

Abstract
SNARE complexes are the core molecular machinery of membrane fusion, a fundamental process that drives inter- and intra- cellular communication and trafficking. One of the questions that remain controversial has been whether and how SNAREs cooperate. Here we show the use of self-assembled DNA-nanostructure rings to template uniform-sized small unilamellar vesicles containing predetermined maximal number of externally facing SNAREs to study the membrane-fusion process. We also incorporated lipid-conjugated complementary ssDNA as tethers into vesicle and target membranes, which enabled bypass of the rate-limiting docking step of fusion reactions and allowed direct observation of individual membrane-fusion events at SNARE densities as low as one pair per vesicle. With this platform, we confirmed at the single event level that after docking of the templated-SUVs to supported lipid bilayers (SBL), 1–2 pairs of SNAREs are sufficient to drive fast lipid mixing. Modularity and programmability of this platform makes it readily amenable to studying more complicated systems where auxiliary proteins are involved.
Graphical abstract
INTRODUCTION
In the extensively investigated model system of synaptic-vesicle fusion, the SNARE (Soluble N-ethylmaleimide-sensitive factor Attachment protein REceptor) complex assembles from three proteins: the plasma-membrane proteins (t-SNAREs), Syntaxin-1 and synaptosome-associated protein of 25 kD (STX1 and SNAP25A), and the vesicular protein (v-SNARE), synaptobrevin-2 (VAMP2).1,2 Formation of the highly thermostable SNARE complex starts from the zippering of the N-termini of t- and v-SNAREs, and then proceeds toward the C-termini in a stage-wise way.3–5
In the past, complexity-reduced in vitro setups have been instrumental in elucidating the mechanism of SNARE-mediated membrane fusion at a molecular level. Typically in such systems, t- and v- SNAREs were reconstituted into separate membranes (e.g. small unilamellar vesicles (SUVs), giant unilamellar vesicles (GUVs), nanodiscs, supported lipid bilayers (SBLs)) where SNARE-driven membrane fusion could be monitored as fluorescence signal changes due to the lipid or content mixing.6–11 Up to date, the estimated number of SNAREs that cooperate to drive membrane fusion has varied from 1 to 30 depending on the biophysical and biochemical characteristics of the particular membrane system. Recent studies that were based on SUV-SUV12 and nanodisc-SUV 13 fusion systems showed that one pair of SNAREs was sufficient to drive fusion, albeit at a reaction rate orders of magnitude slower than what is observed physiologically. On the other hand, two groups independently reported a modified SUV-SBL system11,14 in which individual fusion events were monitored by TIRF microscopy, demonstrating that for a system where membrane fusion requires SNAP25, SNAREs can drive fusion within about 150 ms after docking, in better agreement with observations in vivo. Both groups estimated that ~15 pairs of SNAREs are required to maximize the rate of docking and subsequent lipid mixing.
Despite the elegance of these reconstitution systems, general limitations of the previous studies include the following: (i) a wide distribution of the size of proteo-liposomes prepared by detergent-dialysis or extrusion method; (ii) the lack of precise control of the orientation and the number of SNAREs displayed on SUVs; (iii) slow overall reaction (docking & fusion) rate in the ensemble assays, or undetectable fusion at low protein density (i.e. less than 10 SNAREs per vesicle, corresponding to fewer than 5 facing outwards) in the single-particle SUV-SBL fusion assays, both of which have been attributed to the slow, rate-limiting, SNARE-mediated vesicle docking step.
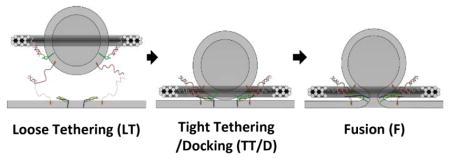
Here we report a novel platform, in which the vesicle size, the number of proteins per vesicle, and the orientation of the SNAREs were rigorously controlled by a nanoscale template generated through DNA self-assembly (Fig. 1). Specifically, we took advantage of a technique termed “DNA origami” that has been used by us and others to produce nanoscale objects with customizable geometry.15–18 Such DNA nanostructures feature programmable rigidity and surface addressability, making them useful as structural templates to organize guest molecules (nucleic acid, lipid, protein, antibody, etc.) with nanometer precision. In addition, we incorporated freely diffusing lipid-conjugated DNA “tethers” into both SUVs (“v-tethers”) and SBL (“t-tethers”) to promote SNARE-independent, more rapid docking (Fig. 1), 19–26 which allowed for the observation of individual fusion events at SNARE densities as low as 1 per vesicle.
Figure 1.

Manufacture of uniform-sized DNA-origami templated SUV that bears pre-determined numbers of VAMP2 (v-SNAREs) and v-tethers. Illustrated is an example of a DNA origami-templated vesicle, where the template displays 4 full length VAMP2 (aa 1–116), 12 ssDNA-lipid anchors. A number of v-tethers, not attached to the template, diffuse freely on the external surface of a vesicle. The detailed preparation procedure is described in the Material and Methods section in the Supplementary Information. Briefly, (f) was first prepared by mixing and incubating individually purified components (c), (d), and (e). After purification, complex (f) was reconstituted into (g) and the final product (h) was obtained. Anti-handles holding DOPE anchors are not shown in (h) cross-section view for clarity. Color codes: Grey annulus, DNA origami-ring containing a total of 16 inner handles, with 4 of “handle i” to direct VAMP2 and 12 of “handle ii” to direct DOPE lipid anchors; Green, full-length VAMP2; Orange, DOPE lipid anchors; Red in different shades, ssDNA with different sequences on templated-SUV, serving as anti-handles and v-tethers (see Table S1 for oligonucleotide sequences).
RESULTS AND DISCUSSION
1. System Design
The DNA nano-template used in this study is a rigid 12-helix-bundle (12hb) ring with inner diameter, outer diameter and thickness of 60 nm, 84 nm, and 7 nm, respectively (Fig. 1). To achieve a ring that organizes a precise number of spatially defined individual SNAREs, we designed sixteen evenly spaced, 42-nt long single-stranded DNA “handles” that all stem from the same inner DNA helix and point toward the center of the ring. Furthermore, we assigned each inner handle with one of two orthogonal DNA sequences (termed handle-i and handle-ii; see Table S1 for sequences), so that it can direct the assembly of either an anti-handle-i conjugated VAMP2 or an anti-handle-ii conjugated DOPE “anchor” through specific handle/anti-handle hybridization. In this way, we could easily vary the number of SNAREs anchored to a ring from 0 to 16, while maintaining constant the total number of hydrophobic linkages between the ring and the vesicle it templates. To mitigate the steric hindrance posed by the ring, we left a 21-nt single-stranded region at the ring-proximal end of each handle so that the anchored molecules (VAMP2 and lipid) could still diffuse within a relatively large area on the vesicle (Fig. 1).
Inspired by the evolutionarily conserved tethering proteins that function prior to the zippering of SNAREs, 27–34 we designed DNA tethers that could bring a vesicle and its target membrane into proximity to prepare for the subsequent SNARE-docking and fusion events (Fig. 1, Table S1). We previously optimized the lengths and density of these DNA tethers in a bulk fusion assay. 35 Specifically, we designed two types of 63-nt long DNA tethers, each with a 42-nt poly-thymidine segment at the membrane-proximal end and a complementary 21-nt segment at the membrane distal end (Fig. 1), to be incorporated into SUVs (v-tethers) and SBL (t-tethers) separately. Thus the hybridization between v-tethers and t-tethers should form a 21-bp DNA duplex that can loosely localize the SUVs on their target SBL. However the long, flexible poly-T domains should act as spacers to prevent tether pairing from driving fusion (Fig. S1). Note that the v-tethers are not anchored to the templating DNA-origami ring.
2. Preparation, characterization, and validation of templated-SUVs
2.1 Preparation
To manufacture nano-templated SUV bearing lipid-conjugated v-tethers, (Fig. 1, details in Suppl. Materials and Methods), ring-VAMP2-DOPE complexes were first produced by mixing and incubating (at 37 °C for 2 hrs) individual purified components: Alexa 647-labeled anti-handle-i-VAMP2, Alexa 488-labeled anti-handle-ii-DOPE, and a 12hb DNA-origami ring. The resultant ring-VAMP2-DOPE complexes were separated from the excessive lipid or VAMP2 conjugated anti-handles by rate-zonal centrifugation using a glycerol gradient. The purified complexes were then reconstituted into SUVs (mole%: DOPC 78.9%, DOPS 15%, PEG2000-DOPE 5%, Rho-DOPE 1%, v-tether-DOPE 0.1%) via detergent dialysis and further purified by isopycnic centrifugation using an iodixanol gradient. Fractions collected with increasing iodixanol concentration were then characterized by electrophoresis using agarose gel containing 0.05% sodium dodecyl sulfate (SDS), which separated the vesicles (in the form of lipid-detergent micelles, labeled by Rhodamine B) and the VAMP2/DOPE-decorated DNA rings (labeled by Alexa 647 and/or Alexa 488). Typical gel images (Fig. 2a) suggested that the majority of the DNA-ring templated liposomes resided in fraction 2 of the iodixanol gradient while most of the ring-less vesicles were enriched in fraction 1. Such a separation was due to the increased buoyant density of the templated liposomes concomitant with the incorporation of the DNA-origami ring.
Figure 2.

Characterization of DNA origami-templated SUVs containing v-tethers.
(A) SDS-agarose gel for the templated vesicles. Gel runs from the top to the bottom as in the images. Fractions obtained after equilibrium density gradient separation (from the Fraction 1, “F1” on the top layer of the density gradient to the Fraction 12 on the bottom layer) were loaded on the gel. As shown in the images, Fraction 1 contained liposomes without ring complexes, Fraction 2 contained the ring-templated SUV, and later fractions contained little amount of detectable substances. Aggregates were absent in all the fractions. Limited by the space, gel images were shown till fraction 4 for each preparation. Fraction 2 (“F2” in gel image) was collected for further measurement in the fusion assay. The color of the product bands gradually changed from pure blue to pure red, with the number of VAMP2 (conjugated to anti-handle i-Alexa647, coded as red) increasing from 0 to 16, and the number of balancing lipid anchors (conjugated to anti handle ii-Alexa488, coded as blue) decreasing from 16 to 0. (B–C) Transmission Electronic Microscopy (TEM) characterization of the final templated vesicles. (B) Representative TEM images with a selection of zoomed-in images (100 nm × 100 nm). Scale bar, 100 nm. (C) Size distribution of the vesicles on ring-templates. The measured mean size of vesicles was 39.1 ± 7.7 nm in diameter (N = 110 vesicles). The solid black line represents a Gaussian fit for the size-distribution histogram. The solid blue line represents a Gaussian fit for the size-distribution histogram for vesicles prepared by standard detergent-dialysis method that contained v-tethers and v-SNAREs with a measured mean diameter of 54.5 ± 16.3 nm (N = 370 vesicles).
2.2 Size & orientation
Negative-stain transmission electron microscopy (TEM) was then applied to characterize the size of the nano-templated SUVs. Representative TEM images (Fig. 2b) revealed that about 80% of the rings carried exactly one vesicle (Fig. S2). We then manually measured the size of each vesicle that appeared to be associated with a DNA template. As shown in Fig. 2c, the size of the templated SUVs showed a narrower size distribution (diameter: 39.1 ± 7.7 nm, mean ± std) than the SUVs prepared in the absence of DNA templates (diameter: 54.5 ± 16.3 nm, mean ± std). These size distributions are consistent with what we previously observed with protein-free vesicles prepared with DNA templates.36
Having precise control of the vesicle size is critical in studying membrane fusion, because the intrinsic curvature of a vesicle closely correlates with its fusion propensity. 37 When a vesicle’s diameter increases from 40 nm to 100 nm, its curvature stress reduces about tenfold, 33 reaching a level similar to that of giant vesicles. This is why a smaller vesicle, whose curvature stress can be released through fusion, is more fusogenic than a larger vesicle. In fact, it has been reported that vesicles with various sizes follow distinct membrane fusion kinetics and require different cooperativity of SNAREs to drive fusion. 12,33,38,39 Therefore, compared to the standard SUVs, templated SUVs with better size homogeneity constitute a more rigorously controlled system to investigate membrane fusion.
The final templated-vesicles should display all the VAMP2 protein in the outer leaflet, which is the correct orientation required to drive membrane fusion; in the other orientation, the DNA handles would be forced to penetrate the membrane, which would be highly unfavorable energetically.
2.3 Number of proteins on templated-SUV
To demonstrate that the nano-templated vesicles carry the desired number of VAMP2, we performed the following two experiments. First, SDS-agarose gel electrophoresis (Fig. 2a) was used as a bulk assay to estimate the number of VAMP2 on a ring. As shown in the gel images in Fig. 2a, the color of the DNA-ring band gradually changed from blue (representing the Alexa 488-labeled anti-handle-DOPE) to red (representing the Alexa 647-labeled anti-handle-VAMP2) when the number of VAMP2 molecules per ring increased from 0 to 16, consistent with the design.
Second, to quantitatively confirm the number of VAMP2 on the templated vesicles at the single-molecule level, we carried out stepwise photobleaching experiments using TIRFM (Fig. 3 and Movie S1). We first deposited templated SUV on a glass coverslip at the density of about 300–500 particles per view (Fig. 3a). Fluorescence-intensity (excited at 647 nm, for more experimental details, see Suppl. Materials and Methods) traces for each particle showed distinct steps during bleaching (Fig. 3b and S11). We then manually counted the number of steps of each selected spot, which revealed the number of Alexa 647 labeled anti-handle-VAMP2 on each ring complex. When a DNA ring enclosed exactly one SUV, which is 80% of the time, the measured bleaching steps also represented the number of VAMP2 per templated vesicle. The distribution of the experimentally determined photobleaching steps per spot matched closely with a theoretical prediction assuming 75% probability for a handle to be occupied by VAMP2 (Fig. 3c–f). We observed that, for each type of ring designed to carry 1–4 VAMP2, fewer than 10% of the spots had more bleaching steps than the designed number of VAMP2 (possibly because two vesicles were too close to each other to be differentiated by TIRF when they were deposited onto the glass coverslip), indicating that the maximum number of VAMP2 has been rigorously controlled by the ring template.
Figure 3.

Single-molecule step-bleaching experiments confirming the number of SNAREs on each vesicle. (A) Selected fluorescent spots (particles in blue circles) for analysis of 4×VAMP2 ring (for an area of 91 μm × 91 μm). (B) Examples of mean fluorescence intensity (area of 6×6 pixels) time courses showing bleaching steps. (C) – (F) Distribution of bleaching steps of rings with different numbers of labeled VAMP2. The observed distributions match calculated distributions assuming 75% yield of binding of VAMP2 to each handle and 10% probability of two spots co-localized with one another during the measurements.
Through the above quality-control experiments, we have demonstrated that the DNA-origami rings served as templates to control the size of SUVs (~40 nm in diameter) and the maximum number of externally orientated VAMP2 per vesicle (ranging from 0 to 16).
3. Fusion of templated-SUV to supported lipid bilayers (SBL)
Next, we studied the functional activity for a series of templated-SUV variants, in which the only independent variable was the number of pre-organized SNAREs (0, 1, 2, 3, 4, 8, or 16) on the outer leaflet of the vesicles (Fig. 1). We applied the SUV-SBL assay setup, developed earlier by our group, 14 with additional assistance from v-tethers that allows direct measurement at low SNARE density (Fig. 4a).
Figure 4.


Templated SUVs fused with SBL after capture mediated by tether pairings. (A) Cartoon illustrating the transition from the Loosely Tethered (LT) stage, to the Tightly Tethered/Docked (TT/D) stage, and to the Fusion (F) stage. For clarity, DOPE anchors on the template are omitted, and only t-SNARE and the t-tethers facing upward are shown. Color codes: Blue, Syntaxin 1; Yellow, SNAP25; Light grey curls, t-tethers. The other colors have the same meaning as in Figure 1. (B) An example of fusion event, in which a vesicle was observed to undergo an LT to TT to F transition sequence. (i) Image series during measurement (11 μm × 11 μm area). (ii) Mean intensity time course of the area (5 μm × 4 μm area exclude other particles). (iii) 2D diffusion traces of the observed vesicle (5 μm × 4 μm area). The vesicle (bright spot) was first captured onto the SBL by tether pairings (noted as entry into the LT state, at time 0 s in (i) and (ii)); then the vesicle diffused on the bilayer (red trace in (iii)) until it was tightly tethered and immobile on the SBL (noted as entry into the TT state, at 2.4 s in (i) and (ii)); after v- and t-SNARE docking (noted as entry into the D state), certain fractions of the populations of these immobile vesicles proceeded to fusion (noted as entry into the F state, at about 6.9 s in (i) and (ii) ) while others remained immobilized until they became invisible due to photobleaching. Note that at the moment of immobilization (i.e. entry into the TT state), a characteristic intensity step-increase was observed, indicating the vesicle was pulled closer to the SBL. 30 (C) Normalized rates for capture of vesicles into the LT state. Tethering of vesicles onto SBL is determined by hybridization of t- and v-tethers, and is independent of the presence or the density of SNAREs. (D) Percentage of the loosely tethered vesicles (LT) that proceeded to the tightly tethered (TT) state as a function of the maximal number of templated VAMP2. No correlation was observed, indicating that immobilization of the vesicles onto the SBL was dominated by the tether pairings and was independent of the presence or the density of SNAREs. (E) Percentage of tightly tethered (TT) vesicles that proceeded to fusion (i.e. fusion competency) as a function of the maximal number of templated VAMP2. In contrast to the two proceeding stages, fusion competency was observed to be dependent on the number of templated SNAREs on the vesicles. Error bars represent S.E.M. in (C) – (E). Detailed statistics are listed in Table S2.
Briefly, in a microfluidic 4-channel setup, SUVs (lipid composition by mole fraction: DOPC 79.2%, DOPS 15%, PEG2000-DOPE 5%, NBD-DOPE 0.8%) containing reconstituted full length t-SNAREs (lipid: protein, mole/mole, 2000:1), and v-tethers (total lipid: oligo-linked DOPE, mole/mole, 200:1) were introduced by flow. These vesicles burst onto the glass coverslip and formed a lipid bilayer (Fig. S3). Bilayers, without visible defects, or patches, and whose fluorescence (from NBD-DOPE) could recover to at least 80% of the original intensity (under continuous 488 nm laser excitation) after photobleaching, were chosen for further measurement. The diffusion coefficient of lipids on the SBLs was measured to be 2.4 ± 0.4 μm2/s (Mean ± SD) (Fig. S3). 9 After rinsing out any leftover unburst t-SUV in solution, we introduced by flow the SUV containing v-tethers and ring-templated v-SNAREs (at ~3 pM SUV concentration). Lipid mixing was then recorded under continuous 532 nm excitation and was evident as delivery of Rho-DOPE fluorophore from the templated-SUVs to, and the subsequent lateral diffusion in, the target membrane (Fig. 4b image series).
3.1 Overall Picture
For a typical event, we observed three sequential stages (Movie S2, Fig. 4a and Fig. 4b). The first stage was identified by the capture of a templated-SUV to the SBL. It was characterized by relatively rapid diffusion of SUVs within the SBL (see below; noted as the loosely tethered state “LT”, Fig. 4b, red segment). The second stage was identified by a dramatic reduction of diffusion on the SBL (noted as the tightly tethered state “TT”, green segment). Lastly, within a few seconds, some of the immobilized vesicles proceeded to fuse with the SBL (noted as the fusion state, “F”, black segment) while the rest were bleached after about 2 mins’ continuous laser excitation and their putative subsequent lipid mixing could not be monitored.
3.2 LT stage
For individual events in the LT stage, the diffusion trajectory of every single particle, i.e. every single vesicle, was tracked, as shown in Fig. 4b (red segment). Based on this trajectory, the mean square displacement (MSD) for each particle as a function of time can then be derived (Fig. S4), and the slope of this line represents the diffusion coefficient of each particle. A histogram of the diffusion coefficients of 109 vesicles (randomly picked, with or without SNAREs; Fig. S4 and S12) was calculated, with an average of 1.4 ± 1.2 μm2/s (Mean ± SD). This value is comparable to those observed for liposomes attached in a supported bilayer through just a single lipid anchor19,21,30 (membrane anchored protein diffuse slower), and does not correlate with the presence of the density of SNAREs on the vesicles, suggesting that loose tethering is caused by hybridization of the first pair of tethers.
We then analyzed the rate for vesicles to be tethered onto the SBL (Fig. 4c) (Suppl. Mat. for Data Analysis), measured as the number of vesicles appearing on a selected membrane area, normalized by the acquisition time, the concentration of templated-SUV, and the area of interest. As demonstrated in Fig. 4c, in the presence of v-tethers, the normalized tethering rate increased ~10 fold over those in the absence of v-tethers, and this rate was not affected by the presence or the density of v-SNAREs on the vesicles. This observation also suggested that the loose tethering rate was solely determined by the tethers (about 10 v-tethers on outer leaflet of vesicles; about 10 fold more t-tethers than t-SNAREs per unit area on the SBL).
Previously, the force-extension curve for an ssDNA has been measured by stretching both ends of ssDNA using dual-beam laser trap instrument, and was found to be well fit by a worm-like chain model.40 Based on this model, we estimated that, before tethering, the length of v- and t-tethers (63 nt long) each samples a distribution with an average extension of ~6 nm, where ~10% of the tethers sample a length of 10 nm at any given time. Because each of the SUVs and the SBL surface are also covered by a 4–5 nm thick PEG brush layer (PEG 2000 at 5 mol%), 9 we roughly estimated that some tether pairs start to engage at an average separation distance of about 25–30 nm (for detailed calculations (see Supplementary). Once the tether pairs fully hybridize (21 nt out of the 63 nt), then 84 nt of ssDNA span the membranes of templated-SUV and the SBL. Following the same method, we estimate that the average separation distance, after tether hybridization is complete, is about 15–20 nm. A Surface Force Apparatus (SFA) experiment demonstrated that t- and v-SNAREs in two opposing bilayers start overlapping (but cannot assemble yet) at a separation distance of about 20 nm, 3 and SNAREpins (SNARE complexes linking two membranes) start to fold when the separation distance is below 8 nm. Therefore according to our model, SNAREpins have not yet engaged at the LT stage.
3.3 LT to TT/D stage
Among the vesicles that were captured on the SBL by tether pairing, (Suppl. Mat. for Data Analysis), about 80% to 90% quickly immobilized on the surface; we refer to this immobilized condition as the TT stage (Fig. 4d). This percentile was independent of the presence or the number of VAMP2 on the templated vesicles, which indicates that the immobilization is also dominated by tether pairings (Fig. 4d). Since t-tethers were about 10-fold more abundant than t-SNAREs on the SBL and the separation distance during the LT stage is modeled by us to be larger than that required for SNAREpin formation, it is very likely that the immobilization was caused by simultaneous hybridization of multiple pairs of tethers. 41 Our argument is as follows: vesicles with a 50 nm diameter, close to a membrane, should be characterized by a diffusion coefficient of ~5 μm2/s, 42 whereas the diffusion coefficient for a free DOPE lipid in a membrane was measured to be ~2.5 μm2/s (Fig. S3C). Therefore, the rate of diffusion of a vesicle linked to a bilayer should be slowed significantly by increasing numbers of linkages. This dependence with the number of tethers is consistent with previous observations.43,44 Furthermore, the engagement of a large number of tethers may draw the vesicle even closer to the bilayer surface, thus slowing down diffusion even more due to increased proximity to the no-slip boundary.
We also observed that immobilization of vesicles was usually accompanied by a step-increase of the fluorescence intensity (Fig. 4b and S13). Within the TIRF evanescent field, the fluorescence intensity of a spot exponentially decays with increasing distance of this spot relative to the focal plane 30 (the TIRF decay length was measured to be 70–100 nm for our microscope). Therefore, the observed step-increase of fluorescence intensity suggested that tethered vesicles were pulled toward the SBL upon immobilization, and, based on the magnitude of the intensity change (average of about 16% increase), we estimated that the templated SUV was pulled closer towards the SBL by about 10–15 nm (detailed estimation, see Suppl. Calculations section). Therefore for our model, at the moment of immobilization, the maximal separation distance decreases from 15–20 nm to less than 5–10 nm, and thus brings SBL and templated SUVs within a distance where t- and v-SNAREs can begin to assemble.
In our model, engagement of cognate SNAREs (noted in our model as docked state, “D”) cannot occur efficiently until after immobilization of the templated-vesicles on the SBL. However, under our current assay conditions, we lack an explicit experimental signature for discriminating between a TT and the presumed D stage. Nonetheless, it is instructive to attempt a rough estimate of the expected time intervals between the TT and the D stages based on literature values, and compare these with our observed transition times from TT to F stages. It has been reported that the kon of the SNARE assembly reaction, measured in solution between the soluble t-SNARE (without Habc domain) and the soluble v-SNARE, is about 5× 105 M−1 s−1.45 Assuming t-SNAREs are freely diffusing and homogeneously distributed on the SBL, and knowing the number of proteins inside the volume covered by a templated-SUV (inner diameter of 60 nm), we can estimate that after immobilization, it takes about 100 ms for one pair of t- and v-SNAREs to assemble (see Suppl. Calculations). According to this estimate, docking should occur within a few frames after entry into the TT stage with our acquisition frequency of 30 ms/frame. It is noticeable that, in our system, instead of soluble t- and v-SNAREs (t-SNARE without Habc domain), membrane reconstituted full length t- SNAREs, including the Habc, H3 and the transmembrane domains, and full length v-SNAREs, were used. It has been reported that the Habc domains slow down the assembly of t- and v-SNAREs, 46 therefore the delay time between TT and D is very likely to be longer than 100 ms in our setup.
3.4 Third, F, stage
Among the tightly tethered vesicles, some proceeded to fuse with SBL and the rest remained docked until they were bleached. In most of the fusion events, vesicles completely disappear after fusion, suggesting that full fusion occurs (i.e. pore opening) where both the outer and the inner leaflets have merged with the supported bilayer (Movie S1). In some rare events (less than 2%), vesicles were still visible after fusion until they were bleached, which resembles the hemifusion process where the outer leaflet merged with the supported bilayer and the inner leaflet remained intact (Movie S3). In contrast to the LT or the TT stages, the percentage of immobilized vesicles that proceeded to lipid mixing depended on the number of VAMP2 on the templated vesicles. It increased from about 3% for SUVs bearing no VAMP2 to about 6% SUVs bearing maximum of one VAMP2, and reached about 12% for SUVs bearing a maximum of two VAMP2 (Fig. 4e). This SNARE-density dependency suggests that (i) the observed lipid mixing reaction is determined by the interactions between t- and v-SNAREs; and (ii) after docking, 1–2 pairs of SNAREs are sufficient to drive SNARE-dependent lipid mixing. Note that SNARE occupancy on the DNA-origami rings is about 75%, and perhaps only a fraction of templated v-SNAREs may be active; therefore our data is consistent with a model where just a single pair of SNAREs already confers greater lipid mixing competence (i.e. enables a larger fraction of vesicles in our assay to fuse following immobilization). Because of the unknown about the fraction of active v-SNAREs in our system, we cannot make a strong conclusion about whether additional SNARE pairs confer even more lipid mixing competence. Additionally, we were able to validate this VAMP2 dependency of lipid mixing in an independent set of experiments we performed at Harvard (we collected the first dataset at Yale). Data generated from both sets of experiments (Supplementary Figs. S6–10) were in excellent qualitative agreement with each other. Consistent with previous efforts, a maximum of 10–15% of docked SUVs were found to be fusion competent even for vesicles bearing >10 SNAREs. We speculate this behavior to be a function of static heterogeneities in our lipid bilayers, e.g. patches of SBL that are resistant to fusion, and a function of an unknown portion of non-functional t-SNAREs in the SBL which sometimes form 2:1 Syntaxin1A/SNAP25 complexes during protein expression. In addition, we note that some SUVs without rings (Fig 2B) can possibly contribute to the observed TT/docking events but cannot fuse with SBL. Thus the estimated fusion efficiency (10–15%) is likely a lower bound of fusion-competent SUVs among docked vesicles.
The activation energy for lipid bilayer fusion has been estimated to be in the range 50–100 kBT. 2,47 Assembly of a single SNAREpin has been estimated to release a total of ~65 kBT of free energy. 4 Therefore, formation of 1–2 pairs of SNARE complexes should be able to release sufficient energy to overcome the energy barrier associated with membrane fusion. Using ensemble assays, SUV-SUV and SUV-nanodisc, 12,13,38 it has been reported that one pair of SNAREs was sufficient to drive membrane fusion. However, the previous ensemble assays were limited because they could not distinguish between fast (i.e. physiologically relevant) and slow fusion. The previous single-particle assay was limited in that vesicle docking was rate-limiting. Using DNA tethers, docking rate was normalized for our system. Therefore, by using a rigorously controlled system, we confirm these results at the single-event level.
The time interval (defined as the delay time) between the SNARE complex formation (the docking stage, D) and fusion (i.e. lipid mixing) of two opposing membranes (the fusion stage, F) characterizes how fast a SNARE-mediated membrane fusion reaction occurs, and is a unique parameter that can be experimentally measured only using the SUV-SBL assay. In the original report of SUV-SBL setup, this delay time was shown to be around 100–150 ms. 14 Distribution histograms of the delay time of the fusion events under our assay conditions (between the TT and the F stages), presented as the survival function of all fusion events (Fig. S5), showed that about 50% of all the fusion events, regardless of the density of SNAREs, occurred within 1 sec. after the templated-vesicles were immobilized on SBL (i.e. entry into the TT stage). Given our calculation that it takes at least 100 ms for a templated SUV to proceed from the TT to the D stages, we estimate that under our assay conditions, after SNAREpin assembly (i.e. docking), fusion reactions occur on the ms time scale, and are roughly as fast as that reported previously. Unfortunately, we were not able to precisely measure the time intervals between the D and the F stages in our assay. Practical solutions to overcome this problem in the future may include developing multiplexed single-molecule techniques that can simultaneously detect docking by single-molecule Fluorescence Resonance Energy Transfer (FRET) (coming from the interactions between t- and v-SNAREs) and the lipid mixing between SUV and SBL.
CONCLUSION
With all these observations, we have demonstrated, at a single-event level, that our DNA-tether assisted templated SUV and SBL membrane fusion system functions as designed. In our model, during a successful fusion event, when a templated SUV arrives within 25–30 nm of the SBL, it is captured and then drawn closer by a single tethering event to a distance of 15–20 nm; after diffusing on the SBL, the vesicle is then immobilized through additional tether pairings, pulling it closer to a distance of less than 10 nm above the SBL; lastly, zippering of t- and v-SNAREs drives membrane fusion (i.e. lipid mixing).
In conclusion, using DNA origami rings as templates for v-SNAREs and lipid-conjugated oligonucleotides as v- and t-tethers, we built a novel system that allowed us to bypass the rate-limiting docking step and directly observe single SNARE-dependent fusion between uniform-sized SUV and SBL at SNARE densities as low as one per vesicle. At a single-event level, using a rigorously controlled system, we confirmed that 1–2 pairs of SNAREs, by themselves, are sufficient to drive fast membrane fusion.
In general, the system we built provides a versatile and powerful platform to investigate membrane fusion by controlling the micro-environment at fusion sites, such as specifically directing SNAREs to spatially asymmetrical positions, or organizing other regulator proteins (e.g. Complexin, Synaptotagmin 1) around the fusion pore. Furthermore, we also provide a general strategy that can be applied to regulating the function of other membrane proteins by templating and organizing them to desired orientations and stoichiometries within a membrane environment.
Supplementary Material
Acknowledgments
We thank Dr. Y Gao for discussions on the crosslinking reactions. This work was supported by US National Institutes of Health (NIH) grant DK027044-39 and Agence Nationale de la Recherche (ANR) ANR-14-1CHN-0022-01 grant to J.E.R., by a National Institutes of Health (NIH) Director’s New Innovator Award (DP2-GM114830), an NIH grant (R21-GM109466) and a Yale University faculty startup fund to C.L., by an NIH Director’s New Innovator Award (DP2-OD004641), an Army Research Office MURI grant (W911NF-12-1-0420), National Science Foundation Expeditions Grant (1317694), and a Wyss Institute for Biologically Inspired Engineering Faculty Award to W.M.S, and an NIH grant R01GM108954 and a Kavli Neuroscience Scholar Award to E.K..
Footnotes
- Materials and Methods sections including detailed experimental procedures.
- Calculations
References
- 1.Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Sollner TH, Rothman JE. Cell. 1998;92:759. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- 2.Sudhof TC, Rothman JE. Science. 2009;323:474. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li F, Pincet F, Perez E, Eng WS, Melia TJ, Rothman JE, Tareste D. Nat Struct Mol Biol. 2007;14:890. doi: 10.1038/nsmb1310. [DOI] [PubMed] [Google Scholar]
- 4.Gao Y, Zorman S, Gundersen G, Xi Z, Ma L, Sirinakis G, Rothman JE, Zhang Y. Science. 2012;337:1340. doi: 10.1126/science.1224492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zorman S, Rebane AA, Ma L, Yang G, Molski MA, Coleman J, Pincet F, Rothman JE, Zhang Y. Elife. 2014;3:e03348. doi: 10.7554/eLife.03348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scott BL, Van Komen JS, Liu S, Weber T, Melia TJ, McNew JA. Methods Enzymol. 2003;372:274. doi: 10.1016/S0076-6879(03)72016-3. [DOI] [PubMed] [Google Scholar]
- 7.Diao J, Su Z, Ishitsuka Y, Lu B, Lee KS, Lai Y, Shin YK, Ha T. Nat Commun. 2010;1:54. doi: 10.1038/ncomms1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diao J, Ishitsuka Y, Lee H, Joo C, Su Z, Syed S, Shin YK, Yoon TY, Ha T. Nat Protoc. 2012;7:921. doi: 10.1038/nprot.2012.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karatekin E, Rothman JE. Nat Protoc. 2012;7:903. doi: 10.1038/nprot.2012.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi L, Howan K, Shen QT, Wang YJ, Rothman JE, Pincet F. Nat Protoc. 2013;8:935. doi: 10.1038/nprot.2013.048. [DOI] [PubMed] [Google Scholar]
- 11.Domanska MK, Kiessling V, Stein A, Fasshauer D, Tamm LK. J Biol Chem. 2009;284:32158. doi: 10.1074/jbc.M109.047381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van den Bogaart G, Holt MG, Bunt G, Riedel D, Wouters FS, Jahn R. Nat Struct Mol Biol. 2010;17:358. doi: 10.1038/nsmb.1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi L, Shen QT, Kiel A, Wang J, Wang HW, Melia TJ, Rothman JE, Pincet F. Science. 2012;335:1355. doi: 10.1126/science.1214984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karatekin E, Di Giovanni J, Iborra C, Coleman J, O’Shaughnessy B, Seagar M, Rothman JE. Proc Natl Acad Sci U S A. 2010;107:3517. doi: 10.1073/pnas.0914723107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rothemund PW. Nature. 2006;440:297. doi: 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- 16.Dietz H, Douglas SM, Shih WM. Science. 2009;325:725. doi: 10.1126/science.1174251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Douglas SM, Dietz H, Liedl T, Hogberg B, Graf F, Shih WM. Nature. 2009;459:414. doi: 10.1038/nature08016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Douglas SM, Marblestone AH, Teerapittayanon S, Vazquez A, Church GM, Shih WM. Nucleic Acids Res. 2009;37:5001. doi: 10.1093/nar/gkp436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshina-Ishii C, Boxer SG. J Am Chem Soc. 2003;125:3696. doi: 10.1021/ja029783+. [DOI] [PubMed] [Google Scholar]
- 20.Pfeiffer I, Hook F. J Am Chem Soc. 2004;126:10224. doi: 10.1021/ja048514b. [DOI] [PubMed] [Google Scholar]
- 21.Benkoski JJ, Hook F. J Phys Chem B. 2005;109:9773. doi: 10.1021/jp044947p. [DOI] [PubMed] [Google Scholar]
- 22.Beales PA, Vanderlick TK. J Phys Chem A. 2007;111:12372. doi: 10.1021/jp075792z. [DOI] [PubMed] [Google Scholar]
- 23.Stengel G, Zahn R, Hook F. J Am Chem Soc. 2007;129:9584. doi: 10.1021/ja073200k. [DOI] [PubMed] [Google Scholar]
- 24.Gunnarsson A, Jonsson P, Marie R, Tegenfeldt JO, Hook F. Nano Lett. 2008;8:183. doi: 10.1021/nl072401j. [DOI] [PubMed] [Google Scholar]
- 25.Jakobsen U, Simonsen AC, Vogel S. J Am Chem Soc. 2008;130:10462. doi: 10.1021/ja8030054. [DOI] [PubMed] [Google Scholar]
- 26.Chan YH, van Lengerich B, Boxer SG. Proc Natl Acad Sci U S A. 2009;106:979. doi: 10.1073/pnas.0812356106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pfeffer SR. Nat Cell Biol. 1999;1:E17. doi: 10.1038/8967. [DOI] [PubMed] [Google Scholar]
- 28.Jahn R, Lang T, Sudhof TC. Cell. 2003;112:519. doi: 10.1016/s0092-8674(03)00112-0. [DOI] [PubMed] [Google Scholar]
- 29.Sudhof TC. Annu Rev Neurosci. 2004;27:509. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- 30.Karatekin E, Tran VS, Huet S, Fanget I, Cribier S, Henry JP. Biophys J. 2008;94:2891. doi: 10.1529/biophysj.107.116756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fernandez-Busnadiego R, Zuber B, Maurer UE, Cyrklaff M, Baumeister W, Lucic V. J Cell Biol. 2010;188:145. doi: 10.1083/jcb.200908082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hutagalung AH, Novick PJ. Physiol Rev. 2011;91:119. doi: 10.1152/physrev.00059.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hernandez JM, Stein A, Behrmann E, Riedel D, Cypionka A, Farsi Z, Walla PJ, Raunser S, Jahn R. Science. 2012;336:1581. doi: 10.1126/science.1221976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hong W, Lev S. Trends Cell Biol. 2014;24:35. doi: 10.1016/j.tcb.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 35.Xu W, Wang J, Rothman JE, Pincet F. Angew Chem Int Ed Engl. 2015;54:14388. doi: 10.1002/anie.201506844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Y, Wang J, Shigematsu H, Xu W, Shih WM, Rothman JE, Lin C. Nat Chem. 2016 doi: 10.1038/nchem.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malinin VS, Lentz BR. Biophys J. 2004;86:2951. doi: 10.1016/S0006-3495(04)74346-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hernandez JM, Kreutzberger AJ, Kiessling V, Tamm LK, Jahn R. Proc Natl Acad Sci U S A. 2014;111:12037. doi: 10.1073/pnas.1407435111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Malsam J, Parisotto D, Bharat TA, Scheutzow A, Krause JM, Briggs JA, Sollner TH. EMBO J. 2012;31:3270. doi: 10.1038/emboj.2012.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith SB, Cui Y, Bustamante C. Science. 1996;271:795. doi: 10.1126/science.271.5250.795. [DOI] [PubMed] [Google Scholar]
- 41.Gambin Y, Lopez-Esparza R, Reffay M, Sierecki E, Gov NS, Genest M, Hodges RS, Urbach W. Proc Natl Acad Sci U S A. 2006;103:2098. doi: 10.1073/pnas.0511026103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kyoung M, Sheets ED. Biophys J. 2008;95:5789. doi: 10.1529/biophysj.108.128934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johnson-Buck A, Jiang S, Yan H, Walter NG. ACS nano. 2014;8:5641. doi: 10.1021/nn500108k. [DOI] [PubMed] [Google Scholar]
- 44.Kocabey S, Kempter S, List J, Xing Y, Bae W, Schiffels D, Shih WM, Simmel FC, Liedl T. ACS nano. 2015;9:3530. doi: 10.1021/acsnano.5b00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pobbati AV, Stein A, Fasshauer D. Science. 2006;313:673. doi: 10.1126/science.1129486. [DOI] [PubMed] [Google Scholar]
- 46.Misura KM, Scheller RH, Weis WI. Nature. 2000;404:355. doi: 10.1038/35006120. [DOI] [PubMed] [Google Scholar]
- 47.Cohen FS, Melikyan GB. J Membr Biol. 2004;199:1. doi: 10.1007/s00232-004-0669-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.