

Abstract
The present study investigated whether atorvastatin antagonizes the visfatin-induced expression of inflammatory mediators in human coronary artery endothelial cells (HCAECs). Several analysis methods, such as reverse transcription-quantitative polymerase chain reaction, western blot analysis and H2DCFDA incubation, were used in the present study. The data showed that atorvastatin decreased the visfatin-induced expression of interleukin (IL)-6 and IL-8 in HCAECs. In addition, atorvastatin inhibited the visfatin-induced expression of intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in HCAECs. In addition, the present study found that atorvastatin inhibited the visfatin-activated nuclear factor-κB (NF-κB) signal pathway by preventing extracellular signal-regulated kinase phosphorylation in HCAECs. Atorvastatin significantly inhibited visfatin-induced NF-κB activity via the upregulation of reactive oxygen species production. Atorvastatin, a visfatin antagonist (FK866) and an NF-κB inhibitor (BAY11-7082) decreased the visfatin-induced expression of inflammatory mediators via the upregulation of NF-κB activation in HCAECs. These results suggest that atorvastatin may inhibit the visfatin-induced upregulation of inflammatory mediators through blocking the NF-κB signal pathway. The findings of the present study provide a potential use for atorvastatin and visfatin in the pathogenesis of HCAEC dysfunction. This knowledge may contribute to the development of novel therapies for atherosclerosis.
Keywords: human coronary artery endothelial cells, atorvastatin, visfatin, nuclear factor-κB
Introduction
Obesity is an independent risk factor for cardiovascular diseases (1). Previous studies have revealed that adipose tissue acts as an endocrine organ, generating a range of secreted factors including leptin, adiponectin, tumor necrosis factor-α (TNF-α), resistin, plasminogen activator inhibitor-1 and visfatin (2–5). These adipocytokines have been found to be expressed in atherosclerotic plaques, modulating the inflammatory processes of atherosclerosis, which implies that adipocytokines are vital for the development of atherosclerosis (6–9).
Atherosclerosis is an inflammatory process that starts with endothelial dysfunction, which is important in the initial pathogenesis of atherosclerotic lesion formation (10,11). Adipokines, including resistin and leptin, are proinflammatory mediators that directly contribute to endothelial dysfunction and atherogenesis (12–15).
Visfatin corresponds to a protein that was previously identified as pre-B cell colony enhancing factor, which is a 52-kDa cytokine expressed in lymphocytes. Visfatin is a newly found visceral-fat-specific adipokine that is predominantly produced in visceral adipose tissue (16,17). Visfatin can upregulate the levels of interleukin (IL)-1β, IL-6, IL-8 and TNF-α in cytokines (18–20). Therefore, visfatin may provide an additional association between adipose tissues and inflammation. Visfatin has previously been found to markedly increase the expression of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) through activating nuclear factor-κB (NF-κB), which directly contributes to endothelial dysfunction (12,13,21). Several studies have reported that the expression of visfatin is increased at plaque rupture sites in patients with coronary artery disease and that visfatin accelerates monocyte adhesion to endothelial cells by upregulating adhesion molecules in vascular endothelial cells (22). This finding suggests a positive association between visfatin foam cell expression, aortic inflammation and plaque destabilization. Emerging evidence has established that visfatin is important for the development of atherosclerosis (20,23,24). However, the molecular mechanism underlying the association of visfatin in the development of atherosclerosis remains unclear.
In addition, previous studies suggest that visfatin can dysregulate the vascular endothelial function in a dose-dependent manner. Visfatin can significantly increase the expression of IL-6, ICAM-1, VCAM-1 and other cytokines in endothelial dysfunction (21). Whether the levels of visfatin in adipose tissue surrounding of the coronary artery are associated with coronary endothelial dysfunction has not yet been studied (25). Visfatin located in the adipose tissue surrounding the coronary artery is presumed to initiate coronary artery endothelial inflammation via the activation of the NF-κB signal pathway, which then leads to coronary endothelial dysfunction and promotes the development and progression of atherosclerosis.
The present study demonstrates, for the first time, that atorvastatin can decrease the visfatin-induced expression of IL-6 and IL-8 in HCAECs. In addition, atorvastatin is found to inhibit the visfatin-induced expression of ICAM-1 and VCAM-1 in HCAECs. The present study also indicates that atorvastatin inhibits the visfatin-activated NF-κB signaling pathway by preventing the phosphorylation of extracellular signal-regulated kinase (ERK) in HCAECs. Atorvastatin is shown to significantly inhibit visfatin-induced NF-κB activity by decreasing the production of reactive oxygen species (ROS). Atorvastatin, FK866 and BAY11-7082 are shown to decrease the visfatin-induced expression of inflammatory mediators via the upregulation of NF-κB activation in HCAECs. These results suggest that atorvastatin may inhibit visfatin-induced upregulation of inflammatory mediators through blocking the NF-κB signal pathway. The findings of the present study provide a potential role of atorvastatin and visfatin in the pathogenesis of HCAEC dysfunction, and this knowledge may contribute to the development of novel therapies for atherosclerosis.
Materials and methods
Cell culture
HCAECs were purchased from ScienCell Research Laboratories (San Diego, CA, USA). Cells were grown in endothelial cell growth medium (ScienCell Research Laboratories). In these experiments, cells were seeded in 0.5 ml complete medium in 24-well plates. The control cells were the vehicle cells, which were created using HCAECs dissolved in dimethyl sulfoxide.
Drugs
Atorvastatin was purchased from the Shanghai Yijing Industrial Co., Ltd. (Shanghai, China), FK866 was purchased from Apexbio Technology LLC (Houston, TX, USA) and BAY11-7082 was purchased from Selleck Chemicals LLC (Houston, TX, USA).
Antibodies
The following antibodies were purchased from Abcam (Cambridge, MA, USA): Anti-VCAM1 rabbit polyclonal antibody against human (predicted: mouse, rat and rabbit; dilution, 1:500–1,000; catalog no., ab106777; localization, membrane); anti-ICAM1 rabbit polyclonal antibody against mouse/human (predicted: rat, chimpanzee and Chinese hamster; dilution, 1:500–1,000; catalog no., ab124759; localization, membrane); anti-IL6 rabbit polyclonal antibody against human/pig (dilution, 1:400–800; catalog no., ab6672; localization, secreted); anti-IL8 rabbit polyclonal against rat/human/cynomolgus monkey/rhesus monkey (dilution, 1:10; catalog no., ab7747; localization, secreted); anti-NF-κB p65 rabbit polyclonal antibody against mouse/rat/chicken/human/Indian muntjac (dilution, 1:400–800; catalog no., ab16502; localization, nucleus); and anti-ERK5 rabbit polyclonal antibody against mouse/rat/human (dilution, 1:500–1,000; catalog no., ab196609; localization, cytoplasm). The anti-phosphorylated-ERK (p-ERK) mouse monoclonal antibody against mouse/rat/human/canine/bovine (dilution, 1:200–500; catalog no., sc-7383; localization, cytoplasm/nucleus) was purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). The anti-β-actin rabbit polyclonal antibody against mouse/human (dilution, 1:100; catalog no., ab16039; localization, cytoplasm) was purchased from Abcam and used as the control. The secondary polyclonal goat anti-rabbit IgG-horseradish peroxidase-conjugated antibody (dilution, 1:1,000–10,000; catalog no., SE12) was purchased from Solarbio (Beijing, China).
RNA isolation and reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted using TRIzol Reagent (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA), and reverse transcription was performed using a PrimeScript RT reagent kit (Takara Bio, Inc., Otsu, Japan), according the manufacturer's instructions. For RT-qPCR analysis, aliquots of complementary DNA were amplified using SYBR Premix Ex Taq (Takara Bio, Inc.). PCR reactions were performed in triplicate with the following conditions: 95°C for 30 sec, then 40 cycles of 95°C for 5 sec, 60°C for 15 sec and 72°C for 10 sec, on an MXP3000 cycler (Stratagene; Agilent Technologies, Inc., Santa Clara, CA, USA) and repeated at least three times. The primers were as follows: VCAM1 forward, 5′-GGGAAGATGGTCGTGATCCTT-3′ and reverse, 5′-TCTGGGGTGGTCTCGATTTTA-3′; ICAM1 forward, 5′-ATGCCCAGACATCTGTGTCC-3′ and reverse, 5′-GGGGTCTCTATGCCCAACAA-3′. Relative messenger RNA (mRNA) levels were calculated using the ΔΔCq method, using β-actin as a control, and expressed as 2(−ΔΔCq) (26).
Measurement of VCAM-1 and ICAM-1 in HCAECs by enzyme-linked immunosorbent assay (ELISA)
The concentration of VCAM-1 and ICAM-1 in conditioned media was measured using Human ELISA Kits (Thermo Fisher Scientific, Inc.), according to the manufacturer's instructions.
Western blot analysis
Total protein of tissues and cells was obtained using radioimmunoprecipitation assay lysis buffer containing a mixture of proteinase inhibitors (Sigma-Aldrich, St. Louis, MO, USA). The protein concentration was determined using BCA protein assay reagent (Beyotime Institute of Biotechnology, Haimen, China). Equivalent amounts of proteins were separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and then transferred onto nitrocellulose membranes (Invitrogen; Thermo Fisher Scientific, Inc.). After being blocked in Tris-buffered saline containing 5% fat-free milk, the membranes were incubated with the aforementioned VCAM-1, ICAM-1, IL-6, IL-8, NF-κB p65, ERK or p-ERK primary antibodies at 4°C overnight, washed using phosphate-buffered saline (PBS)-Tween buffer [0.01 mol/l PBS (pH 7.2–7.4); 0.2 mol/l NaH2PO4 19 ml; 0.2 mol/l Na2HPO4 81 ml; NaCl 17 g; 2,000 ml ddH2O; 20% Tween 20] and then incubated with horseradish peroxidase-conjugated secondary antibody at room temperature for 2 h. A β-actin antibody was used as a loading control. Signals were detected on X-ray film using the Pierce ECL detection system (Thermo Fisher Scientific, Inc.).
Measurement of ROS production
ROS production was measured by using 2′,7′-diclorofluorescein diacetate (H2DCFDA; Invitrogen; Thermo Fisher Scientific, Inc,). In total, 4 HCAECs were incubated in the absence or presence of 50 ng/ml visfatin, with or without 50 nM FK866, 50 µM BAY11-0782 or 10 µM atorvastatin, for 24 h. The HCAECs were then incubated with H2DCFDA (10 µM) for 30 min at 37°C. Fluorescence images were obtained using a fluorescence microscope (BX-51; Olympus Corporation, Tokyo, Japan). The fluorescence intensity was measured using Image J software version 2.1.4.7 (imagej.nih.gov/ij/; National Institutes of Health, Bethesda, MD, USA); then the mean fluorescence was calculated and normalized to the control value.
Statistical analysis
Statistical analysis was performed with SPSS 13.0 for Windows (SPSS Inc., Chicago, IL, USA). The Pearson χ2 test or Fisher's exact test was used to compare qualitative variables, and the Student's t-test for quantitative variables. All statistical tests were two-sided, and a P-value of <0.05 was considered to indicate a statistically significant difference.
Results
Visfatin improves the production of cytokines in HCAECs, but BAY11-7082 and atorvastatin can antagonize this process
IL-6 and IL-8 have been previously reported to be involved in the pathogenesis of obesity, insulin resistance and atherosclerosis (27,28). Therefore, the present study aimed to examine whether visfatin, FK866, BAY11-7082 and atorvastatin could affect cytokine and adhesion molecule gene expression in HCAECs. Compared with the control, HCAECs incubated in the presence of visfatin caused a significant increase in IL-6 and IL-8 mRNA expression. However, the IL-6 and IL-8 mRNA levels of the visfatin group were reduced in the presence of FK866, BAY11-7082 and atorvastatin (Fig. 1A and B). An ELISA was used to measure the levels of IL-6 and IL-8 released from HCAECs. Compared with the control group, visfatin group showed significant increases in the release of IL-6 and IL-8. However, levels of IL-6 and IL-8 in the FK866, BAY11-7082 and atorvastatin groups were decreased compared with the visfatin group and increased compared with the control group (Fig. 1C and D). Overall, these results suggest that FK866, BAY11-7082 and atorvastatin decrease the visfatin-induced upregulation of IL-6 and IL-8 in HCAECs.
Figure 1.

Reverse transcription-quantitative polymerase chain reaction assays revealed the effect of atorvastatin on visfatin-induced inflammation. Human coronary artery endothelial cells were incubated in the absence or presence of 50 ng/ml visfatin, with or without 50 nM FK866, 50 µM BAY11-0782 or 10 µM atorvastatin, for 24 h. (A) Atorvastatin downregulated visfatin-induced IL-6 gene expression (P-values from left to right vs. control: 0.0081, 0.0045, 0.0233 and 0.0057). (B) Atorvastatin decreased visfatin-induced IL-8 gene expression (P-values from left to right vs. control: 0.0096, 0.0037, 0.0043 and 0.0031). (C) Atorvastatin downregulated visfatin-induced IL-6 release (P-values from left to right vs. control: 0.0078, 0.0022, 0.0783 and 0.0382). (D) Atorvastatin inhibited visfatin-induced IL-8 release (P-values from left to right vs. control: 0.0094, 0.0032, 0.0039 and 0.0035). Significant differences were determined using Student's t-test; *P<0.05, **P<0.01 vs. visfatin control. IL-6, interleukin-6; IL-8, interleukin-8.
Atorvastatin inhibits the visfatin-induced expression of adhesion molecules in HCAECs
The expression of adhesion molecules, including ICAM-1 and VCAM-1, on the endothelial cell surface is an initial step in atherogenesis (29). ICAM-1 and VCAM-1 can mediate leukocyte adhesion to the endothelium during inflammation. Therefore, the present study investigated whether visfatin could induce the expression of ICAM-1 and VCAM-1 in HCAECs. The data revealed that visfatin strongly increased ICAM-1 and VCAM-1 mRNA levels compared with the control group. However, the ICAM-1 and VCAM-1 mRNA levels in the visfatin group were decreased in the presence of FK866, BAY11-7082 and atorvastatin (Fig. 2A and B). Western blot analysis assays also indicated that the treatment of HCAECs with visfatin increased ICAM-1 and VCAM-1 protein levels compared with control the group. However, the ICAM-1 and VCAM-1 protein levels of the visfatin group were decreased in the presence of FK866, BAY11-7082 and atorvastatin (Fig. 2C and D). Overall, these results suggest that atorvastatin inhibited the visfatin-induced expression of ICAM-1 and VCAM-1 in HCAECs.
Figure 2.

Atorvastatin and signaling inhibitors decreased visfatin-induced VCAM-1 and ICAM-1 expression in HCAECs. HCAECs were incubated in the absence or presence of 50 ng/ml visfatin, with or without 50 nM FK866, 50 µM BAY11-0782 or 10 µM atorvastatin, for 24 h. (A) RT-qPCR assays revealed that atorvastatin downregulated visfatin-induced VCAM-1 gene expression (P-values from left to right vs. control: 0.0068, 0.0015, 0.0021 and 0.0017). (B) RT-qPCR assays revealed that atorvastatin decreased visfatin-induced ICAM-1 gene expression (P-values from left to right vs. control: 0.0073, 0.0011, 0.0026 and 0.0019). (C) Western blotting assays revealed that atorvastatin downregulated the visfatin-upregulated VCAM-1 protein level (P-values from left to right vs. control: 0.0089, 0.0016, 0.0023 and 0.0018). (D) Western blotting assays revealed that atorvastatin inhibited the visfatin-upregulated ICAM-1 protein level (P-values from left to right vs. control: 0.0097, 0.0014, 0.0020 and 0.0016). Total cell lysates were separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblots were analyzed with anti-VCAM-1, anti-ICAM-1 and anti-β-actin antibodies. β-actin was measured as an internal control. Significant differences were determined using Student's t-test, **P<0.01. VCAM-1, vascular adhesion molecule-1; ICAM-1, intercellular adhesion molecule-1; HCAECs, human coronary artery endothelial cells; RT-qPCR, reverse transcription-quantitative polymerase chain reaction.
Visfatin induces the expression of cytokines and adhesion molecules through the NF-κB signaling pathway, but atorvastatin can antagonize the process
Numerous cytokines, such as IL-1, IL-6, IL-8 and TNF-α, and cell adhesion molecules, such as CD4, CD8, CD22, CD28, CTLA-4, ICOS, ICAM-1, CD80 and CD86, can induce gene expression through the NF-κB signaling pathway (30). NF-κB is a potent proinflammatory nuclear transcription factor, and the activation of NF-κB is a major part of the initiation and amplification of inflammatory responses. NF-κB activity can be modulated via phosphorylation by mitogen-activated protein kinases (MAPKs), including ERK1/2 (31–33). In addition, visfatin has been shown to promote angiogenesis through the activation of the ERK1/2 in endothelial cells. Therefore, the present study investigated whether visfatin could activate NF-κB activity via the ERK signaling pathway in HCAECs.
Western blot analyses indicated that the treatment of HCAECs with visfatin increased p-ERK and p65 protein levels, but did not increase ERK protein levels, compared with the control group. However, the protein levels of p-ERK and p65 in the visfatin group were reduced in the presence of FK866, BAY11-7082 and atorvastatin (Fig. 3A). The present study then aimed to determine whether visfatin could enhance the nuclear accumulation of p65 protein. A western blot analysis of p65 demonstrated that there was no significant difference in the cytoplasm protein levels of p65 compared with control cells; however, the nuclear protein levels of p65 significantly increased with the treatment of HCAECs with visfatin. However, this effect was inhibited in the presence of FK866, BAY11-7082 and atorvastatin (Fig. 3B). Overall, the results suggest that visfatin upregulates the expression of p-ERK and p65 and promotes the translocation of p65 to the nucleus in HCAECs, but that atorvastatin could antagonize this effect.
Figure 3.

Effect of atorvastatin on the visfatin-induced NF-κB signaling pathway in HCAECs. (A) Western blotting assays revealed that visfatin increased p-ERK and p65 protein levels, but did not increase ERK protein levels compared with the control group. However, FK866, BAY11-7082 and atorvastatin inhibited visfatin-induced NF-κB activity (P-values from left to right vs. controls: 0.0082, 0.0055, 0.0047 and 0.0058; 0.0090, 0.0061, 0.0053 and 0.0037; 0.0632, 0.0689, 0.0620 and 0.0627). (B) Cell fractionation assays revealed that atorvastatin decreased the visfatin-increased nuclear p65 protein levels in HCAECs (P-values from left to right vs. control, left image: 0.0728, 0.0675, 0.0622 and 0.0530; right image: 0.0082, 0.0056, 0.0024 and 0.0037). Significant differences were determined using Student's t-test, *P<0.05, **P<0.01. NF-κB, nuclear factor-κB; HCAECs, human coronary artery endothelial cells; p-ERK, phosphorylated-extracellular signal-regulated kinase; ERK, extracellular signal-regulated kinase; CE, cytoplasmic extract; NE, nuclear extract.
Visfatin promotes NF-κB activation associated with ROS production, but atorvastatin can antagonize the process
Visfatin has been previously found to upregulate NF-κB activity, indicating that visfatin may induce the expression of pro-inflammatory and adhesion molecules (34). ROS are known to be produced by cytokines, growth factors and vasoactive agents, and contribute to the intracellular signaling cascades associated with inflammatory response (35). ROS can induce the activation of the NF-κB signaling pathway by modifying the activation of one or more of the kinase enzymes involved in NF-κB activation cascades (36). Therefore, the present study investigated whether visfatin could induce ROS generation in HCAECs. The H2DCFDA assay was used to assess the effect of visfatin on ROS generation. Visfatin significantly increased ROS generation in HCAECs; however the visfatin-induced ROS generation was decreased in the presence of FK866, BAY11-7082 and atorvastatin (Fig. 4). Overall, these results suggest that atorvastatin decreased visfatin-induced ROS generation in HCAECs.
Figure 4.
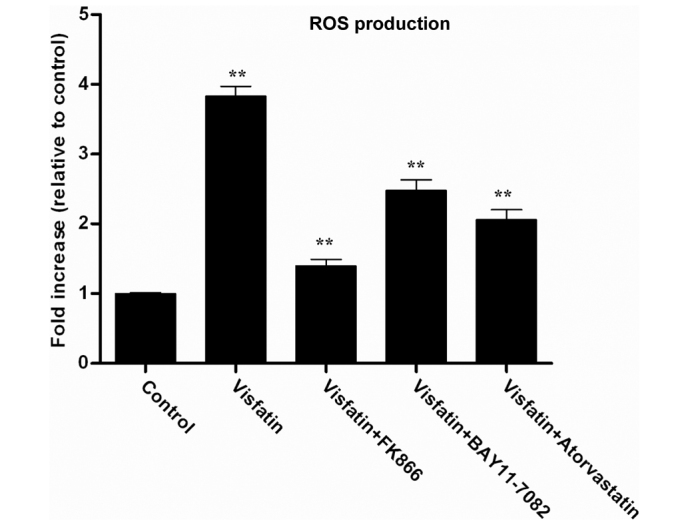
H2DCFDA incubation revealed the effect of atorvastatin on visfatin-induced ROS generation in HCAECs. ROS production in HCAECs was determined by fluorescence staining using H2DCFDA. HCAECs was incubated in the absence or presence of 50 ng/ml visfatin, with or without 50 nM FK866, 50 µM BAY11-0782 or 10 µM atorvastatin, for 24 h. Fluorescent intensity was measured using Image J software (version 2.1.4.7) and then averaged and shown as fold increase relative to the control (n=3 independent experiments). P-values from left to right vs. control: 0.0085, 0.0017, 0.0036 and 0.0024. Significant differences were determined using Student's t test, **P<0.01. ROS, reactive oxygen species; HCAECs, human coronary artery endothelial cells.
Discussion
Obesity is a significant risk factor for cardiovascular diseases, including atherosclerosis and hypertension (37,38). Atherosclerosis is an inflammatory process that begins with systemic endothelial dysfunction, a pivotal event in the early pathogenesis of atherosclerotic lesion formation (10,11). Adipokines regulate the endothelial expression of cell adhesion molecules and chemoattractant chemokines, one of the early important steps in atherosclerosis (14,15). Previous studies have reported that the expression of visfatin is increased at plaque rupture sites in patients with coronary artery disease, which suggests a possible role for visfatin in the development of atherosclerosis (21,25). The present study demonstrated that visfatin increased the production of IL-6 and IL-8 in HCAECs. The treatment of HCAECs with visfatin resulted in the upregulation of ICAM-1 and VCAM-1 expression. Using western blotting assays, the treatment of HCAECs with visfatin was found to result in increased p-ERK and p65 protein levels and decreased ERK protein levels compared with the control group. In addition, visfatin enhanced p65 protein nuclear accumulation in HCAECs. Finally, visfatin was shown to induce ICAM-1 and VCAM-1 expression through the NF-κB signal pathway and promote NF-κB activation, in association with ROS production. However, atorvastatin was indicated to antagonize the function of visfatin in HCAECs. Overall, these results suggest that atorvastatin may be vital for the prevention and therapy of atherosclerosis.
IL-6 and IL-8 have been reported to be involved in the pathogenesis of obesity, insulin resistance and atherosclerosis (27,28). In the present study, visfatin was found to induce the production of IL-6 and IL-8 in HCAECs. This finding is in agreement with previous studies, which showed that visfatin induced the production of the IL-1β, IL-6, IL-8 and TNF-α cytokines (18–20). However, for the first time, the present study found that atorvastatin could antagonize the visfatin-promoted production of IL-6 and IL-8. The expression of adhesion molecules such as VCAM-1 and ICAM-1 on the endothelial cell surface is the initial step in atherogenesis (29). A previous study reported that the upregulation of ICAM-1 and VCAM-1 has been found in human atherosclerotic plaques, and was associated with upregulated leukocyte recruitment and accumulation (21). The present study demonstrated that visfatin increased ICAM-1 and VCAM-1 mRNA and protein levels in HCAECs, but that atorvastatin could antagonize this visfatin effect. These results suggest that atorvastatin can use anti-inflammatory actions to attenuate the dysfunction of HCAECs and atherosclerosis.
NF-κB can induce the expression of numerous cytokines and cell adhesion molecules (32). NF-κB activity can be modulated through phosphorylation by MAPKs, including ERK1/2, JNK and p38 (30). The present study demonstrated that visfatin upregulated p-ERK and p65 protein levels, suggesting that visfatin could activate p65 via activation of the MAPK/ERK signaling pathway. Visfatin also promoted the translocation of p65 to the nucleus in HCAECs, but atorvastatin could antagonize this effect. In the present study, visfatin upregulated NF-κB activity, which indicates that visfatin induced the expression of inflammatory and adhesion molecules, possibly through the NF-κB signaling pathway. ROS were produced by cytokines, growth factors and vasoactive agents, which contribute to the intracellular signaling cascades associated with inflammatory response (39). ROS induced NF-κB activation by modifying the activation of one or more of the kinase enzymes in the NF-κB activation cascades (40,41). In the present study, visfatin significantly increased ROS generation in HCAECs, but the visfatin-induced ROS generation was reduced in the presence of FK866, BAY11-7082 and atorvastatin. These results suggest that the inhibition of the visfatin-induced expression of cytokines and cell adhesion molecules by atorvastatin may be mediated by the downregulation of NF-κB activation due to decreased ROS production.
In conclusion, the present study indicates that visfatin causes in the dysfunction of HCAECs by upregulating the expression of IL-6, IL-8, ICAM-1 and VCAM-1, probably through activation of the NF-κB pathway. The activation of the NF-κB pathway may be mainly due to visfatin-induced p-ERK expression and ROS generation. However, atorvastatin can antagonize the visfatin-promoted development of atherosclerosis, suggesting that atorvastatin may be vital for the prevention and therapy of atherosclerosis.
References
- 1.Rashid MN, Fuentes F, Touchon RC, Wehner PS. Obesity and the risk for cardiovascular disease. Prev Cardiol. 2003;6:42–47. doi: 10.1111/j.1520-037X.2003.01358.x. [DOI] [PubMed] [Google Scholar]
- 2.Ajuwon KM, Spurlock ME. Adiponectin inhibits LPS-induced NF-kappaB activation and IL-6 production and increases PPARgamma2 expression in adipocytes. Am J Physiol Regul Integr Comp Physiol. 2005;288:R1220–R1225. doi: 10.1152/ajpregu.00397.2004. [DOI] [PubMed] [Google Scholar]
- 3.Anfossi G, Russo I, Doronzo G, Pomero A, Trovati M. Adipocytokines in atherothrombosis: Focus on platelets and vascular smooth muscle cells. Mediators Inflamm. 2010;2010:174341. doi: 10.1155/2010/174341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kawanami D, Maemura K, Takeda N, Harada T, Nojiri T, Imai Y, Manabe I, Utsunomiya K, Nagai R. Direct reciprocal effects of resistin and adiponectin on vascular endothelial cells: A new insight into adipocytokine-endothelial cell interactions. Biochem Biophys Res Commun. 2004;314:415–419. doi: 10.1016/j.bbrc.2003.12.104. [DOI] [PubMed] [Google Scholar]
- 5.Lau DC, Dhillon B, Yan H, Szmitko PE, Verma S. Adipokines: Molecular links between obesity and atheroslcerosis. Am J Physiol Heart Circ Physiol. 2005;288:H2031–H2041. doi: 10.1152/ajpheart.01058.2004. [DOI] [PubMed] [Google Scholar]
- 6.Berg AH, Scherer PE. Adipose tissue, inflammation and cardiovascular disease. Circ Res. 2005;96:939–949. doi: 10.1161/01.RES.0000163635.62927.34. [DOI] [PubMed] [Google Scholar]
- 7.Guerre-Millo M. Adipose tissue and adipokines: For better or worse. Diabetes Metab. 2004;30:13–19. doi: 10.1016/S1262-3636(07)70084-8. [DOI] [PubMed] [Google Scholar]
- 8.Sethi JK, Vidal-Puig A. Visfatin: The missing link between intra-abdominal obesity and diabetes? Trends Mol Med. 2005;11:344–347. doi: 10.1016/j.molmed.2005.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu ZH, Zhao SP. Adipocyte: A potential target for the treatment of atherosclerosis. Med Hypotheses. 2006;67:82–86. doi: 10.1016/j.mehy.2005.12.042. [DOI] [PubMed] [Google Scholar]
- 10.Blankenberg S, Barbaux S, Tiret L. Adhesion molecules and atherosclerosis. Atherosclerosis. 2003;170:191–203. doi: 10.1016/S0021-9150(03)00097-2. [DOI] [PubMed] [Google Scholar]
- 11.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 12.Kougias P, Chai H, Lin PH, Yao Q, Lumsden AB, Chen C. Effects of adipocyte-derived cytokines on endothelial functions: Implication of vascular disease. J Surg Res. 2005;126:121–129. doi: 10.1016/j.jss.2004.12.023. [DOI] [PubMed] [Google Scholar]
- 13.Kralisch S, Sommer G, Stangl V, Köhler U, Kratzsch J, Stepan H, Faber R, Schubert A, Lössner U, Vietzke A, et al. Secretory products from human adipocytes impair endothelial function via nuclear factor kappaB. Atherosclerosis. 2008;196:523–531. doi: 10.1016/j.atherosclerosis.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 14.Cooke JP, Oka RK. Does leptin cause vascular disease? Circulation. 2002;106:1904–1905. doi: 10.1161/01.CIR.0000036864.14101.1B. [DOI] [PubMed] [Google Scholar]
- 15.Kougias P, Chai H, Lin PH, Lumsden AB, Yao Q, Chen C. Adipocyte-derived cytokine resistin causes endothelial dysfunction of porcine coronary arteries. J Vasc Surg. 2005;41:691–698. doi: 10.1016/j.jvs.2004.12.046. [DOI] [PubMed] [Google Scholar]
- 16.Samal B, Sun Y, Stearns G, Xie C, Suggs S, McNiece I. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol Cell Biol. 1994;14:1431–1437. doi: 10.1128/MCB.14.2.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sommer G, Garten A, Petzold S, Beck-Sickinger AG, Blüher M, Stumvoll M, Fasshauer M. Visfatin/PBEF/Nampt: Structure, regulation and potential function of a novel adipokine. Clin Sci (Lond) 2008;115:13–23. doi: 10.1042/CS20070226. [DOI] [PubMed] [Google Scholar]
- 18.Moschen AR, Kaser A, Enrich B, Mosheimer B, Theurl M, Niederegger H, Tilg H. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J Immunol. 2007;178:1748–1758. doi: 10.4049/jimmunol.178.3.1748. [DOI] [PubMed] [Google Scholar]
- 19.Lee WJ, Wu CS, Lin H, Lee IT, Wu CM, Tseng JJ, Chou MM, Sheu WH. Visfatin-induced expression of inflammatory mediators in human endothelial cells through the NF-kappaB pathway. Int J Obes (Lond) 2009;33:465–472. doi: 10.1038/ijo.2009.24. [DOI] [PubMed] [Google Scholar]
- 20.Guzik TJ, Mangalat D, Korbut R. Adipocytokines-novel link between inflammation and vascular function? J Physiol Pharmacol. 2006;57:505–528. [PubMed] [Google Scholar]
- 21.Kim SR, Bae YH, Bae SK, Choi KS, Yoon KH, Koo TH, Jang HO, Yun I, Kim KW, Kwon YG, et al. Visfatin enhances ICAM-1 and VCAM-1 expression through ROS-dependent NF-kappaB activation in endothelial cells. Biochim Biophys Acta. 2008;1783:886–895. doi: 10.1016/j.bbamcr.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 22.Buldak RJ, Gowarzewski M, Buldak L, Skonieczna M, Kukla M, Polaniak R, Zwirska-Korczala K. Viability and oxidative response of human colorectal HCT-116 cancer cells treated with visfatin/eNampt in vitro. J Physiol Pharmacol. 2016;66:557–566. [PubMed] [Google Scholar]
- 23.Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 24.Kim SR, Bae SK, Choi KS, Park SY, Jun HO, Lee JY, Jang HO, Yun I, Yoon KH, Kim YJ, et al. Visfatin promotes angiogenesis by activation of extracellular signal-regulated kinase 1/2. Biochem Biophys Res Commun. 2007;357:150–156. doi: 10.1016/j.bbrc.2007.03.105. [DOI] [PubMed] [Google Scholar]
- 25.Dahl TB, Yndestad A, Skjelland M, Øie E, Dahl A, Michelsen A, Damås JK, Tunheim SH, Ueland T, Smith C, et al. Increased expression of visfatin in macrophages of human unstable carotid and coronary atherosclerosis: Possible role in inflammation and plaque destabilization. Circulation. 2007;115:972–980. doi: 10.1161/CIRCULATIONAHA.106.665893. [DOI] [PubMed] [Google Scholar]
- 26.Kotsopoulos J, Zhang S, Akbari M, Salmena L, Llacuachaqui M, Zeligs M, Sun P, Narod SA. BRCA1 mRNA levels following a 4–6-week intervention with oral 3,3′-diindolylmethane. Br J Cancer. 2014;111:1269–1274. doi: 10.1038/bjc.2014.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simonini A, Moscucci M, Muller DW, Bates ER, Pagani FD, Burdick MD, Strieter RM. IL-8 is an angiogenic factor in human coronary atherectomy tissue. Circulation. 2000;101:1519–1526. doi: 10.1161/01.CIR.101.13.1519. [DOI] [PubMed] [Google Scholar]
- 28.Yudkin JS, Kumari M, Humphries SE, Mohamed-Ali V. Inflammation, obesity, stress and coronary heart disease: Is interleukin-6 the link? Atherosclerosis. 2000;148:209–214. doi: 10.1016/S0021-9150(99)00463-3. [DOI] [PubMed] [Google Scholar]
- 29.Takahashi M, Ikeda U, Masuyama J, Kitagawa S, Kasahara T, Shimpo M, Kano S, Shimada K. Monocyte-endothelial cell interaction induces expression of adhesion molecules on human umbilical cord endothelial cells. Cardiovasc Res. 1996;32:422–429. doi: 10.1016/0008-6363(96)00085-5. [DOI] [PubMed] [Google Scholar]
- 30.Han SG, Newsome B, Hennig B. Titanium dioxide nanoparticles increase inflammatory responses in vascular endothelial cells. Toxicology. 2013;306:1–8. doi: 10.1016/j.tox.2013.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adya R, Tan BK, Chen J, Randeva HS. Nuclear factor-kappaB induction by visfatin in human vascular endothelial cells: Its role in MMP-2/9 production and activation. Diabetes Care. 2008;31:758–760. doi: 10.2337/dc07-1544. [DOI] [PubMed] [Google Scholar]
- 32.Adya R, Tan BK, Punn A, Chen J, Randeva HS. Visfatin induces human endothelial VEGF and MMP-2/9 production via MAPK and PI3K/Akt signalling pathways: Novel insights into visfatin-induced angiogenesis. Cardiovasc Res. 2008;78:356–365. doi: 10.1093/cvr/cvm111. [DOI] [PubMed] [Google Scholar]
- 33.Collins T, Read MA, Neish AS, Whitley MZ, Thanos D, Maniatis T. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappaB and cytokine-inducible enhancers. FASEB J. 1995;9:899–909. [PubMed] [Google Scholar]
- 34.Said RS, El-Demerdash E, Nada AS, Kamal MM. Resveratrol inhibits inflammatory signaling implicated in ionizing radiation-induced premature ovarian failure through antagonistic crosstalk between silencing information regulator 1 (SIRT1) and poly(ADP-ribose) polymerase 1 (PARP-1) Biochem Pharmacol. 2016;103:140–150. doi: 10.1016/j.bcp.2016.01.019. [DOI] [PubMed] [Google Scholar]
- 35.Görlach A, Bertram K, Hudecova S, Krizanova O. Calcium and ROS: A mutual interplay. Redox Biol. 2015;6:260–271. doi: 10.1016/j.redox.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chaudhari N, Talwar P, Parimisetty A, d'Hellencourt Lefebvre C, Ravanan P. A molecular web: endoplasmic reticulum stress, inflammation, and oxidative stress. Front Cell Neurosci. 2014;8:213. doi: 10.3389/fncel.2014.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shirai K. Obesity as the core of the metabolic syndrome and the management of coronary heart disease. Curr Med Res Opin. 2004;20:295–304. doi: 10.1185/030079903125003008. [DOI] [PubMed] [Google Scholar]
- 38.Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444:875–880. doi: 10.1038/nature05487. [DOI] [PubMed] [Google Scholar]
- 39.Kim YR, Koh HJ, Kim JS, Yun JS, Jang K, Lee JY, Jung JU, Yang CS. Peptide inhibition of p22phox and Rubicon interaction as a therapeutic strategy for septic shock. Biomaterials. 2016;101:47–59. doi: 10.1016/j.biomaterials.2016.05.046. [DOI] [PubMed] [Google Scholar]
- 40.Ruiz-Ojeda FJ, Gomez-Llorente C, Aguilera CM, Gil A, Rupérez AI. Impact of 3-amino-1,2,4-triazole (3-AT)-derived increase in hydrogen peroxide levels on inflammation and metabolism in human differentiated adipocytes. PLoS One. 2016;11:e0152550. doi: 10.1371/journal.pone.0152550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Satoh R, Kakugawa K, Yasuda T, Yoshida H, Sibilia M, Katsura Y, Levi B, Abramson J, Koseki Y, Koseki H, et al. PLoS Genet. 2016;12:e1005776. doi: 10.1371/journal.pgen.1005776. [DOI] [PMC free article] [PubMed] [Google Scholar]