle or TKI (25 mg/kg per day) for 9 days. Data are means ± SEM (n = 5 to 7 mice per group). **P < 0.01, control versus TKI (two-tailed Student’s t test). (H) Schematic depicting NSCLC cells expressing EGFR proteins, wherein JAK2 bridges SOCS5-dependent EGFR degradation, and inhibition or reduction of JAK2 uncouples SOCS5 from EGFR, effectively increasing EGFR abundance on the cell surface.
The turnover of EGFR occurs in a ligand-dependent and ligand-independent manner and is regulated through a physical complex with proteins that modify EGFR, leading to its ubiquitination by E3 ubiquitin ligases (29). Ligand-independent degradation of EGFR is regulated in part through suppressor of cytokine signaling (SOCS) proteins SOCS4 and SOCS5. These highly homologous SOCS boxes containing proteins bind constitutively to EGFR, together with elongins B/C, the cullin family of ubiquitin ligases and ring box proteins, to enhance EGFR ubiquitination and degradation of the receptor in a c-Cbl–independent manner (30–32). Additionally, SOCS5 can bind to JAK1 and JAK2 (33). We hypothesized that JAK2 may form a complex between SOCS4/5 and EGFR as has been described for JAK2-SOCS3-gp130 (34–37). To examine the effect of JAK inhibition on the interaction between SOCS4/5 and EGFR, we used the in situ proximity ligase assay (PLA or Duolink), which can measure associations between proteins including those that are weak and transient. Notably, this approach does not allow one to differentiate between direct or indirect associations. We chose this technique because the SOCS4/5 proteins are labile and difficult to detect by standard biochemical methods. We found that JAK2 and both SOCS4 and SOCS5 are constitutively associated with the EGFR, but these interactions were abrogated with JAK inhibition (Fig. 3C and fig. S4B). We then asked if EGFR activity was required for an association between SOCS5 and JAK2 with the EGFR. We treated PC-9R cells with a T790M mutant–specific TKI, and by PLA, we demonstrated that both SOCS5 and JAK2 were no longer associated with EGFR (fig. S4C). Additionally, by coimmunoprecipitation (co-IP) using antibodies against EGFR and JAK2, we found that JAK2 interacted with EGFR. Inhibition of JAK1/2 activity with a JAKi reduced the association between JAK2 and EGFR (Fig. 3D and fig. S4D). Additionally, by depleting JAK2 abundance through the use of short hairpin RNA (shRNA), we observed a reduced association between SOCS4/5 and EGFR, as assessed by PLA (Fig. 3E and fig. S4E). In agreement with a previous study showing that SOCS5 mediates the ubiquitination of EGFR and enhances its degradation (31), we found that disrupting the SOCS5-EGFR association with a JAKi reduced the amount of ubiquitinated EGFR in H1650 cells (Fig. 3D). To determine whether SOCS5 played a role in regulating EGFR abundance, we reduced SOCS5 levels by siRNA in H1975 and PC-9R cells. We observed an increase in total EGFR and pEGFR abundance, which was not further increased by JAK inhibition (fig. S4F). Similarly, reducing SOCS5 abundance with shRNA increased the abundance of total EGFR and pEGFR in H1975 cells compared to control cells (H1975-Csh). Compared to that in controls, treating H1975-SOCS5sh cells with TKI (erlotinib) reduced the abundance of pEGFR and enhanced growth inhibition both in cultured cells and in xenografts (Fig. 3, F and G, and fig. S4, G to I). Thus, a reduction of SOCS5 was sufficient to reverse TKi resistance, similar to that seen with the combination of JAKi and TKI. Finally, the association between JAK2 and EGFR was intact in H1975-SOCS5sh cells when assessed by PLA (fig. S4J). In summary, these data suggest that activated JAK2 reduces EGFR abundance by coupling the negative regulatory SOCS4/5 proteins to EGFR (Fig. 3H).
JAK2 inhibition promotes erlotinib-sensitive heterodimers between wild-type and mutant EGFR
Given that JAK inhibition (and/or a reduction in SOCS5) increased EGFR abundance and downstream signaling (as assessed by the abundance of pERK), we next asked how this increase could paradoxically restore sensitivity to TKIs rather than promote resistance. It has been shown that ligand-induced EGFR internalization is more rapid for wild-type EGFR than mutant EGFR (38–40). Here, we examined EGF-mediated EGFR internalization and determined that it occurred more rapidly in JAKi-treated cells compared to vehicle-treated cells (Fig. 4A and fig. S5A), suggesting that JAKi may increase the abundance of specifically wild-type EGFR. We hypothesized that JAKi promotes the formation of heterodimers between wild-type and mutant EGFR, the activity of which can be repressed by TKI; thus, we next determined whether wild-type EGFR could indeed form heterodimers with mutant EGFR. Unfortunately, no antibodies exist that discriminate wild-type from mutant EGFR; therefore, to test our hypothesis, we used H1975 (L858R + T790M mutant) cells that overexpressed a Myc-tagged wild-type EGFR (41) and found that JAKi increased the association between Myc-tagged wild-type EGFR and the L858R + T790M EGFR in these cells (Fig. 4B). Furthermore, we showed that JAK inhibition increased the abundance of markers indicating wild-type EGFR signaling in an NSCLC line (H292) that overexpressed only wild-type EGFR (fig. S5B). Next, we selected H1975 cells enriched for wild-type EGFR (“wild type–dominant subclone”) by treating cells with WZ4002 (42), a T790M mutant–specific TKI. Conversely, we selected for H1975 cells enriched for the L858R/T790M EGFR (“mutant-dominant subclone”) by treating cells with erlotinib to target wild-type EGFR, as previously described (43) (Fig. 4C). Treatment of wild type–dominant cells with a JAKi led to an increase in the abundance of pEGFR, EGFR, and pERK (Fig. 4C, fig. S5C). Cotreatment of EGFR wild type–dominant cells with JAKi and erlotinib led to a greater drug synergism as compared to the parental H1975 cells (CI, 0.26 versus 0.40) (Fig. 4C). On the other hand, cotreatment of mutant-dominant cells with JAKi and erlotinib did not overcome resistance to erlotinib (CI, 0.91 versus 0.40) (Fig. 4C). In conclusion, these data support our hypothesis that JAK inhibition leads to enhanced heterodimer formation between wild-type and TKI-resistant mutant EGFR.
Fig. 4. Enhancement of EGFR-ERK signaling by JAK inhibition is mediated through heterodimerization between wild-type/mutant EGFR, which is abrogated by TKI.
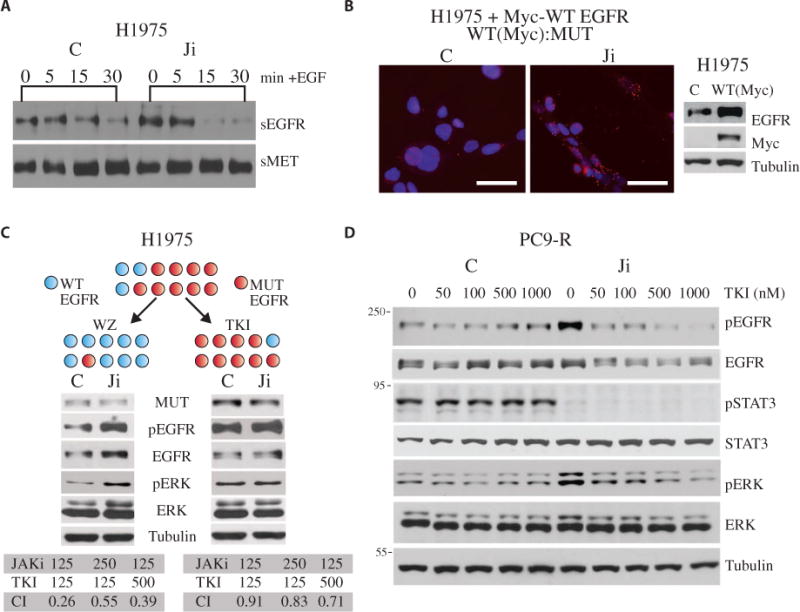
(A) Serum-starved H1975 cells were pretreated with AZD1480 or control (C) for 1 hour, and EGF ligand was added for the indicated times. Surface proteins were biotinylated, precipitated with avidin resin beads, and analyzed by Western blot for surface EGFR (sEGFR) and c-MET (sMET). (B) Duolink staining (left) for wild-type (WT) EGFR (Myc) and mutant L858R EGFR (MUT) interaction in H1975 cells expressing Myc-tagged WT EGFR protein and treated with AZD1480 or control for 1 hour. Scale bars, 50 μm. Western blot (right) in lysates from H1975 parental cells (control) and cells expressing Myc-tagged WT EGFR were analyzed for EGFR, Myc-tagged protein, and tubulin. (C) Top, experimental schematic in which H1975 cells expressing both the WT (blue sphere) and EGFR-L858R/T790M gatekeeper mutant (red sphere) proteins are depicted as a single cell expressing variable amounts of each. H1975 cells were treated with either TKI (0.2 μM) or the T790M-specific inhibitor WZ4002 (WZ; 25 nM) for 30 days, and selected populations are depicted by their relative expression of WT and mutant EGFR per cell. Extracts from WZ4002- and TKI-selected cells treated with either control or AZD1480 were then analyzed by Western blot as indicated (note that EGFR detects both WT and mutant). The growth inhibitory effects of AZD1480 (JAKi) in combination with erlotinib (TKI) are shown below as representative CIs. (D) Western blotting as indicated in lysates from serum-starved PC-9R cells treated with AZD1480 for 1 hour and then EGF for 30 min in the presence of erlotinib at the indicated concentrations. Blots in (A), (C), and (D) are representative of three experiments each and are quantified in figs. S5, A and C, and S6, respectively.
We next asked whether the JAKi-dependent increase in wild-type EGFR abundance and activity could be blocked by the TKI erlotinib. Although wild-type EGFR is sensitive to TKI, it requires higher concentrations to inhibit its activity compared to mutant EGFR. We postulated that the wild-type/mutant EGFR heterodimers would thus require higher concentrations of erlotinib as compared to cells expressing erlotinib-sensitive mutant EGFR to inhibit signaling. pEGFR and pERK abundance in control TKI-resistant PC-9R and H1975 cells was unaffected by erlotinib, whereas in JAKi-treated cells, pEGFR and pERK abundance was markedly attenuated by increasing concentrations of TKI (Fig. 4D and figs. S5D and S6A). In summary, our data suggest that JAK2 inhibition induced an increase in EGFR abundance through the loss of SOCS4/5-mediated degradation, leading to the formation of wild-type/mutant, TKI-sensitive heterodimers (Fig. 5).
Fig. 5. Working model: JAK inhibition enhances TKI-sensitive EGFR signaling and growth inhibition in NSCLC.
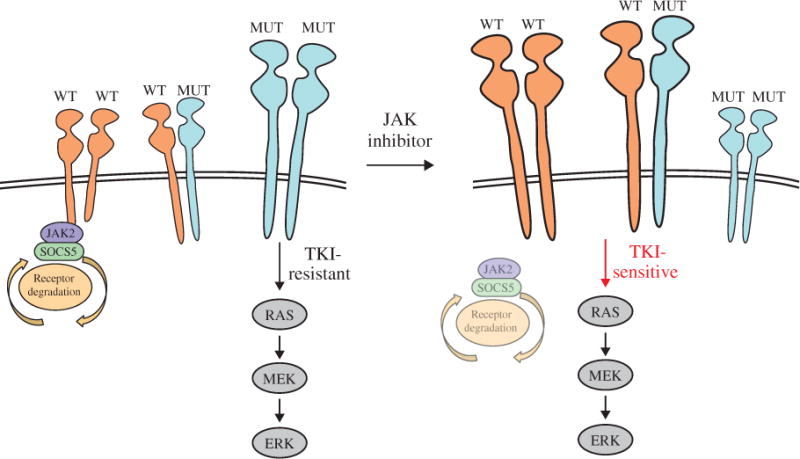
Schematic depicting TKI-resistant NSCLC cells expressing both WT and gatekeeper mutant EGFR proteins, which form homodimers or heterodimers. At steady state, JAK2 promotes SOCS5-dependent EGFR degradation. TKI-resistant, homodimeric mutant EGFR is the principle driver of the RAS–MEK (mitogen-activated protein kinase kinase)–ERK signaling cascade (in bold). Inhibition or reduction of JAK2 uncouples SOCS5 from EGFR, effectively increasing TKI-sensitive, wild-type EGFR homodimers/heterodimers and signaling (in bold). Although the role of EGFR WT:MUT heterodimer signaling has not been clearly defined, we hypothesize that it may regulate TKI sensitivity. This working model may explain the synergistic actions observed between the EGFR-targeted TKI and JAKi on NSCLC tumor growth.
DISCUSSION
Constitutive activation of oncogenic signaling pathways in cancers has led to the development of targeted inhibitors. Erlotinib and gefitinib, TKIs of EGFR, both induce clinical responses in up to 90% of patients with EGFR-mutant lung adenocarcinoma, yet these responses endure, on average, less than 1 year (2). Efforts to understand the mechanisms of resistance have revealed significant cross-inhibition between mitogenic pathways, such that blockade of one pathway relieves the negative feedback on another resulting in a relative “insensitivity” to the targeting pharmacologic agent (44–48). In the case of EGFR-mutant NSCLC, an estimated 60% of tumors acquire a gatekeeper mutation that impairs TKI binding (5). To date, clinical trials of drugs intended to restore sensitivity to tumors with acquired resistance have been unsuccessful.
Aberrant regulation of the IL-6–JAK–STAT3 signaling pathway is a critical mediator of tumorigenesis, which has similarly led to the development of targeted inhibitors (17). Here, we found that high pSTAT3 abundance persists in TKI-resistant NSCLC primary tumors, suggesting that pSTAT3 signaling may play a role in mediating TKI resistance. Consistent with our observations, Yao et al. showed that IL-6–activated gp130/JAK/STAT3 signaling decreased TKI sensitivity in resistant NSCLC H1650 cells that have no T790M gatekeeper mutation or other known resistance mechanisms (10). Another publication by Li et al. suggested that a STAT3/Bcl2/Bcl-XL survival pathway may be required for early adaptive and late acquired TKI resistance, in an erlotinib-selected resistant NSCLC cell line (49). Additionally, we showed that JAK inhibition alone reduced the growth of NSCLC xenografts (14). These data support the therapeutic potential of JAKi for the treatment of solid tumors as monotherapy (12–14, 18, 20).
Although JAK inhibition alone could decrease the growth of TKI-resistant cells, we were surprised that combined treatment with JAKi and TKI could overcome resistance in TKI-resistant cells and xenografts. Specifically, PC-9R and H1975 cells expressing the TKI-resistant gatekeeper EGFR and semi–TKI-resistant H1650 cells were rendered sensitive to a TKI when treated in combination with a JAKi. We then examined the mechanisms underlying this synergism.
We first determined that either JAK2 depletion or inhibition of activity enhanced EGFR/RAS/MEK/ERK signaling through an increase in membrane EGFR protein abundance in the absence of ligand. Several non–ligand-dependent inhibitors of EGFR have been identified, including leucine-rich repeats and immunoglobulin-like domains protein 1 (LRIG1), receptor-associated late transducer (RALT; also known as MIG6 and ERRFI1), SOCS4, and SOCS5. Ectopic overexpression of SOCS5 and SOCS4 was shown to induce the degradation of EGFR in a ligand-and c-Cbl–independent manner and through an elongin B/C–dependent process (6, 30, 31, 50). Although structural studies have suggested that the SH2 domains of SOCS4 and SOCS5 proteins may bind to the phosphorylated Tyr1068 of the EGFR, no functional studies have demonstrated this (6, 30, 31, 36). More recently, SOCS5 was shown to inhibit JAK1 and JAK2 through its N-terminal domain (33). Additionally, Shc-1 was identified as a potential substrate for SOCS5, suggesting that this protein can negatively regulate multiple tyrosine kinases (33). Here, our data suggest that JAK2 bridges a ternary complex between SOCS5 and EGFR. Specifically, pharmacologically inhibiting JAK2 or knocking down its protein abundance led to a loss of SOCS5 association with the EGFR, resulting in attenuated ubiquitination of EGFR and the consequent up-regulation of EGFR surface expression and downstream RAS-pERK signaling. Similarly, a reduction of SOCS5 in TKI-resistant H1975 cells also led to an increase in pEGFR, which was inhibited by TKI (specifically, erlotinib). Notably, the loss of SOCS5 had no impact on the association between JAK2 and EGFR. Conversely, enforced expression of activated JAK2 could attenuate EGFR signaling possibly through a mechanism involving JAK2-mediated increased SOCS4/5 recruitment to the EGFR. In support of this hypothesis, Harada et al. observed that in a TKI-resistant PC-9 cell clone, increased pJAK2 abundance was associated with reduced total EGFR and pEGFR abundance compared to that in the parental line (23). Finally, the JAK2-EGFR association is likely dependent on activated EGFR because pharmacological inhibition of EGFR resulted in the loss of both EGFR-JAK2 and EGFR-SOCS5 interactions. In summary, these data suggest that activated EGFR is negatively regulated by activated JAK2 in part by bringing SOCS5 to EGFR, leading to its degradation (51, 52).
The resultant increase in EGFR expression and activity translated into restoration of sensitivity to TKIs was surprising. We determined that JAK inhibition led to increased levels of heterodimers between mutant EGFR and wild-type EGFR, which were sensitive to TKIs. Inhibition of specifically wild-type EGFR in these heterodimers could explain how JAK inhibition conferred sensitivity to otherwise resistant tumors. About 30% of tumors resistant to erlotinib lack known alterations (namely, T790M mutation or MET amplification); this phenotype is exemplified in the H1650 cell line (5). We observed the greatest growth inhibitory synergy between the JAK2 inhibitor and erlotinib in H1650 cells. We hypothesize that the JAK2-regulated increase in EGFR expression and activity enhances the dependence or “addiction” to this signaling pathway. We propose that increased EGFR signaling activates negative feedback loops suppressing other pathways that normally participate in promoting survival and proliferation. The chronic inhibition of compensatory pathways leads to the EGFR-addicted phenotype. This hypothesis has been proposed for other diseases such as estrogen receptor (ER)– and HER2-positive breast cancers, in which the expression and activity of these drivers of disease are positively correlated with response to targeted inhibitors. For example, modifiers of ER abundance such as inhibitors of the phosphatidylinositol 3-kinase–mammalian target of rapamycin pathway result in synergistic antiproliferative effects when given in combination with endocrine therapy, leading to a survival advantage in patients (53–55). Our results demonstrate crosstalk between the JAK and EGFR oncogenic signaling pathways, thus providing a molecular rationale for combination JAK- and EGFR-targeted tyrosine kinase inhibitor therapies in patients that exhibit innate or acquired resistance to EGFR-targeted TKIs, a group that ultimately includes all patients with EGFR-mutant lung adenocarcinoma.
MATERIALS AND METHODS
Human tumor specimens
Biopsy specimens were obtained from patients starting TKI therapy and those who had developed progression of disease on continuous TKI therapy. These specimens were subsequently analyzed for EGFR kinase–activating mutations/second T790M mutation, and assessment of MET gene copy number alterations was carried out by standard protocols as previously described (22).
Drugs
The JAK1/2 inhibitor AZD1480 was provided by AstraZeneca. The type II JAK2 inhibitor BBT594 was provided by Novartis. INCB18424 and EGFR TKI erlotinib were purchased from Chemietek. Pan-JAKi P6 was purchased from Calbiochem. The EGFR T790M–specific inhibitor WZ4002 was purchased from Selleck Chemicals. For in vitro experiments, the inhibitors were dissolved in 100% dimethyl sulfoxide (DMSO) to prepare a 10 mM stock and stored at −20°C. For in vivo experiments, the indicated inhibitors were formulated daily in purified, sterile water supplemented with 0.5% methyl cellulose and 0.1% Tween 80.
Cell lines
Cell lines were grown in RPMI 1640 medium. PC-9 and PC-9R cell lines were previously described (43); H1975, H1650, and H292 cell lines were purchased from the American Type Culture Collection (ATCC). H1975 cells overexpressing Myc-tagged wild-type EGFR were generated by retroviral infection of pMSCVpuro-IRES-EGFR-Myc/His into H1975 cells, and stable clones were isolated by puromycin selection (41, 56).
Animals
C57/B6 and athymic nude mice were purchased from Taconic and Harlan. Mice harboring the CCSP-rtTA and tet-regulated EGFRL858R+T790M transgenes (“C/L858R + T790M”) were developed by W. Pao. All experiments involving animals were approved by the Memorial Sloan Kettering Cancer Center Institutional Animal Care and Use Committee. Cell lines were subcutaneously implanted in athymic mice for PC-9, H1975, and H1650 xenograft tumors in a 1:1 mixture of Matrigel (BD Biosciences) and culture medium. Tumor-bearing mice were randomized on the basis of tumor volume before the treatment, which was initiated when average tumor volume was about 100 mm3. All inhibitors were given orally by gavage: AZD1480, 30 mg/kg, twice a day for H1975 and H1650, and 20 mg/kg daily for PC-9R; erlotinib, 25 mg/kg, once a day for H1975, H1650, and PC-9R. Tumors were measured every 3 to 4 days, and tumor volumes were calculated by the formula L × W2 × π/6, where L is the tumor length and W the width. Three hours before sacrifice, the animals were given the last dose of drug treatment.
Cell lysate preparation, Western blotting, and densitomeric analysis
Adherent cells were lysed in cell lysis buffer, and Western blot analysis was performed as previously described (8). Cells were treated with the proteasome inhibitor MG132 (N-carbobenzyloxy-L-leucyl-L-leucyl-L-leucinal) (5 μM) for 16 hours before the lysis of cells for the analysis of SOCS4 and SOCS5 proteins. Antibodies used for Western blotting were as follows: anti-pSTAT3 (Y705), total STAT3, pERK (p44/42, T202/Y204), pAKT (S473), total AKT, pS6 (S240/244), S6, pEGFR (Y1068), total EGFR, L858R-EGFR– or del19-EGFR–specific antibodies, anti-JAK2 (Cell Signaling Technology), and anti-JAK1 (BD Biosciences); anti–total ERK, EGFR, Myc, ubiquitin and horseradish peroxidase (HRP)–conjugated anti-rabbit IgG, HRP-conjugated anti-mouse IgG, HRP-conjugated anti-rabbit IgG, SOCS4, and SOCS5 (Santa Cruz Biotechnology); anti–c-MET (Invitrogen); and anti–α-tubulin (Sigma-Aldrich). Densitometric quantification of high-resolution immunoblot images was performed using an analysis program, ImageJ (developed by W. Rasband, Research Services Branch of the National Institute of Mental Health). Analyses of the phosphorylation status of RTK and ErbB family members were performed using the RayBio Human RTK and EGFR Phosphorylation Array kits according to the manufacturer’s instruction (RayBiotech).
RAS activation assay
The status of activated RAS was measured using the RAS Activation Assay Kit according to the manufacturer’s instruction (US Biological). Briefly, the cells were treated by AZD1480 as indicated, washed twice with ice-cold phosphate-buffered saline (PBS), and lysed with lysis buffer. The activated/guanosine 5′-triphosphate–bound RAS was then pulled down by agarose bead–conjugated RAS-binding domain of Raf-1, electrophoresed by SDS–polyacrylamide gel electrophoresis gel, and probed with an anti-RAS antibody.
DNA constructs
pBabe-YFP-JAK2/V617F, an active form of human JAK2 expression construct, was generated by excising a wild-type JAK2 complementary DNA from pEF-YFP-JAK2 (57) using Age I and Spe I, Klenow filled, and ligated into the SnaB I site of pBabe-puro. A polymerase chain reaction–based mutagenesis of converting JAK2 amino acid 617 valine to phenylalanine was subsequently performed using DNA polymerase Pfu and primer sets 5′-GAATTATGGTGTCTGTTTCTGTGGAGAGGAGAAC-3′ and 5′-GTTCTCCTCTCCACAGAAACAGACACCATAATTC-3′.
Cell transfection, RNA interference, and lentiviral infections
H1975, H1650, and PC-9 cells were transfected by siRNAs (25 nM) with HiPerFect (Qiagen) according to the manufacturer’s protocol. Silencer validated JAK2, SOCS4, and SOCS5 siRNAs and nonsilencing scramble siRNA were purchased from Qiagen. Cells were plated in a six-well plate and incubated for 36 hours after siRNA transfection, and cell lysates were collected for Western blotting. The plasmids pBabe-puro and pBabe-JAK2/V617F were transiently transfected into PC-9 cells with the SuperFect reagent (Qiagen). Forty-eight hours later, cell lysates were collected for Western blotting. JAK2 shRNA, SOCS5 shRNA lentiviral, and control constructs were purchased from Sigma-Aldrich, and infections into H1975 followed by selection with puromycin (2 μg/ml) were performed according to the manufacturer’s instructions.
Flow cytometry
Cells were resuspended in 0.5 ml of ice-cold 1% bovine serum albumin (BSA)–PBS. Cell apoptosis was determined by labeling with annexin V and propidium iodide (PI). Samples were analyzed using a BD FACSCalibur cytometer, and data were analyzed using FlowJo software. Under unfixed conditions, cells that were annexin V(−) and PI(−) were considered viable cells. Cells that were annexin V(+) and PI(−) were considered early-stage apoptotic cells. Cells that were annexin V(+) and PI(+) were considered late-stage apoptotic cells.
Duolink fluorescence staining
In situ protein-protein interactions were measured by using the Duolink II Red fluorescence staining kit according to the manufacturer’s instruction (Olink Bioscience). Cells were fixed in 4% paraformaldehyde (PFA), permeabilized with 0.2% Triton X-100 in PBS, and probed with primary antibodies against the proteins of interest and isotype controls (1:100 to 1:200) at 4°C overnight. The kit-provided PLA probes were subsequently added, and signal amplification was performed. Imaging analyses were conducted using a Zeiss fluorescence microscope. SOCS4 and SOCS5 antibodies were purchased from Santa Cruz Biotechnology.
Cell viability assays
The effects of AZD1480 and other inhibitors on in vitro cell growth were assayed using the MTT Cell Proliferation Assay kit (ATCC). In brief, cells were plated in 96-well plates in five replicates in RPMI plus 10% fetal bovine serum and allowed to attach for 24 hours before the addition of DMSO as control or the indicated drugs. After 6 days, with medium replenishment at day 3, cells were first incubated with MTT until a purple precipitate was visible (in about 3 hours), which was then solubilized by detergent and quantified by absorbance at 570 nm with a reference wavelength of 670 nm.
Coimmunoprecipitation
The cells treated with AZD1480 or control DMSO were first lysed by whole-cell lysis buffer without detergent, followed by preclearing using control IgG/serum and protein A and GammaBind G Sepharose beads (protein A/G beads, Pharmacia). For EGFR IP, 1 μg of antibody was used per 200 μg of protein extract; for JAK2 IP, 5 μl of antiserum was used. Co-IPs used 1 mg of precleared protein extract, which was incubated with the antibody/antiserum overnight at 4°C, before adding a 1:1 mix of protein A/G beads for another 2 hours. The beads were washed five times with lysis buffer before boiling in sample buffer and being subjected to Western blot analysis.
Biotinylation and precipitation of cell surface proteins
Cells were serum-starved overnight and pretreated with AZD1480 or control DMSO for 1 hour, and the cell surface proteins were then biotinylated with Sulfo-NHS-LC-Biotin (0.2 mg/ml; Pierce) in PBS for 30 min at 4°C. Unreacted biotin was quenched and removed by washing twice with ice-cold PBS containing 0.1 M glycine and twice with ice-cold PBS. For precipitation of surface biotinylated proteins, cells were lysed with lysis buffer, and the cell lysate was incubated with NeutrAvidin Plus UltraLink Resin (Pierce) and equilibrated in PBS at 4°C for 1 hour. The resin was then washed twice with PBS, and the levels of cell surface EGFR and c-MET were analyzed by Western blotting.
Statistical and CI analyses
The effectiveness of the inhibitors used in this study, alone and in combination, was analyzed by using CalcuSyn software (Biosoft). The CI was calculated according to the Chou-Talalay method (58). CI < 0.9 indicates synergy, CI between 0.9 and 1.1 is addictive, and CI > 1.1 indicates antagonism between the two drugs. The determination of the in vitro effects of drugs on cell cycle and apoptosis was achieved by three independent experiments. Data were analyzed by Student’s t test, and statistical significance was set at P < 0.05.
Surface EGFR labeling using Alexa Fluor 488–EGF
Cells were seeded at 0.5 × 106 and grown on coverslips in six-well plates before being treated with 1 μM AZD1480 or control DMSO for 1 hour. The coverslips were then chilled on ice for 10 min and then incubated with Alexa Fluor 488–EGF (0.5 μg/ml; Molecular Probes) in serum-free medium with 1% BSA at 4°C for 1 hour for surface EGFR labeling. The cells were subsequently washed twice with cold PBS, fixed in PFA, and viewed by fluorescence imaging.
Immunohistochemistry and immunofluorescence analyses
Tumor xenografts were fixed immediately after removal in a 4% PFA solution for 24 hours at 4°C followed by standard paraffin embedding and sectioning. Slides underwent deparaffinization, antigen retrieval, and immunohistochemistry and immunofluorescence analyses as previously described (8). Semiquantitative analysis of pSTAT3 was performed (for human specimens, xenografts, and mouse transgenic tumors) by considering both the intensity (0, negative; 1, low; 2, moderate; 3, strong) and the percentage of cells displaying a positive signal (0, negative; 1, 0 to 5%; 2, 5 to 25%; 3, 25 to 50%; 4, 50 to 75%; 5, 75 to 100%), and the respective indices were multiplied to each other to calculate a score value of 0, 1+ (low), 2+ (moderate), or 3+ (high).
Supplementary Material
Fig. S1. STAT3 activation and inhibition in TKI-resistant NSCLC.
Fig. S2. JAK2 inhibition increases the abundance of pEGFR and pERK in NSCLC.
Fig. S3. JAK2 inhibition leads to rapid activation of EGFR/RAS/ERK signaling.
Fig. S4. JAK2 links SOCS5 to EGFR, regulating sensitivity to TKI.
Fig. S5. Quantification of Western blots in Fig. 4, A and C.
Fig. S6. Quantification of Western blots in Fig. 4D.
Acknowledgments
We thank T. Radimerski from Novartis for BBT-594. We also thank the members of our laboratories for helpful discussions and the members of the Memorial Sloan Kettering Cancer Center Animal Imaging, Antitumor Assessment and Molecular Cytology core facilities (in particular, H. Zhao, J. Qiu, A. Barlas, S. Fujisawa, and V. D. Gueorguiev). We would also like to thank L. Regales for providing assistance with the EGFR transgenic mice. We thank C. Haan for providing pEF-YFP-JAK2 and AstraZeneca for providing AZD1480.
Funding: Our work was supported by NIH grants U54 CA148967 (J.F.B. and G.A.-B.), R01 CA87637 (J.F.B.), and P30 CA008748 (J.F.B.); the Charles and Marjorie Holloway Foundation (J.F.B.); the Sussman Family Fund (J.F.B.); the Lerner Foundation (J.F.B.); Astra Zeneca (J.F.B.); the Manhasset Women’s Coalition Against Breast Cancer (J.F.B.); the New York State USBC Women’s Bowling Association (J.F.B.); Uniting Against Lung Cancer grant (S.P.G.); the Children’s Cancer and Blood Foundation (D.L.); the Manning Foundation (D.L.); the Hartwell Foundation (D.L.); the Pediatric Oncology Experimental Therapeutics Investigators Consortium (D.L.); the Stavros S. Niarchos Foundation (D.L.); the Champalimaud Foundation (D.L.); the Nancy C. and Daniel P. Paduano Foundation (D.L.); the Mary Kay Foundation (D.L.); the American Hellenic Educational Progressive Association, 5th District (D.L.); the Malcolm Hewitt Wiener Foundation (D.L.); the George Best Costacos Foundation (D.L.); National Cancer Institute grant R01CA 098234-01 (D.L.); Susan G. Komen for the Cure (D.L.); Physical Sciences-Oncology Centers training grant NCI-U54-CA143836 (D.L.); and the Beth C. Tortolani Foundation (J.F.B. and D.L.).
Footnotes
SUPPLEMENTARY MATERIALS
Author contributions: D.L. and J.F.B. contributed to study design, data interpretation, and manuscript preparation. S.P.G., L.A.D., Q.C., G.A.-B., K.M.-T., M.L., N.M., T.C., S.H.L., R.V., E.B., D.S., E.d.S., and E.S. contributed to study design and data acquisition and analysis. S.P.G., L.A.D., Q.C., T.C., and E.S. also contributed to manuscript preparation. M.A. and L.N. contributed to study design and manuscript preparation. R.L.L. and M.Z. contributed to study design. H.Z. characterized the wild-type allele in TKI-resistant cells. W.P. and M.R.B. produced H1975-EGFRmyc cells. D.H. provided AZD1480. L.M. performed the statistical analysis.
Competing interests: The authors declare that they have no competing interests. D.H. was an employee of AstraZeneca and holds stock. W.P. is an employee of Roche. M.Z. is an employee of AstraZeneca and has equity holdings. M.A. is on the advisory committee for AstraZeneca. M.L. is on the Afatinib targeted therapy advisory committee for National Comprehensive Cancer Network/Boehringer Ingelheim. J.F.B. is a consultant for Roche/Genentech.
Data and materials availability: A patent related to this manuscript is pending. A material transfer agreement is required for AZD1480 (J.F.B.) and H1975-EGFRmyc cells (M.R.B.).
REFERENCES AND NOTES
- 1.Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359:1367–1380. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia-Gomez R, Pallares C, Sanchez JM, Porta R, Cobo M, Garrido P, Longo F, Moran T, Insa A, De Marinis F, Corre R, Bover I, Illiano A, Dansin E, de Castro J, Milella M, Reguart N, Altavilla G, Jimenez U, Provencio M, Moreno MA, Terrasa J, Muñoz-Langa J, Valdivia J, Isla D, Domine M, Molinier O, Mazieres J, Baize N, Garcia-Campelo R, Robinet G, Rodriguez-Abreu D, Lopez-Vivanco G, Gebbia V, Ferrera-Delgado L, Bombaron P, Bernabe R, Bearz A, Artal A, Cortesi E, Rolfo C, Sanchez-Ronco M, Drozdowskyj A, Queralt C, de Aguirre I, Ramirez JL, Sanchez JJ, Molina MA, Taron M, Paz-Ares L. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–246. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 3.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, Nishiwaki Y, Ohe Y, Yang JJ, Chewaskulyong B, Jiang H, Duffield EL, Watkins CL, Armour AA, Fukuoka M. Gefitinib or carboplatin–paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 4.Oxnard GR, Arcila ME, Chmielecki J, Ladanyi M, Miller VA, Pao W. New strategies in overcoming acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer. Clin Cancer Res. 2011;17:5530–5537. doi: 10.1158/1078-0432.CCR-10-2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M, Riely GJ. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 Patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19:2240–2247. doi: 10.1158/1078-0432.CCR-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Avraham R, Yarden Y. Feedback regulation of EGFR signalling: Decision making by early and delayed loops. Nat Rev Mol Cell Biol. 2011;12:104–117. doi: 10.1038/nrm3048. [DOI] [PubMed] [Google Scholar]
- 7.Vecchione L, Jacobs B, Normanno N, Ciardiello F, Tejpar S. EGFR-targeted therapy. Exp Cell Res. 2011;317:2765–2771. doi: 10.1016/j.yexcr.2011.08.021. [DOI] [PubMed] [Google Scholar]
- 8.Gao SP, Mark KG, Leslie K, Pao W, Motoi N, Gerald WL, Travis WD, Bornmann W, Veach D, Clarkson B, Bromberg JF. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J Clin Invest. 2007;117:3846–3856. doi: 10.1172/JCI31871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Song L, Rawal B, Nemeth JA, Haura EB. IL6/JAK1 pathway inhibition downregulates STAT3 and inhibits tumor growth in lung cancer. Mol Cancer Ther. 2011;10:481–494. doi: 10.1158/1535-7163.MCT-10-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yao Z, Fenoglio S, Gao DC, Camiolo M, Stiles B, Lindsted T, Schlederer M, Johns C, Altorki N, Mittal V, Kenner L, Sordella R. TGF-β IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc Natl Acad Sci USA. 2010;107:15535–15540. doi: 10.1073/pnas.1009472107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiu HC, Chou DL, Huang CT, Lin WH, Lien TW, Yen KJ, Hsu JTA. Suppression of Stat3 activity sensitizes Gefitinib-resistant non small cell lung cancer cells. Biochem Pharmacol. 2011;81:1263–1270. doi: 10.1016/j.bcp.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 12.Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, Buettner R, Proia D, Kowolik CM, Xin H, Armstrong B, Bebernitz G, Weng S, Wang L, Ye M, McEachern K, Chen H, Morosini D, Bell K, Alimzhanov M, Ioannidis S, McCoon P, Cao ZA, Yu H, Jove R, Zinda M. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16:487–497. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xin H, Herrmann A, Reckamp K, Zhang W, Pal S, Hedvat M, Zhang C, Liang W, Scuto A, Weng S, Morosini D, Cao ZA, Zinda M, Figlin R, Huszar D, Jove R, Yu H. Antiangiogenic and antimetastatic activity of JAK inhibitor AZD1480. Cancer Res. 2011;71:6601–6610. doi: 10.1158/0008-5472.CAN-11-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murakami T, Takigawa N, Ninomiya T, Ochi N, Yasugi M, Honda Y, Kubo T, Ichihara E, Hotta K, Tanimoto M, Kiura K. Effect of AZD1480 in an epidermal growth factor receptor-driven lung cancer model. Lung Cancer. 2014;83:30–36. doi: 10.1016/j.lungcan.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 15.Lee JH, Park KS, Alberobello AT, Kallakury B, Weng MT, Wang Y, Giaccone G. The Janus kinases inhibitor AZD1480 attenuates growth of small cell lung cancers in vitro and in vivo. Clin Cancer Res. 2013;19:6777–6786. doi: 10.1158/1078-0432.CCR-13-1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dolgin E. Companies hope for kinase inhibitor JAKpot. Nat Rev Drug Discov. 2011;10:717–718. doi: 10.1038/nrd3571. [DOI] [PubMed] [Google Scholar]
- 17.Seavey MM, Dobrzanski P. The many faces of Janus kinase. Biochem Pharmacol. 2012;83:1136–1145. doi: 10.1016/j.bcp.2011.12.024. [DOI] [PubMed] [Google Scholar]
- 18.McFarland BC, Ma JY, Langford CP, Gillespie GY, Yu H, Zheng Y, Nozell SE, Huszar D, Benveniste EN. Therapeutic potential of AZD1480 for the treatment of human glioblastoma. Mol Cancer Ther. 2011;10:2384–2393. doi: 10.1158/1535-7163.MCT-11-0480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gheeya JS, Chen QR, Benjamin CD, Cheuk AT, Tsang P, Chung JY, Metaferia BB, Badgett TC, Johansson P, Wei JS, Hewitt SM, Khan J. Screening a panel of drugs with diverse mechanisms of action yields potential therapeutic agents against neuroblastoma. Cancer Biol Ther. 2009;8:2386–2395. doi: 10.4161/cbt.8.24.10184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corcoran RB, Contino G, Deshpande V, Tzatsos A, Conrad C, Benes CH, Levy DE, Settleman J, Engelman JA, Bardeesy N. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res. 2011;71:5020–5029. doi: 10.1158/0008-5472.CAN-11-0908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Riely GJ, Pao W, Pham D, Li AR, Rizvi N, Venkatraman ES, Zakowski MF, Kris MG, Ladanyi M, Miller VA. Clinical course of patients with non–small cell lung cancer and epidermal growth factor receptor exon 19 and exon 21 mutations treated with gefitinib or erlotinib. Clin Cancer Res. 2006;12:839–844. doi: 10.1158/1078-0432.CCR-05-1846. [DOI] [PubMed] [Google Scholar]
- 22.Arcila ME, Oxnard GR, Nafa K, Riely GJ, Solomon SB, Zakowski MF, Kris MG, Pao W, Miller VA, Ladanyi M. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res. 2011;17:1169–1180. doi: 10.1158/1078-0432.CCR-10-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harada D, Takigawa N, Ochi N, Ninomiya T, Yasugi M, Kubo T, Takeda H, Ichihara E, Ohashi K, Takata S, Tanimoto M, Kiura K. JAK2-related pathway induces acquired erlotinib resistance in lung cancer cells harboring an epidermal growth factor receptor-activating mutation. Cancer Sci. 2012;103:1795–1802. doi: 10.1111/j.1349-7006.2012.02363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gendreau SB, Ventura R, Keast P, Laird AD, Yakes FM, Zhang W, Bentzien F, Cancilla B, Lutman J, Chu F, Jackman L, Shi Y, Yu P, Wang J, Aftab DT, Jaeger CT, Meyer SM, De Costa A, Engell K, Chen J, Martini JF, Joly AH. Inhibition of the T790M gatekeeper mutant of the epidermal growth factor receptor by EXEL-7647. Clin Cancer Res. 2007;13:3713–3723. doi: 10.1158/1078-0432.CCR-06-2590. [DOI] [PubMed] [Google Scholar]
- 25.Sawai A, Chandarlapaty S, Greulich H, Gonen M, Ye Q, Arteaga CL, Sellers W, Rosen N, Solit DB. Inhibition of Hsp90 down-regulates mutant epidermal growth factor receptor (EGFR) expression and sensitizes EGFR mutant tumors to paclitaxel. Cancer Res. 2008;68:589–596. doi: 10.1158/0008-5472.CAN-07-1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang W, Li Q, Yamada T, Matsumoto K, Matsumoto I, Oda M, Watanabe G, Kayano Y, Nishioka Y, Sone S, Yano S. Crosstalk to stromal fibroblasts induces resistance of lung cancer to epidermal growth factor receptor tyrosine kinase inhibitors. Clin Cancer Res. 2009;15:6630–6638. doi: 10.1158/1078-0432.CCR-09-1001. [DOI] [PubMed] [Google Scholar]
- 27.Regales L, Balak MN, Gong Y, Politi K, Sawai A, Le C, Koutcher JA, Solit DB, Rosen N, Zakowski MF, Pao W. Development of new mouse lung tumor models expressing EGFR T790M mutants associated with clinical resistance to kinase inhibitors. PLOS One. 2007;2:e810. doi: 10.1371/journal.pone.0000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJP, Boggon TJ, Wlodarska I, Clark JJ, Moore S, Adelsperger J, Koo S, Lee JC, Gabriel S, Mercher T, D’Andrea A, Fröhling S, Döhner K, Marynen P, Vandenberghe P, Mesa RA, Tefferi A, Griffin JD, Eck MJ, Sellers WR, Meyerson M, Golub TR, Lee SJ, Gilliland DG. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 29.Sorkin A, Goh LK. Endocytosis and intracellular trafficking of ErbBs. Exp Cell Res. 2009;315:683–696. doi: 10.1016/j.yexcr.2008.07.029. [DOI] [PubMed] [Google Scholar]
- 30.Kario E, Marmor MD, Adamsky K, Citri A, Amit I, Amariglio N, Rechavi G, Yarden Y. Suppressors of cytokine signaling 4 and 5 regulate epidermal growth factor receptor signaling. J Biol Chem. 2005;280:7038–7048. doi: 10.1074/jbc.M408575200. [DOI] [PubMed] [Google Scholar]
- 31.Nicholson SE, Metcalf D, Sprigg NS, Columbus R, Walker F, Silva A, Cary D, Willson TA, Zhang JG, Hilton DJ, Alexander WS, Nicola NA. Suppressor of cytokine signaling (SOCS)-5 is a potential negative regulator of epidermal growth factor signaling. Proc Natl Acad Sci USA. 2005;102:2328–2333. doi: 10.1073/pnas.0409675102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Calvisi DF, Ladu S, Gorden A, Farina M, Lee JS, Conner EA, Schroeder I, Factor VM, Thorgeirsson SS. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J Clin Invest. 2007;117:2713–2722. doi: 10.1172/JCI31457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Linossi EM, Chandrashekaran IR, Kolesnik TB, Murphy JM, Webb AI, Willson TA, Kedzierski L, Bullock AN, Babon JJ, Norton RS, Nicola NA, Nicholson SE. Suppressor of Cytokine Signaling (SOCS) 5 utilises distinct domains for regulation of JAK1 and interaction with the adaptor protein Shc-1. PLOS One. 2013;8:e70536. doi: 10.1371/journal.pone.0070536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kershaw NJ, Murphy JM, Liau NPD, Varghese LN, Laktyushin A, Whitlock EL, Lucet IS, Nicola NA, Babon JJ. SOCS3 binds specific receptor–JAK complexes to control cytokine signaling by direct kinase inhibition. Nat Struct Mol Biol. 2013;20:469–476. doi: 10.1038/nsmb.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang S, Park K, Turkson J, Arteaga CL. Ligand-independent phosphorylation of Y869 (Y845) links mutant EGFR signaling to stat-mediated gene expression. Exp Cell Res. 2008;314:413–419. doi: 10.1016/j.yexcr.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bullock AN, Rodriguez MC, Debreczeni JÉ, Songyang Z, Knapp S. Structure of the SOCS4-ElonginB/C complex reveals a distinct SOCS box interface and the molecular basis for SOCS-dependent EGFR degradation. Structure. 2007;15:1493–1504. doi: 10.1016/j.str.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sutherland JM, Keightley RA, Nixon B, Roman SD, Robker RL, Russell DL, McLaughlin EA. Suppressor of cytokine signaling 4 (SOCS4): Moderator of ovarian primordial follicle activation. J Cell Physiol. 2012;227:1188–1198. doi: 10.1002/jcp.22837. [DOI] [PubMed] [Google Scholar]
- 38.Hendriks BS, Griffiths GJ, Benson R, Kenyon D, Lazzara M, Swinton J, Beck S, Hickinson M, Beusmans JM, Lauffenburger D, de Graaf D. Decreased internalisation of erbB1 mutants in lung cancer is linked with a mechanism conferring sensitivity to gefitinib. Syst Biol (Stevenage) 2006;153:457–466. doi: 10.1049/ip-syb:20050108. [DOI] [PubMed] [Google Scholar]
- 39.Lazzara MJ, Lane K, Chan R, Jasper PJ, Yaffe MB, Sorger PK, Jacks T, Neel BG, Lauffenburger DA. Impaired SHP2-mediated extracellular signal-regulated kinase activation contributes to gefitinib sensitivity of lung cancer cells with epidermal growth factor receptor-activating mutations. Cancer Res. 2010;70:3843–3850. doi: 10.1158/0008-5472.CAN-09-3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walsh AM, Lazzara MJ. Regulation of EGFR trafficking and cell signaling by Sprouty2 and MIG6 in lung cancer cells. J Cell Sci. 2013;126:4339–4348. doi: 10.1242/jcs.123208. [DOI] [PubMed] [Google Scholar]
- 41.Red Brewer M, Yun C-H, Lai D, Lemmon MA, Eck MJ, Pao W. Mechanism for activation of mutated epidermal growth factor receptors in lung cancer. Proc Natl Acad Sci USA. 2013;110:E3595–E3604. doi: 10.1073/pnas.1220050110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, Cortot AB, Chirieac L, Iacob RE, Padera R, Engen JR, Wong KK, Eck MJ, Gray NS, Jänne PA. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462:1070–1074. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chmielecki J, Foo J, Oxnard GR, Hutchinson K, Ohashi K, Somwar R, Wang L, Amato KR, Arcila M, Sos ML, Socci ND, Viale A, de Stanchina E, Ginsberg MS, Thomas RK, Kris MG, Inoue A, Ladanyi M, Miller VA, Michor F, Pao W. Optimization of dosing for EGFR-mutant non–small cell lung cancer with evolutionary cancer modeling. Sci Transl Med. 2011;3:90ra59. doi: 10.1126/scitranslmed.3002356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chandarlapaty S. Negative feedback and adaptive resistance to the targeted therapy of cancer. Cancer Discov. 2012;2:311–319. doi: 10.1158/2159-8290.CD-12-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, Rosen N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19:58–71. doi: 10.1016/j.ccr.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Serra V, Scaltriti M, Prudkin L, Eichhorn PJA, Ibrahim YH, Chandarlapaty S, Markman B, Rodriguez O, Guzman M, Rodriguez S, Gili M, Russillo M, Parra JL, Singh S, Arribas J, Rosen N, Baselga J. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene. 2011;30:2547–2557. doi: 10.1038/onc.2010.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, Brown RD, Pelle PD, Dias-Santagata D, Hung KE, Flaherty KT, Piris A, Wargo JA, Settleman J, Mino-Kenudson M, Engelman JA. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF-mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2:227–235. doi: 10.1158/2159-8290.CD-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 49.Li R, Hu Z, Sun SY, Chen ZG, Owonikoko TK, Sica GL, Ramalingam SS, Curran WJ, Khuri FR, Deng X. Niclosamide overcomes acquired resistance to erlotinib through suppression of STAT3 in non–small cell lung cancer. Mol Cancer Ther. 2013;12:2200–2212. doi: 10.1158/1535-7163.MCT-13-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Linossi EM, Babon JJ, Hilton DJ, Nicholson SE. Suppression of cytokine signaling: The SOCS perspective. Cytokine Growth Factor Rev. 2013;24:241–248. doi: 10.1016/j.cytogfr.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamashita T, Kamada H, Kanasaki S, Maeda Y, Nagano K, Abe Y, Inoue M, Yoshioka Y, Tsutsumi Y, Katayama S, Inoue M, Tsunoda S. Epidermal growth factor receptor localized to exosome membranes as a possible biomarker for lung cancer diagnosis. Pharmazie. 2013;68:969–973. [PubMed] [Google Scholar]
- 52.Singh B, Coffey RJ. Trafficking of epidermal growth factor receptor ligands in polarized epithelial cells. Annu Rev Physiol. 2014;76:275–300. doi: 10.1146/annurev-physiol-021113-170406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baselga J, Semiglazov V, van Dam P, Manikhas A, Bellet M, Mayordomo J, Campone M, Kubista E, Greil R, Bianchi G, Steinseifer J, Molloy B, Tokaji E, Gardner H, Phillips P, Stumm M, Lane HA, Dixon JM, Jonat W, Rugo HS. Phase II randomized study of neoadjuvant everolimus plus letrozole compared with placebo plus letrozole in patients with estrogen receptor–positive breast cancer. J Clin Oncol. 2009;27:2630–2637. doi: 10.1200/JCO.2008.18.8391. [DOI] [PubMed] [Google Scholar]
- 54.Bosch A, Li Z, Bergamaschi A, Ellis H, Toska E, Prat A, Tao JJ, Spratt DE, Viola-Villegas NT, Castel P, Minuesa G, Morse N, Rodón J, Ibrahim Y, Cortes J, Perez-Garcia J, Galvan P, Grueso J, Guzman M, Katzenellenbogen JA, Kharas M, Lewis JS, Dickler M, Serra V, Rosen N, Chandarlapaty S, Scaltriti M, Baselga J. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor–positive breast cancer. Sci Trans Med. 2015;7:283ra251. doi: 10.1126/scitranslmed.aaa4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jerusalem G, Bachelot T, Barrios C, Neven P, Di Leo A, Janni W, de Boer R. A new era of improving progression-free survival with dual blockade in postmenopausal HR+, HER2− advanced breast cancer. Cancer Treat Rev. 2015;41:94–104. doi: 10.1016/j.ctrv.2014.12.011. [DOI] [PubMed] [Google Scholar]
- 56.Yang S, Qu S, Perez-Tores M, Sawai A, Rosen N, Solit DB, Arteaga CL. Association with HSP90 inhibits Cbl-mediated down-regulation of mutant epidermal growth factor receptors. Cancer Res. 2006;66:6990–6997. doi: 10.1158/0008-5472.CAN-06-1042. [DOI] [PubMed] [Google Scholar]
- 57.Behrmann I, Smyczek T, Heinrich PC, Schmitz-Van de Leur H, Komyod W, Giese B, Müller-Newen G, Haan S, Haan C. Janus kinase (Jak) subcellular localization revisited: The exclusive membrane localization of endogenous Janus kinase 1 by cytokine receptor interaction uncovers the Jak.receptor complex to be equivalent to a receptor tyrosine kinase. J Biol Chem. 2004;279:35486–35493. doi: 10.1074/jbc.M404202200. [DOI] [PubMed] [Google Scholar]
- 58.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. STAT3 activation and inhibition in TKI-resistant NSCLC.
Fig. S2. JAK2 inhibition increases the abundance of pEGFR and pERK in NSCLC.
Fig. S3. JAK2 inhibition leads to rapid activation of EGFR/RAS/ERK signaling.
Fig. S4. JAK2 links SOCS5 to EGFR, regulating sensitivity to TKI.
Fig. S5. Quantification of Western blots in Fig. 4, A and C.
Fig. S6. Quantification of Western blots in Fig. 4D.