

Abstract
Human hepatocellular carcinoma (HCC) is one of the most common types of cancer and has a very poor prognosis; thus, the development of effective therapies for the treatment of advanced HCC is of high clinical priority. In the present study, the anti-oncogenic effect of combined knockdown of c-Myc expression and ectopic restoration of deleted in liver cancer 1 (DLC1) expression was investigated in human liver cancer cells. Expression of c-Myc in human HCC cells was knocked down by stable transfection with a Myc-specific short hairpin (sh) RNA vector. DLC1 expression in Huh7 cells was restored by adenovirus transduction, and the effects of DLC1 expression and c-Myc knockdown on Ras homolog gene family, member A (RhoA) levels, cell proliferation, soft agar colony formation and cell invasion were measured. Downregulation of c-Myc or re-expression of DLC1 led to a marked reduction in RhoA levels, which was associated with decreases in cell proliferation, soft agar colony formation and invasiveness; this inhibitory effect was augmented with a combination of DLC1 transduction and c-Myc suppression. To determine whether liver cell-specific delivery of DLC1 was able to enhance the inhibitory effect of c-Myc knockdown on tumor growth in vivo, DLC1 vector DNA complexed with galactosylated polyethylene glycol-linear polyethyleneimine was administered by tail vein injection to mice bearing subcutaneous xenografts of Huh7 cells transfected with shMyc or control shRNA. A cooperative inhibitory effect of DLC1 expression and c-Myc knockdown on the growth of Huh7-derived tumors was observed, suggesting that targeted liver cell delivery of DLC1 and c-Myc shRNA may serve as a possible gene therapy modality for the treatment of human HCC.
Keywords: deleted in liver cancer 1, c-Myc, hepatocellular carcinoma, Ras homolog gene family guanosine triphosphatase, tumorigenesis
Introduction
Human hepatocellular carcinoma (HCC) is one of the most common and lethal types of cancer, and its incidence is increasing worldwide (1,2). As numerous cases of HCC are diagnosed at later stages when the prognosis is poor, the development of effective therapy for advanced and metastatic HCC is a high clinical priority (1,2). HCC is a heterogeneous disorder with a complex pattern of genetic alterations, and increasing our knowledge of the specific genes and signaling pathways involved in the pathogenesis of HCC is critical for identifying novel clinical tools for diagnosis, classification and targeted therapy of the disease (3,4).
Despite the heterogeneity of genomic alterations in HCC, certain chromosomes or chromosomal sites are more frequently deleted or amplified, resulting in deregulation of crucial genes that may ultimately trigger malignant transformation of healthy hepatocytes (5). Chromosome 8 exhibits a highly recurrent pattern of DNA copy number loss on the short arm (8p21-p22) and gain on the long arm (8q22-q24) (5,6), and these genetic alterations are associated with HCC subclasses with a less favorable outcome (7). The 8p and 8q sites correspond with the loci of the tumor suppressor gene deleted in liver cancer 1 (DLC1) and the c-Myc proto-oncogene, respectively (8,9).
The DLC1 gene encodes a RhoGTPase activating protein (RhoGAP) that is downregulated in a number of types of human cancer and inhibits the growth and metastasis of tumor cells (10,11). The c-Myc oncoprotein is a helix-loop-helix transcription factor that regulates genes involved in promoting tumorigenesis, cell proliferation and stem cell maintenance (12). Evidence accumulated in recent years clearly implicates the DLC1 and Myc genes in the pathogenesis of HCC in humans and in mouse models (13–17). The opposing effects of DLC1 and Myc in the initiation and progression of HCC suggest that therapeutics simultaneously targeting signaling pathways governing the functions of these genes may be beneficial in the treatment of HCC (18).
Despite existing evidence implicating DLC1 and Myc alterations in HCC, the combined role of the DLC1 and Myc proteins in the initiation and progression of HCC has not been addressed to the best of our knowledge. Based on these considerations, the present study investigated the effects of DLC1 transduction and c-Myc knockdown in Huh7 cells, a metastatic HCC cell line with low DLC1 expression. It was revealed that this combined approach resulted in a robust cooperative inhibition of tumor cell growth in vitro, and evidence was also provided that liver cell targeting of DLC1 expression may cooperate with c-Myc knockdown to suppress tumor growth in vivo.
Materials and methods
Cell culture and transfection
The human SK-Hep1 HCC cell line and the prostate cancer 22Rv1 cell line were obtained from the American Type Culture Collection (Manassas, VA, USA), and the Huh7 HCC line (originally from the Japanese Collection of Research Bioresources Cell Bank) was donated by Dr Curtis Harris (National Cancer Institute, NIH, Bethseda, MD, USA). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen™, Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 10% (v/v) fetal calf serum (HyClone™; Thermo Fisher Scientific, Inc., Logan, UT, USA) at 37°C in a humidified 5% CO2 atmosphere. Transient transfections were performed with Lipofectamine® 2000 (Invitrogen™; Thermo Fisher Scientific, Inc.) according to the manufacturer's instructions. Cells stably expressing short hairpin (sh)RNA specific for human c-Myc (catalogue no., sc-29226-SH) or control shRNA (catalog no., sc-108060) were generated by transfection with plasmid vectors obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA) and selected with 2 µg/ml puromycin.
DLC1 expression vectors
Adenovirus particles expressing the wild-type human DLC1 open reading frame (NM_006094) or LacZ were prepared as previously described (19) and used for transduction of cultured cells. For analysis of tumor formation in vivo, DLC1 was expressed using the plasmid vector pLXSN-DLC (20), which was prepared by cloning the DLC1 open reading frame into pLXSN (catalog no, 631509; Clontech Laboratories, Inc., Mountainview, CA, USA).
Ras homolog gene family, member A (RhoA) activity analysis
Active RhoA levels were measured using an enzyme-linked immunosorbent assay (ELISA)-based G-LISA assay (Cytoskeleton, Inc., Denver, CO, USA), which employs the isolated RhoGTPase binding domain of rhotekin to trap active GTP-bound RhoA, according to the manufacturer's protocol. Briefly, following 24 h of serum-starvation, cells were treated with calpeptin (100 µg/ml for 10 min; Cytoskeleton, Inc.). Cells were collected and lysed using the G-LISA kit cell lysis buffer, and 20 µg of cell lysates from each sample were incubated in 96-well plates coated with the isolated rhotekin Rho-binding domain. The level of RhoA activation was detected using indirect immunodetection followed by a colorimetric reaction measured with absorbance at 490 nm using a microplate reader (Bio-Tek Instruments, Inc., Winooski, VT, United States), according to the manufacturer's protocol.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell proliferation assay
Cells were seeded into 96-well plates and infected with adenovirus expressing DLC1 (Ad-DLC1) or control virus expressing LacZ (Ad-LacZ), at multiplicity of infection 25. The MTT (Roche Diagnostics, Indianapolis, IN, USA) assay was used to test cell proliferation as previously described (19).
Matrigel invasion assay
The invasion assay was performed with a modified Boyden chamber to evaluate the ability of tumor cells to migrate through Matrigel. Briefly, 5×104 cells were seeded onto BD BioCoat Matrigel-coated invasion chamber inserts (BD Biosciences, San Diego, CA). The medium in the upper compartment was serum-free DMEM, and DMEM with 10% fetal calf serum was used as a chemoattractant in the lower compartment. Following 24 h of culture, cells on the upper surface of the membrane were removed by wiping with a cotton-tipped swab, and migrated cells on the lower surface of the membrane were fixed with 4% paraformaldehyde and stained using crystal violet. Images of 5 randomly selected fields of the stained cells in the central region of the membrane under a light microscope were captured and cells were counted at magnification, ×40.
Western blotting
Cells were lysed with M-PER Mammalian Protein Extraction Reagent (Pierce; Thermo Fisher Scientific, Inc.) supplemented with proteinase inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA). Cell lysate proteins, 30 µg per lane, were resolved on a 4–12% gradient SDS-PAGE gel (Invitrogen; Thermo Fisher Scientific, Inc.) and transferred to nitrocellulose membranes (Invitrogen; Thermo Fisher Scientific, Inc.). The membrane was blocked with 5% non-fat milk (Santa Cruz Biotechnology, Inc.) and probed with mouse monoclonal anti-DLC1 (1:500; catalog no., 612020; BD Biosciences, Franklin Lakes, NJ, USA), mouse monoclonal anti-c-Myc (1:500; catalog no., sc-40; Santa Cruz Biotechnology, Inc.), rabbit polyclonal anti-RhoA (1:400; catalog no., sc-179; Santa Cruz Biotechnology, Inc.) and mouse monoclonal anti-actin (1:1,000; catalog no., sc-8432; Santa Cruz Biotechnology, Inc.) primary antibodies, followed by incubation with bovine anti-mouse IgG-horseradish peroxidase (HRP) (1:2,000; catalog no., sc-2380) or bovine anti-rabbit IgG-HRP (1:2,000; catalog no., sc-2379) (Santa Cruz Biotechnology, Inc.) secondary antibodies. Bound antibodies were visualized on X-ray film with the SuperSignal West Pico Chemiluminescent substrate (Pierce; Thermo Fisher Scientific, Inc.).
Soft agar colony formation assay
Colony formation in soft agar was performed to evaluate anchorage-independent growth as described previously (21). Following three weeks of growth, the average number of colonies was calculated by counting in six randomly selected fields under a light microscope at magnification, ×40.
Gal-PEG3500-LPEI mediated gene expression
Galactosylated polyethylene glycol-linear polyethyleneimine (Gal-PEG3500-LPEI; molecular weight, 144,000; Gal-PEG substitution, 6%) was synthesized by Combinix, Inc. (Mountain View, CA, USA) and prepared as 10 or 5 µg/µl stock solution in distilled water. The ratio of polymer amine to DNA phosphate (N/P) was calculated based on subunits of 43 and 330 g/mol for PEI and DNA. Cells were seeded into 96-well plates at a density of 5×103 cells/well and grown overnight at 37°C prior to Gal-PEG3500-LPEI mediated transfections. Briefly, 500 ng of luciferase reporter plasmid DNA (pPK-CMV-F4; Promokine; PromoCell GmbH, Heidelberg, Germany) was mixed with 3.3 mg of Gal-PEG3500-LPEI in 100 µl transfection buffer [5% (w/v) D(+)-glucose, 25 mM HEPES, 150 mM NaCl] according to the N/P ratio calculated. Following 20 min of incubation at room temperature, transfection mixtures were applied to cells. Luciferase activity was measured 48 h later using the Luciferase Assay System (catalog no., E1500; Promega Corporation, Madison, WI, USA), according to the manufacturer's protocol.
In vivo tumorigenicity assay
The animal studies were performed according to protocols approved by the Animal Care and Use Committee of the National Institutes of Health. The mice were maintained on a 12 hr light/dark cycle at 24°C with 20–70% humidity, receiving food and bottled water ad libitum. To establish subcutaneous tumors, twenty 6-week-old male BALB/c nude mice (NCI at Frederick, Frederick, MD, USA) were randomly divided into 4 groups and injected with 5×106 exponentially growing Huh7 or Huh7-shMyc cells. A total of 50 µg of pLXSN-DLC1 plasmid DNA (20) or of pLXSN vector DNA were mixed with Gal-PEG3500-LPEI as mentioned previously and administrated weekly by tail veil injection for 3 weeks. A digital caliper was used to measure tumor size and average tumor volume was calculated using the following standard formula: Tumor volume = (width2 × length) × 0.5.
Statistical analysis
Data are presented as the mean ± standard deviation from triplicate experiments. Statistical significance was analyzed by Student's t-test using Microsoft Excel 2010 (Microsoft Corporation, Redmond, WA, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
DLC1 transduction and c-Myc knockdown inhibit tumor cell proliferation
The present study generated SK-Hep1 (DLC1 positive) and Huh7 (low DLC1) cells stably transfected with a plasmid expressing c-Myc shRNA, and it was validated that expression of c-Myc protein was decreased in these cell lines compared to cells transfected with control shRNA (Fig. 1A). A soft agar assay revealed that downregulation of c-Myc resulted in a marked reduction in the colony formation of SK-Hep1 cells (P=0.012; Fig. 1B). To examine the effect of combined DLC1 transduction and c-Myc knockdown on growth inhibition, the present study employed an MTT proliferation assay and soft agar assay using Huh7 cells. The results revealed that restoration of DLC1 expression and downregulation of Myc resulted in ~60 and ~25% reductions in cell growth, respectively, compared to parental Huh7 cells following a five-day incubation (Fig. 1C). The inhibitory effect was augmented to nearly 75% in cells with combined DLC1 transduction and Myc knockdown, suggesting a cooperative effect on inhibition of tumor cell growth. DLC1 expression and Myc knockdown also had a cooperative effect on the inhibition of soft agar colony formation (Fig. 1D).
Figure 1.

DLC1 transduction and Myc knockdown synergistically inhibit hepatocellular carcinoma cell growth. (A) Immunoblots of cell lysates probed with anti-DLC1, anti-Myc and anti-actin antibodies. Left panel, SK-Hep-1 cells stably expressing shRNA (shMyc) or control shRNA (shCtrl); right panel, Huh7 cells stably expressing shMyc or control shRNA and infected with adenovirus expressing DLC1 or LacZ. (B) Quantification of soft agar colony formation of shCtrl or shMyc SK-Hep1 cells (left panel; P=0.012) and images of colonies from one of three independent experiments (right panel). (C) MTT cell proliferation assay of shCtrl or shMyc Huh7 cells following transduction with DLC1 or LacZ adenovirus; downregulation of Myc and/or restoration of DLC1 inhibited Huh7 cell growth. (D) Quantification of soft agar colony formation by Huh7 cells treated as in (A). Left panel, shCtrl/LacZ vs. shMyc/DLC1, P=0.00003; shMyc/DLC1 vs. shMyc/LacZ, P=0.0025; shMyc/DLC1 vs shCtrl/DLC1, P=0.006. Right panel, representative images of the soft agar colonies. Scale bar=50 µm. **P<0.05;***P<0.01. DLC1, deleted in liver cancer 1; shRNA, short hairpin RNA; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; OD, optical density.
DLC1 transduction and c-Myc knockdown attenuate invasion
Subsequently, using a Boyden chamber assay, the effect of combined DLC1 transduction and Myc knockdown on Huh7 tumor cell invasion was tested. As shown in Fig. 2, restoration of DLC1 expression and downregulation of c-Myc resulted in a significant decrease in invasion (P=0.00002) compared to control cells, and the combination of the two treatments demonstrated a further reduction in cells invading through the membrane (P=0.003 vs. DLC1, and P=0.0001 vs. downregulated c-Myc). These observations indicate that c-Myc knockdown cooperates with DLC1 and enhances its oncosuppressive activity.
Figure 2.

DLC1 transduction and Myc knockdown inhibit cell invasiveness. (A) Representative images from one of three independent experiments; cells invading through Matrigel are stained. (B) Quantification of Matrigel invasion of shMyc- or shCtrl-Huh7 cells transduced with DLC1 or LacZ adenovirus. **P<0.01; ***P<0.001. DLC1, deleted in liver cancer 1; sh, short hairpin.
Effect of DLC1 restoration and Myc knockdown on RhoA activation
As DLC1 is a negative regulator of RhoA, and c-Myc also affects RhoA expression, the present study examined whether attenuated RhoA activation contributes to the synergistic inhibitory effect of combined DLC1 transduction and c-Myc knockdown on cell growth and invasion. As shown in Fig. 3A, knockdown of c-Myc in Huh7 also led to downregulation of RhoA protein levels, which is consistent with a previous report that c-Myc is able to control RhoA expression (22). By using an ELISA-based G-LISA assay, it was observed that DLC1 and c-Myc knockdown efficiently inhibited RhoA activation. Notably, DLC1 demonstrated a slightly stronger inhibitory effect on RhoA activation with downregulated Myc (Fig. 3B), suggesting that reduced levels of active RhoA may contribute to the cooperative effect of DLC1 transduction and downregulation of c-Myc on the inhibition of tumor cell growth and invasiveness.
Figure 3.
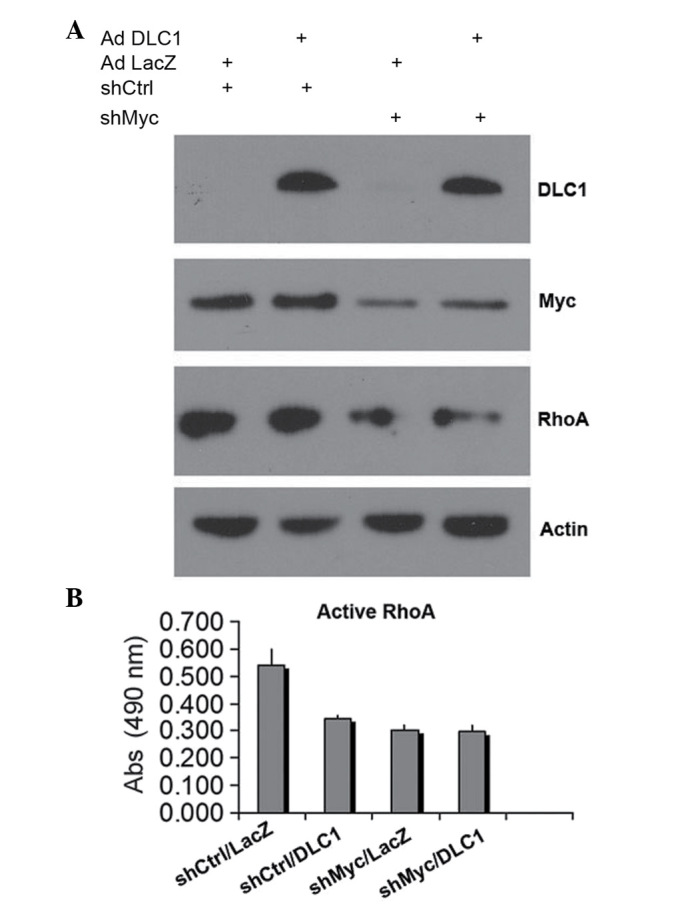
DLC1 transduction and Myc knockdown decrease RhoA activity. (A) Immunoblot analysis of RhoA protein levels in extracts from shMyc or shCtrl Huh7 cells transduced with DLC1 adenovirus (Ad DLC1) or LacZ adenovirus (Ad LacZ). (B) Colorimetric assay of active, guanosine-5′-triphosphate-bound RhoA in shMyc- or shCtrl-transfected Huh7 cells transduced with DLC1 or LacZ adenovirus. Abs, absorbance at 490 nm; DLC1, deleted in liver cancer 1; RhoA, Ras homolog gene family, member A; sh, short hairpin.
DLC1 restoration and c-Myc knockdown inhibit tumor growth in vivo
To test the in vivo effect of combined knockdown of c-Myc expression and ectopic restoration of DLC1 expression on tumor growth, the present study used Gal-PEG3500-LPEI, a DNA carrier system for liver cell-specific targeting via uptake by the hepatic asialoglycoprotein (ASGP) receptor (23), to deliver DLC1 plasmid DNA to HCC xenografts in nude mice. Initially, in vitro transient transfection assays were performed with luciferase reporter plasmid DNA to test the specificity of the transfection reagent. As shown in Fig. 4A, Gal-PEG3500-LPEI mediated strong luciferase activity in Huh7 liver cancer cells but not in 22Rv1 prostate cancer cells, consistent with previous reports that Huh7 cells express the ASGP receptor (24). To test the effectiveness of the transfection reagent in vivo, Huh7-shMyc and control Huh7 cells were implanted into nude mice, and Gal-PEG3500-LPEI mixed with pLXSN-DLC1 or pLXSN vector DNA was administered via tail vein injection weekly, for a total of three weeks. Knockdown of either c-Myc or restoration of DLC1 had an inhibitory effect on Huh7 cell growth in vivo, as the size of the tumors was reduced by 50% at the end point of the experiment (Fig. 4B). Coordinated ectopic expression of DLC1 and knockdown of c-Myc demonstrated a stronger inhibitory effect as compared to either DLC1 or c-Myc knockdown alone (Fig. 4B), which is consistent with the in vitro results of the present study.
Figure 4.
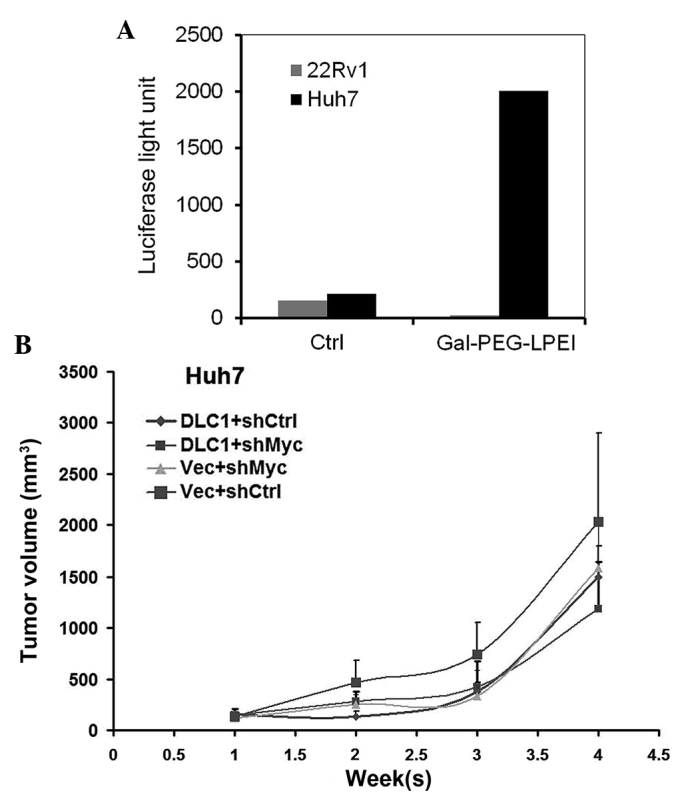
DLC1 transduction and Myc knockdown inhibit Huh7 cell growth in vivo. (A) Luciferase activity in Huh7 and 22Rv1 cell lysates following transfection in vitro with a luciferase reporter plasmid mixed with Gal-PEG3500-LPEI or with transfection buffer alone (Ctrl). (B) Comparison of in vivo growth of xenograft tumors derived from shCtrl-Huh7 or shMyc-Huh7 cells following tail vein injection of pLXSN-DLC1 (DLC1) or pLXSN vector (Vec) DNA mixed with Gal-PEG3500-LPEI. DLC1, deleted in liver cancer 1; Ctrl, control; sh, short hairpin; vec, vector; Gal-PEG3500-LPEI/Gal-PEG-LPEI, galactosylated polyethylene glycol-linear polyethyleneimine.
Discussion
Overexpression of Myc and/or loss of the DLC1 tumor suppressor protein are frequent events in HCC. A number of studies have revealed that DLC1 expression is able to inhibit the growth of human cancer cells in culture and in xenograft models (10,11). In the present study, the anti-oncogenic effect of combining knockdown of c-Myc expression and ectopic restoration of DLC1 expression was investigated in liver cancer cells. Downregulation of c-Myc or re-expression of DLC1 led to a marked reduction in the level of active RhoA, which was associated with decreased colony formation, invasion and cell proliferation. This inhibitory effect was augmented with a combination of DLC1 transduction and c-Myc knockdown, suggesting a cooperative effect on tumor cell growth inhibition.
Hepatocellular carcinoma is frequently diagnosed at an advanced stage, when few therapeutic options are available to the majority of HCC patients (1). Currently the tyrosine kinase inhibitor sorafenib is the only systemic treatment that is able to prolong survival in certain HCC patients, and novel effective therapeutic strategies and regimens are urgently required (25,26). As c-Myc is a key regulator of cell proliferation, apoptosis and differentiation, and is overexpressed in a number of types of cancer, there is much interest in the development of pharmaceutical agents that target Myc (27,28). The observation that DLCl knockdown is able to cooperate with c-Myc overexpression to promote tumor formation in a mouse model of liver cancer (16) suggests that a combination of Myc inhibition and DLC1 upregulation may be a beneficial treatment strategy.
The cooperative oncosuppressive effect of a coordinated increase in DLC1 expression and reduction in c-Myc expression in Huh7 cells may stem from their effects on Rho family GTPases, which transduce signals involved in cell proliferation and migration (29). Rho activity is increased in a number of human malignancies, including HCC (30). The catalytic RhoGAP domain of DLC1 can stimulate the GTPase activity of RhoA, RhoB, RhoC and cell division control protein 42 homolog, converting them to the inactive GDP-bound form (31,32). The negative regulatory effect of DLC1 on cell growth and metastasis largely relies on its RhoGAP activity, although RhoGAP-independent mechanisms may also contribute to the oncosuppressive action of DLC1 (32–34). Indeed, the present study observed that the effect of DLC1 on inhibiting cell growth and invasion in shMyc cells was greater than the effect of DLC1 on decreasing RhoA levels in shMyc cells, suggesting that RhoGAP-independent mechanisms are involved. Evidence has indicated that cross-talk between c-Myc and RhoA influences their expression. Myc RNA and protein levels were increased in 3T3 cells transformed with constitutively active RhoA (35). Myc is part of a complex that binds to the RhoA promoter, and Myc-induced RhoA transcription promotes cell migration and invasion (22). Thus, RhoA may be one of the crucial c-Myc target genes involved in tumorigenesis, and the efficacy of anti-Myc therapies could be enhanced by agents directed against the RhoA signaling pathway.
In conclusion, the current study presents experimental evidence that concurrent ectopic expression of DLC1 tumor suppressor protein and silencing of Myc expression leads to a cooperative oncosuppressive effect on Huh7 liver cancer cells in vitro and in vivo. These findings may stimulate further studies to examine whether shRNA-induced suppression of c-Myc expression, in combination with targeted liver cell-specific delivery of DLC1, may serve as a potential gene therapy modality for HCC.
Acknowlegments
The present study was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, NIH.
References
- 1.Tinkle CL, Haas-Kogen D. Hepatocellular carcinoma: Natural history, current management, and emerging tools. Biologics. 2012;6:207–219. doi: 10.2147/BTT.S23907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marquardt JU, Thorgeirsson SS. SnapShot: Hepatocelluar carcinoma. Cancer Cell. 2014;25:550.e1. doi: 10.1016/j.ccr.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 3.Takai A, Dang HT, Wang XW. Identification of drivers from cancer genome diversity in hepatocellular carcinoma. Int J Mol Sci. 2014;15:11142–11160. doi: 10.3390/ijms150611142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pinyol R, Nault JC, Quetglas IM, Zucman-Rossi J, Llovet JM. Molecular profiling of liver tumors: Classification and clinical translation for decision making. Semin Liver Dis. 2014;34:363–375. doi: 10.1055/s-0034-1394137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moinzadeh P, Breuhahn K, Stützer H, Schirmacher P. Chromosome alterations in human hepatocellular carcinomas correlate with aetiology and histological grade - results of an explorative CGH meta-analysis. Br J Cancer. 2005;92:935–941. doi: 10.1038/sj.bjc.6602448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chochi Y, Kawauchi S, Nakao M, Furuya T, Hashimoto K, Oga A, Oka M, Sasaki K. A copy number gain of the 6p arm is linked with advanced hepatocellular carcinoma: An array-based comparative genomic hybridization study. J Pathol. 2009;217:677–684. doi: 10.1002/path.2491. [DOI] [PubMed] [Google Scholar]
- 7.Katoh H, Ojima H, Kokubu A, Saito S, Kondo T, Kosuge T, Hosoda F, Imoto I, Inazawa J, Hirohashi S, Shibata T. Genetically distinct and clinically relevant classification of hepatocellular carcinoma: Putative therapeutic targets. Gastroenterology. 2007;133:1475–1486. doi: 10.1053/j.gastro.2007.08.038. [DOI] [PubMed] [Google Scholar]
- 8.Yuan BZ, Miller MJ, Keck CL, Zimonjic DB, Thorgeirsson SS, Popescu NC. Cloning, characterization, and chromosomal localization of a gene frequently deleted in human liver cancer (DLC-1) homologous to rat RhoGAP. Cancer Res. 1998;58:2196–2199. [PubMed] [Google Scholar]
- 9.Neel BG, Jhanwar SC, Chaganti RS, Hayward WS. Two human c-onc genes are located on the long arm of chromosome 8. Proc Natl Acad Sci USA. 1982;79:7842–7846. doi: 10.1073/pnas.79.24.7842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Durkin ME, Yuan BZ, Zhou X, Zimonjic DB, Lowy DR, Thorgeirsson SS, Popescu NC. DLC-1: A Rho GTPase-activating protein and tumour suppressor. J Cell Mol Med. 2007;11:1185–1207. doi: 10.1111/j.1582-4934.2007.00098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lukasik D, Wilczek E, Wasiutynski A, Gornicka B. Deleted in liver cancer protein family in human malignancies (Review) Oncol Lett. 2011;2:763–768. doi: 10.3892/ol.2011.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roessler S, Long EL, Budhu A, Chen Y, Zhao X, Ji J, Walker R, Jia HL, Ye QH, Qin LX, et al. Integrative genomic identification of genes on 8p associated with hepatocellular carcinoma progression and patient survival. Gastroenterology. 2012;142:957–966. doi: 10.1053/j.gastro.2011.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoshida Y, Nijman SM, Kobayashi M, Chan JA, Brunet JP, Chiang DY, Villanueva A, Newell P, Ikeda K, Hashimoto M, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009;69:7385–7392. doi: 10.1158/0008-5472.CAN-09-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaposi-Novak P, Libbrecht L, Woo HG, Lee YH, Sears NC, Coulouarn C, Conner EA, Factor VM, Roskams T, Thorgeirsson SS. Central role of c-Myc during malignant conversion in human hepatocarcinogenesis. Cancer Res. 2009;69:2775–2782. doi: 10.1158/0008-5472.CAN-08-3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xue W, Krasnitz A, Lucito R, Sordella R, Vanaelst L, Cordon-Cardo C, Singer S, Kuehnel F, Wigler M, Powers S, et al. DLC1 is a chromosome 8p tumor suppressor whose loss promotes hepatocellular carcinoma. Genes Dev. 2008;22:1439–1444. doi: 10.1101/gad.1672608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beer S, Zetterberg A, Ihrie RA, McTaggart RA, Yang Q, Bradon N, Arvanitis C, Attardi LD, Feng S, Ruebner B, et al. Developmental context determines latency of MYC-induced tumorigenesis. PLoS Biol. 2004;2:e332. doi: 10.1371/journal.pbio.0020332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zimonjic DB, Popescu NC. Role of DLC1 tumor suppressor gene and MYC oncogene in pathogenesis of human hepatocellular carcinoma: Potential prospects for combined targeted therapeutics (review) Int J Oncol. 2012;41:393–406. doi: 10.3892/ijo.2012.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guan M, Tripathi V, Zhou X, Popescu NC. Adenovirus-mediated restoration of expression of the tumor suppressor gene DLC1 inhibits the proliferation and tumorigenicity of aggressive, androgen-independent human prostate cancer cell lines: Prospects for gene therapy. Cancer Gene Ther. 2008;15:371–381. doi: 10.1038/cgt.2008.13. [DOI] [PubMed] [Google Scholar]
- 20.Yuan BZ, Zhou X, Durkin ME, Zimonjic DB, Gumundsdottir K, Eyfjord JE, Thorgeirsson SS, Popescu NC. DLC-1 gene inhibits human breast cancer cell growth and in vivo tumorigenicity. Oncogene. 2003;22:445–450. doi: 10.1038/sj.onc.1206064. [DOI] [PubMed] [Google Scholar]
- 21.Yang X, Popescu NC, Zimonjic DB. DLC1 interaction with S100A10 mediates inhibition of in vitro cell invasion and tumorigenicity of lung cancer cells through a RhoGAP-independent mechanism. Cancer Res. 2011;71:2916–2925. doi: 10.1158/0008-5472.CAN-10-2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan CH, Lee SW, Li CF, Wang J, Yang WL, Wu CY, Wu J, Nakayama KI, Kang HY, Huang HY, et al. Deciphering the transcriptional complex critical for RhoA gene expression and cancer metastasis. Nat Cell Biol. 2010;12:457–467. doi: 10.1038/ncb2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Du H, Zhai G. Recent advances in active hepatic targeting drug delivery system. Curr Drug Targets. 2014;15:573–599. doi: 10.2174/1389450115666140309232100. [DOI] [PubMed] [Google Scholar]
- 24.Treichel U, Meyer Zum Büschenfelde KH, Stockert RJ, Poralla T, Gerken G. The asialoglycoprotein receptor mediates hepatic binding and uptake of natural hepatitis B virus particles derived from viraemic carriers. J Gen Virol. 1994;75:3021–3029. doi: 10.1099/0022-1317-75-11-3021. [DOI] [PubMed] [Google Scholar]
- 25.Reig M, Darnell A, Forner A, Rimola J, Ayuso C, Bruix J. Systemic therapy for hepatocellular carcinoma: The issue of treatment stage migration and registration of progression using the BCLC-refined RECIST. Semin Liver Dis. 2014;34:444–455. doi: 10.1055/s-0034-1394143. [DOI] [PubMed] [Google Scholar]
- 26.Mikhail S, Cosgrove D, Zeidan A. Hepatocellular carcinoma: Systemic therapies and future perspectives. Expert Rev Anticancer Ther. 2014;14:1205–1218. doi: 10.1586/14737140.2014.949246. [DOI] [PubMed] [Google Scholar]
- 27.Albihn A, Johnsen JI, Henriksson MA. MYC in oncogenesis and as a target for cancer therapies. Adv Cancer Res. 2010;107:163–224. doi: 10.1016/S0065-230X(10)07006-5. [DOI] [PubMed] [Google Scholar]
- 28.McKeown MR, Bradner JE. Therapeutic strategies to inhibit MYC. Cold Spring Harb Perspect Med. 2014;4:a014266. doi: 10.1101/cshperspect.a014266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jaffe AB, Hall A. Rho GTPases: Biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 30.Grise F, Bidaud A, Moreau V. Rho GTPases in hepatocellular carcinoma. Biochim Biophys Acta. 2009;1795:137–151. doi: 10.1016/j.bbcan.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 31.Wong CM, Lee JM, Ching YP, Jin DY, Ng IO. Genetic and epigenetic alterations of DLC-1 gene in hepatocellular carcinoma. Cancer Res. 2003;63:7646–7651. [PubMed] [Google Scholar]
- 32.Healy KD, Hodgson L, Kim TY, Shutes A, Maddileti S, Juliano RL, Hahn KM, Harden TK, Bang YJ, Der CJ. DLC-1 suppresses non-small cell lung cancer growth and invasion by RhoGAP-dependent and independent mechanisms. Mol Carcinog. 2008;47:326–337. doi: 10.1002/mc.20389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wong CM, Yam JW, Ching YP, Yau TO, Leung TH, Jin DY, Ng IO. Rho GTPase-activating protein deleted in liver cancer suppresses cell proliferation and invasion in hepatocellular carcinoma. Cancer Res. 2005;65:8861–8868. doi: 10.1158/0008-5472.CAN-05-1318. [DOI] [PubMed] [Google Scholar]
- 34.Qian X, Li G, Asmussen HK, Asnaghi L, Vass WC, Braverman R, Yamada KM, Popescu NC, Papageorge AG, Lowy DR. Oncogenic inhibition by a deleted in liver cancer gene requires cooperation between tensin binding and Rho-specific GTPase-activating protein activities. Proc Natl Acad Sci USA. 2007;104:9012–9017. doi: 10.1073/pnas.0703033104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berenjeno IM, Núñez F, Bustelo XR. Transcriptomal profiling of the cellular transformation induced by Rho subfamily GTPases. Oncogene. 2007;26:4295–4305. doi: 10.1038/sj.onc.1210194. [DOI] [PMC free article] [PubMed] [Google Scholar]