

Abstract
Inflammatory myofibroblastic tumor (IMT), which was previously known as inflammatory pseudotumor, is characterized by myofibroblastic spindle cells accompanied by inflammatory infiltrates. IMT is a rare lesion of unclear etiology, which induces non-specific clinical symptoms. The present case report describes a 74-year-old female patient with recurrent IMT, which was successfully re-resected 30 months subsequent to initial surgical removal. The patient presented with left hydroureteronephrosis and a 10-cm paravertebral mass which, upon surgery, was found to involve the descending colon. Successful, complete en bloc re-resection was achieved, and at 24 months follow-up, the patient remained clinically free of disease. Complete surgical extirpation, where feasible, is the recommended treatment for primary and recurrent IMT lesions.
Keywords: retroperitoneal inflammatory myofibroblastic tumor, recurrent, surgery
Introduction
Inflammatory myofibroblastic tumor (IMT) was formerly known as inflammatory pseudotumor, and is a lesion characterized by myofibroblastic spindle cells accompanied by inflammatory infiltrates. Although the lung is the most common site of occurrence, IMT may arise in diverse extra-pulmonary locations. The etiology of IMT is unclear; however, the anaplastic lymphoma receptor tyrosine kinase gene may play a role in its pathogenesis (1). The clinical presentation of IMT is non-specific and may include fever, weight loss, malaise and anemia. Retroperitoneal IMT usually grows slowly and may present as a solid abdominal mass, accompanied by abdominal pain and weight loss. Gastrointestinal and urinary symptoms may occur with an enlarging expansile mass (2). There are no specific radiographical features for IMT. For abdominal IMT, a solid mass abutting and compressing various organs is the usual presentation. Retroperitoneal IMT is rare, with only 12 cases reported in English literature to date (2–10). The present study describes a case of retroperitoneal IMT, which recurred 30 months following initial surgery, and where re-resection was successfully performed. Written informed consent was obtained from the patient for the publication of the present study.
Case report
A 74-year-old female initially presented with a 10-cm retroperitoneal mass, discovered incidentally during cholecystectomy. The mass was resected via a midline transperitoneal incision. Histopathological examination identified the lesion as IMT. Patient history included a left pulmonary lobectomy for bronchiectasis 20-years previously, and hypertension for 40-years. Subsequently, the patient presented with a palpable left lower-quadrant abdominal mass 30 months later. Hematological and biochemical parameters, including carcinoembryonic antigen and α-fetoprotein expression, were all within normal limits. Abdominal computerized tomography (CT) with contrast revealed an 8×7.5×8.5 cm, well-defined heterogeneous mass with uneven enhancement (Fig. 1), associated with left hydroureteronephrosis. There was no regional lymphadenopathy. Metastatic workup, including a chest CT scan and radionuclide bone scan, was negative.
Figure 1.
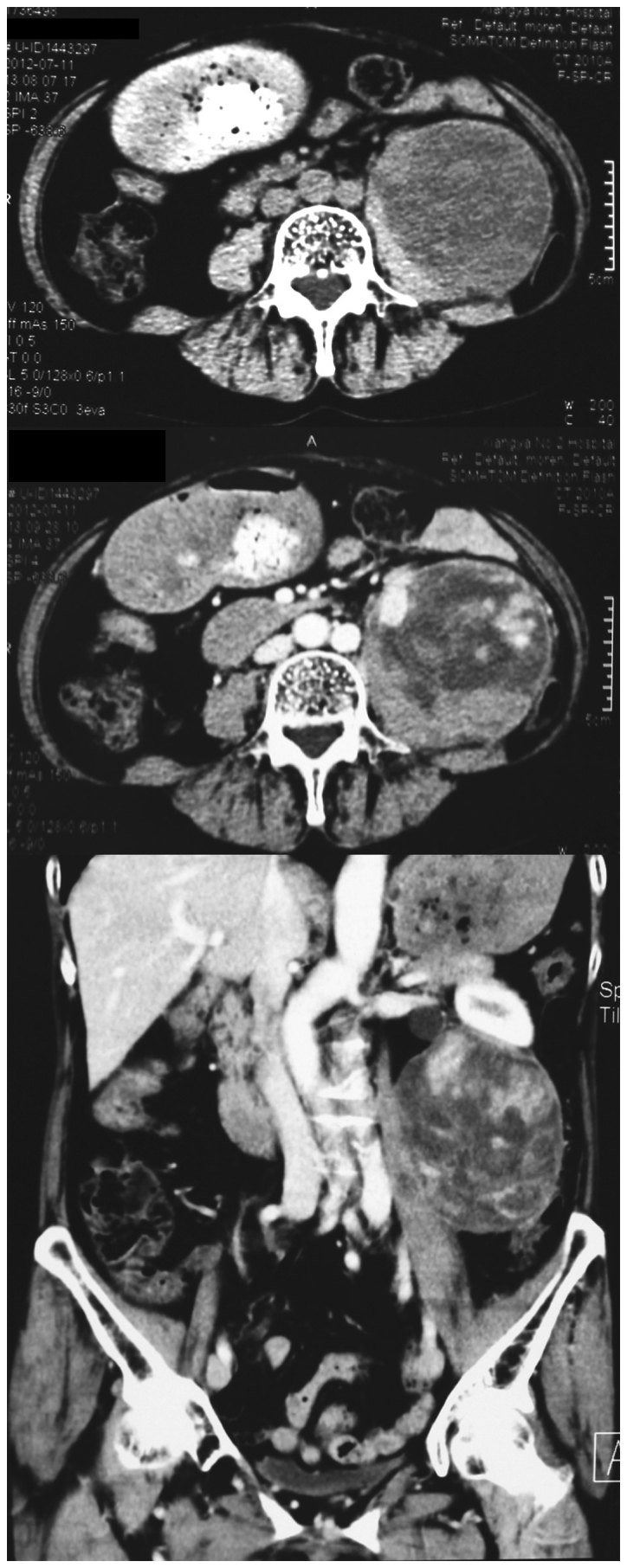
Computed tomography scan revealed a well-defined, heterogeneous lesion with uneven enhancement, occupying the left retroperitoneal region and measuring ~7.5×8.5×8 cm.
Following multidisciplinary consultation, laparotomy was performed via a left subcostal incision. A 10-cm mass was identified, which was compressing the kidney and proximal ureter and adherent to the descending colon. The kidney and ureter were successfully dissected free but partial left colectomy was required to facilitate en bloc removal of the tumor mass. The postoperative course was uneventful. Gross histopathological examination revealed the tumor to be encapsulated, with a grey-whitish cut surface (Fig. 2). Microscopically, the tumor demonstrated proliferation of spindle cells and infiltration of lymphocytes, plasma cells and eosinophils (Fig. 3). Immunohistochemistry revealed diffuse positivity for vimentin, CD68 and desmin; focal staining for myogenin and Ki-67; and negativity for CD34, cytokeratin, smooth muscle actin and S100.
Figure 2.
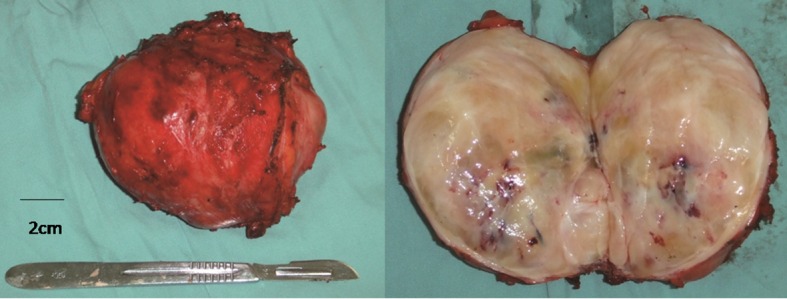
The resected tumor was an encapsulated mass with a grey-whitish cut surface.
Figure 3.
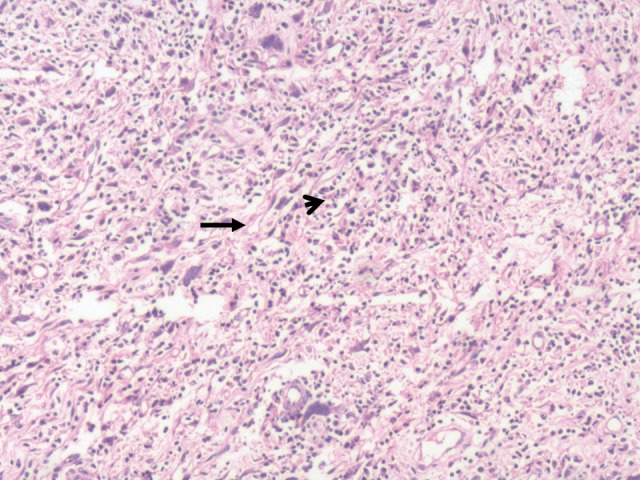
Microscopically the tumor exhibited proliferation of spindle cells (arrow) and infiltration of lymphocytes, plasma cells and eosinophils (arrowhead). Hematoxylin and eosin staining; magnification, ×100.
The multidisciplinary treatment team did not recommend postoperative adjuvant therapy. The patient has remained free of local or distant recurrence 24 months subsequent to re-resection, at the time of the present report.
Discussion
Inflammatory myofibroblastic tumor (IMT) is a distinct lesion, composed of myofibroblastic spindle cells with intermediate biological potential, accompanied by an inflammatory infiltration of plasma cells, lymphocytes and eosinophils (5,6). The most common sites of IMT are the lungs and soft-tissue viscera in children and young adults. The etiology is unclear, although trauma, surgery, inflammation and infection by Epstein-Barr virus or human herpes virus have been proposed as potential causative factors (11). Furthermore, chromosomal rearrangement involving the ALK gene results in activation of a tyrosine kinase receptor and may induce abnormal expression. Immunohistochemically, ~50% of IMTs are positive for ALK (12), suggesting that the ALK gene may have a role in the pathogenesis of IMT (1).
The clinical presentation of IMT is non-specific and may include fever, weight loss, malaise, anemia, thrombocytosis, polyclonal hyper-globulinemia and/or elevated erythrocyte sedimentation rate (2). Frequently, IMT induces anatomical site-specific presentations. Retroperitoneal IMT typically grows slowly and may present as a solid abdominal mass, accompanied by abdominal pain and weight loss. Gastrointestinal and urinary symptoms, particularly bowel and ureteral obstruction, respectively, may occur with an enlarging expansile mass (2). The patient in the present report was asymptomatic with the primary tumor, while the recurrence presented with a palpable mass but an unremarkable hematological and biochemical profile.
There are no radiographic features specific to IMT. In abdominal IMT, a solid mass abutting and compressing various organs is the typical presentation, frequently accompanied by obstruction or invasion of the affected organ(s). CT and magnetic resonance imaging may identify a homogeneous or heterogeneous, hypodense or isodense lesion. Enhancement may be variable and may reveal central necrosis or fibrosis (13).
The diagnosis of IMT mainly occurs via pathological examination. Macroscopically, IMT may be firm, fleshy or gelatinous, with a white or tan cut surface. The tumors vary in size, reaching up to 20 cm in the greatest dimension. Histologically, IMT is characterized by spindle cell proliferation with a prominent inflammatory infiltrate. IMT lesions are also positive for vimentin, smooth muscle actin, muscle-specific actin and occasionally, desmin and cytokeratin (12).
IMT is a neoplasm with ‘intermediate biological potential’ with a propensity for local recurrence, while rarely metastasizing. In a series of 84 cases of extrapulmonary IMT, recurrence was reported in 25% and distal metastasis in only 5% of cases (12). Histopathological features alone may not be sufficient for the prediction of malignant transformation, as tumor size, cellularity, mitotic activity and the presence of necrosis are not significantly correlated with risk of metastasis. However, nuclear atypia and ganglion-like cells may indicate more aggressive cellular behavior. ALK-positive tumors have a notably low risk of metastasis, and ALK reactivity has not been found to correlate with recurrence (12). Compared with other extra-pulmonary lesions, retroperitoneal IMTs tend to be more aggressive. Of the 12 reported cases in the English literature, summarized in Table I, 3 cases developed metastases and one exhibited local recurrence, with follow-up periods varying from 3 to 80 months (2–10).
Table I.
Published cases of retroperitoneal inflammatory myofibroblastic tumor.
| Reference | Age, years | Gender | Size, cm | Treatment | Outcome, time-point |
|---|---|---|---|---|---|
| Coffin et al (3) | 15 | M | NA | Diagnosed and treated as immunoblastic lymphoma 2 months after biopsy | NED, 80 mo |
| 11 | F | 8 | Resection, local recurrences, four re-excisions; regression with xanthogranulomatous histology | NED, 22 mo | |
| Przkora et al (4) | 22 | M | NA | Ibuprofen 400 mg bid | SD, 12 mo |
| Attili et al (5) | 46 | F | 16×13.6×2.1 | Complete resection | NED, 12 mo |
| Mali et al (10) | 12 | M | 3.5 | Complete resection | NED, 6 mo |
| Coffin et al (6) | 14 | F | 22; liver metastasis | NA | NA |
| 17 mo | F | 6; lung metastasis | NA | AWD, 9 mo | |
| 14 | NA | 19; bone metastasis | NA | NA | |
| Koirala et al (7) | 52 | M | 12.5×10.5 | Complete resection | NA |
| Ziadi et al (8) | 41 | M | NA | Partial resection | DOD, 3 mo |
| Chatzikokol et al (2) | 68 | F | 3.3×4.5×4.5 | En bloc resection of the upper section of the adrenal gland, tumor and spleen | NA |
| Tao et al (9) | 14 | F | 3.3×4.5×4.5 | Resection failed; postoperative chemotherapy and diclofenac sodium | NED, 36 mo |
mo, months; M, male; F, female; NED, no evidence of disease; NA, not available; AWD, alive with disease; DOD, died of disease.
The mainstay of management of IMT, as previously reported, is by surgical excision for definitive or palliative treatment (2–10). Incomplete tumor removal frequently results in local recurrence (3), which was likely the case in the present patient. The patient underwent a second successful re-resection, with no recurrence at 30 months follow-up. However, this follow-up period is relatively short and vigilant monitoring is required to facilitate rapid detection of further recurrence.
Tao et al (9) reported an unresectable retroperitoneal IMT, which was successfully treated with methotrexate, cisplatin and diclofenac sodium, facilitating maintenance of a clinical ‘free of disease’ status for 3 years. Przkora et al (4) also reported a case of unreseactable IMT, treated with anti-inflammatory agent, ibuprofen, where the patient was ‘stabilized’ for 12 months. The possible role of the ALK gene in IMT pathogenesis highlights the potential for use of a tyrosine-kinase inhibitor in a neoadjuvant or adjuvant capacity (1). Furthermore, a study reported a sustained partial response to the ALK-inhibitor, crizotinib, in a patient with an ALK-translocated IMT (14). The role of radiotherapy in IMT is unknown, although it may have potential benefits, particularly in unresectable cases. To date, there is no established protocol for the treatment of IMT, due to its rarity (9,15).
Retroperitoneal IMT is a rare entity with unclear etiology and non-specific clinical symptoms. Precise diagnosis mainly occurs via histopathological analysis. Complete surgical excision, where feasible, is the recommended treatment. In addition, complete surgical re-resection is also recommended for the treatment of recurrent lesions.
References
- 1.Cessna MH, Zhou H, Sanger WG, Perkins SL, Tripp S, Pickering D, Daines C, Coffin CM. Expression of ALK1 and p80 in inflammatory myofibroblastic tumor and its mesenchymal mimics: A study of 135 cases. Mod Pathol. 2002;15:931–938. doi: 10.1097/01.MP.0000026615.04130.1F. [DOI] [PubMed] [Google Scholar]
- 2.Chatzikokolis S, Troupis TG, Michalinos A, Bafaloukas N, Filippidis T, Gennimatas V. Retroperitoneal inflammatory myofibroblastic tumor. Am Surg. 2012;78:E190–E191. [PubMed] [Google Scholar]
- 3.Coffin CM, Watterson J, Priest JR, Dehner LP. Extrapulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor). A clinicopathologic and immunohistochemical study of 84 cases. Am J Surg Pathol. 1995;19:859–872. doi: 10.1097/00000478-199508000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Przkora R, Bolder U, Schwarz S, Jauch KW, Spes J, Andreesen R, Mackensen A. Regression of nonresectable inflammatory myofibroblastic tumours after treatment with nonsteroidal anti-inflammatory drugs. Eur J Clin Invest. 2004;34:320–321. doi: 10.1111/j.1365-2362.2004.01333.x. [DOI] [PubMed] [Google Scholar]
- 5.Attili SV, Chandra CR, Hemant DK, Bapsy PP, RamaRao C, Anupama G. Retroperitoneal inflammatory myofibroblastic tumor. World J Surg Oncol. 2005;3:66. doi: 10.1186/1477-7819-3-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coffin CM, Hornick JL, Fletcher CD. Inflammatory myofibroblastic tumor: Comparison of clinicopathologic, histologic and immunohistochemical features including ALK expression in atypical and aggressive cases. Am J Surg Pathol. 2007;31:509–520. doi: 10.1097/01.pas.0000213393.57322.c7. [DOI] [PubMed] [Google Scholar]
- 7.Koirala R, Shakya VC, Agrawal CS, Khaniya S, Pandey SR, Adhikary S, Pathania OP. Retroperitoneal inflammatory myofibroblastic tumor. Am J Surg. 2010;199:e17–e19. doi: 10.1016/j.amjsurg.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 8.Ziadi S, Trimeche M, Mestiri S, Boujelbene N, Mokni M, Sriha B, Korbi S. Retroperitoneal myofibroblastic inflammatory tumor. Tunis Med. 2011;89:400–401. [PubMed] [Google Scholar]
- 9.Tao YL, Wang ZJ, Han JG, Wei P. Inflammatory myofibroblastic tumor successfully treated with chemotherapy and nonsteroidals: A case report. World J Gastroenterol. 2012;18:7100–7103. doi: 10.3748/wjg.v18.i47.7100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mali VP, Tan HC, Loh D, Prabhakaran K. Inflammatory tumour of the retroperitoneum - a case report. Ann Acad Med Singapore. 2005;34:632–635. [PubMed] [Google Scholar]
- 11.Gómez-Román JJ, Sánchez-Velasco P, Ocejo-Vinyals G, Hernández-Nieto E, Leyva-Cobián F, Val-Bernal JF. Human herpesvirus-8 genes are expressed in pulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor) Am J Surg Pathol. 2001;25:624–629. doi: 10.1097/00000478-200105000-00009. [DOI] [PubMed] [Google Scholar]
- 12.Gleason BC, Hornick JL. Inflammatory myofibroblastic tumours: Where are we now? J Clin Pathol. 2008;61:428–437. doi: 10.1136/jcp.2007.049387. [DOI] [PubMed] [Google Scholar]
- 13.Aptel S, Gervaise A, Fairise A, Henrot P, Leroux A, Guillemin F, Laurent V, Régent D. Abdominal inflammatory myofibroblastic tumour. Diagn Interv Imaging. 2012;93:410–412. doi: 10.1016/j.diii.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 14.Butrynski JE, D'Adamo DR, Hornick JL, Dal Cin P, Antonescu CR, Jhanwar SC, Ladanyi M, Capelletti M, Rodig SJ, Ramaiya N, et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med. 2010;363:1727–1733. doi: 10.1056/NEJMoa1007056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chavez C, Hoffman MA. Complete remission of ALK-negative plasma cell granuloma (inflammatory myofibroblastic tumor) of the lung induced by celecoxib: A case report and review of the literature. Oncol Lett. 2013;5:1672–1676. doi: 10.3892/ol.2013.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]