

Abstract
Non-alcoholic fatty liver disease (NAFLD) is a fast-growing silent epidemic that is present in both developed and developing countries. Initially thought as a benign deposition of lipids in the liver, it now has been shown to be a major risk factor for type II diabetes and one of the leading causes of cirrhosis. Recent findings suggest that dysregulation of mitochondrial homeostasis and autophagy play critical roles in the hepatocyte injury and insulin resistance of NAFLD. Thyroid hormone (TH) is a major stimulator of hepatic autophagy and mitochondrial function. Decreased TH action has been associated with NAFLD in man. In this review, we highlight some of the new discoveries that demonstrate the roles of TH in hepatic mitochondrial homeostasis via mitophagy and their implications for NAFLD.
Keywords: Autophagy, Liver, Mitophagy, NAFLD, Thyroid hormones, ULK1
Background
Non-alcoholic fatty liver disease (NAFLD) is initiated by lipid accumulation in hepatocytes [1, 2] that leads to a spectrum of liver dysfunction ranging from excess lipid storage in the liver (hepatosteatosis) to progressive non-alcoholic steatohepatitis (NASH), that in turn, increases the risk for cirrhosis and hepatocellular cancer. NAFLD occurs in 25–35 % of the general US. population, and its prevalence is estimated to be 60–80 % in patients with type II diabetes milletus (DM) and obesity [2]. In addition to causing hepasteatosis and inflammation within the liver, NAFLD also can have profound metabolic effects by inducing hepatic insulin resistance [3]. Additionally, defects in β-oxidation of fatty acids and lipotoxicity owing to intracellular over-accumulation of fatty acids and their toxic metabolites are thought to play important roles in the pathogenesis of NAFLD [1, 2]. The progression in NAFLD often leads to insulin resistance, increased hepatic glucose production, and worsened glycemic control in diabetic patients, resulting in a vicious cycle that further exacerbates the manifestations and complications of diabetes. Unfortunately, little is known about hormonal regulation of hepatosteatosis and gluconeogenesis in NAFLD or the roles of hormones in disease progression. Moreover, although there are many drug therapy options for treating hyperglycemia in diabetes; currently, there are no effective drug treatments for NAFLD.
Thyroid hormones (THs: T3, T4) promote the oxidation of fatty acids within the liver, so it is possible that impaired TH action in the liver may contribute to NAFLD. Indeed, it recently has been shown that the incidence of NAFLD is doubled in patients with hypothyroidism [4] with approximately 15 % patients affected. Additionally, T3 and several TH analogs can ameliorate NAFLD in rodents that are fed high fat diet (HFD) [5, 6]. At the genomic level, many of the genes that have altered expression in NAFLD are regulated by TH [7], further supporting the notion that defects in TH signaling may promote hepatosteatosis and hepatic damage. Two recent studies also have shown that thyroid hypofunction occurs with higher frequency in both young and elderly adults with NAFLD [8, 9].
T3 stimulates the conversion of triglycerides to free fatty acids for delivery into the mitochondria by increasing mRNA expression and activities of hepatic lipases [10]. While this process is well described, it is possible that other cellular pathways may be involved in the delivery of stored triglycerides from lipid droplets to mitochondria. Recently, autophagy has been shown to promote cell survival during nutrient deprivation and upon exposure to inflammatory or pro-apoptotic stimuli [11, 12]. Moreover, autophagy has been implicated in the direct catabolism of fatty acids through “lipophagy” [13] and inhibition of autophagy leads to the development of fatty liver and insulin resistance [14]. We previously showed that T3 stimulated hepatic fatty acid oxidation through lipophagy [15]. We and others also have showed that T3 and TH analogs can decrease hepatosteatosis in cell culture and in rodent models [5, 6, 15]. The accompanying increase in oxidative phosphorylation leads to increased mitochondrial reactive oxygen species (ROS) production that can cause mitochondrial damage and cell death [16].
TH stimulates mitophagy and mitochondrial biogenesis
The major mechanisms for mitochondrial repair are mitochondrial fusion, fission, and mitochondrial autophagy or “mitophagy” [17]. To determine the occurrence of mitophagy, we used a tandem-tagged RFP-EGFP chimeric plasmid, pAT016, encoding a mitochondria targeting signal sequence fused in-frame with RFP and EGFP genes (tandem tagged Mito-mRFP-EGFP). RFP and GFP have different stabilities in an acidic environment [18]. The GFP signal is quenched at lower pH whereas RFP can be visualized in acidic autolysosomes; thus, increased RFP/red-only fluorescence in the lysosomes indicates completion of the mitophagic process (Fig. 1a). Using this assay, we observed that T3 increased autolysosome-resident mitochondria (red fluorescent dots without any green fluorescence) at concentrations as low as 1 nM and as early as 24 h (Fig. 1b, c). We then conducted studies of autophagy in primary mouse hepatocytes. T3 induced autophagic flux and mitophagy in primary mouse hepatocytes observed mitochondrial protein accumulation and the presence of autophagosome-resident mitochondria by electron microscopy (Fig. 1d, e). These findings demonstrate that these cell-autonomous effects occurred in primary hepatic cells with normal TRβ expression. Similar effects for autophagic flux also were seen in cell culture and in mice treated with the lysosomal inhibitor, chloriquine.
Fig. 1.
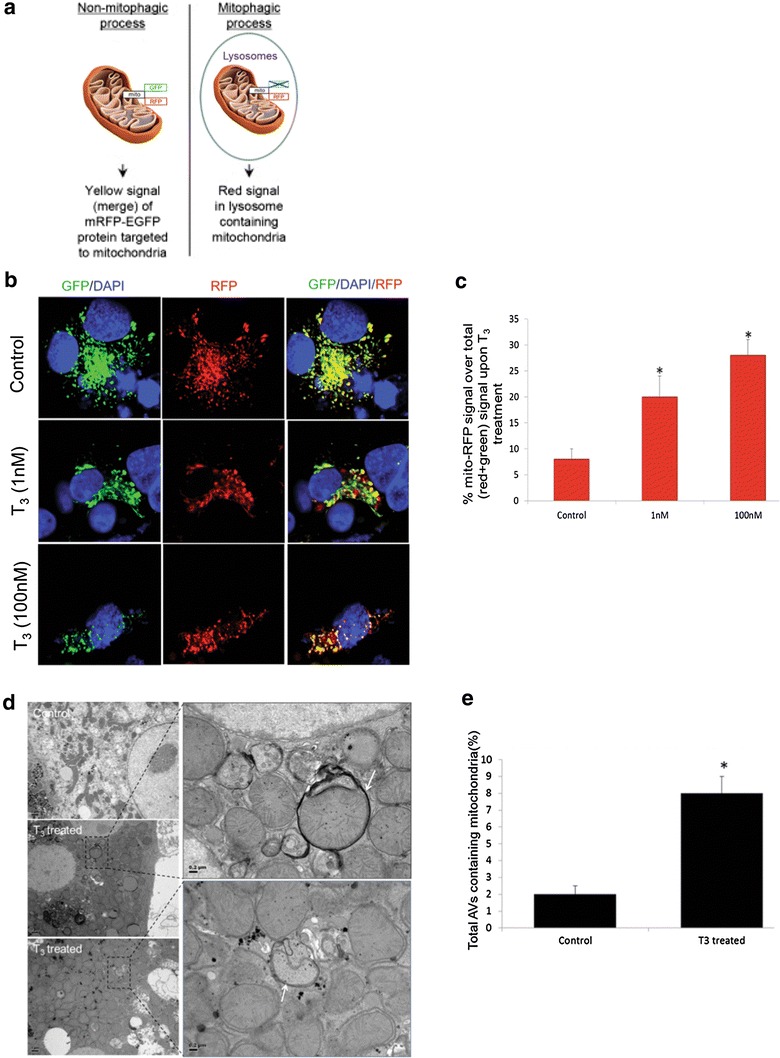
T3 increases autophagy and mitophagy in hepatic cells. a Model showing how mitochondria-specific mRFP-GFP protein detects mitophagy. b Monitoring mitophagic flux using dual fluorescence p-mito-mRFP-EGFP reporter (pAT016) in HepG2 cells. Lysosomal delivery of the tandem fusion protein Mito-mRFP-EGFP along with entire mitochondria results in differential quenching and degradation of the two individual fluorochromes, thus allowing for visual analysis of mitophagic flux. TRβ1-HepG2 cells transiently expressing Mito-mRFP-EGFP were treated with 1 nM or 100 nM T3 for 48 h followed by visualization using confocal microscopy (40× magnification). Nuclei were stained with DAPI (blue). In the images, fluorescence signals indicates the expression of Mito-mRFP-EGFP targeting mitochondria: yellow color no mitophagy or normal cytosolic mitochondria, red color mitophagy or mitochondria inside lysosomes. c Quantitative analysis of the RFP (red-only) fluorescence to denote % mitophagy was done. Quantification of images (at least 20 transfected cells per each sample in 3 different fields) was conducted with ImageJ software. Bars represent the mean of the respective individual ratios ± SD (*p < 0.05). d Electron micrograph of primary mouse hepatocytes treated with T3. EM of untreated control and T3-treated (100 nM/24 h) mouse hepatocytes showing increased mitophagy (Denoted by arrows showing autophagosomes containing mitochondria) under T3 treatment. Scale bar 1 µm and in enlarged figures are 0.2 µm. e Bar graphs showing % of autophagosomes (AVs) containing mitochondria in control and T3-treated primary mouse hepatocytes based on EM micrograph images. Scoring was done by counting 10–15 different autophagic vesicles in 5 random fields per condition (n = 3, *p < 0.05.
Adapted from Ref. [16], Figs. 4 and 6
Mitochondrial translocation of the autophagic machinery is required for mitophagy so we measured the levels of autophagic proteins in purified mitochondrial fractions that were verified to be free from cytosolic and lysosomal contamination (Fig. 2a). T3 treatment increased the localization of Ubiquitin-like protein 1 (ULK1), p62, and LC3II within the mitochondrial fraction of HepG2 cells. Dynamin 1-like protein (Drp1), a protein associated with mitochondrial fission and mitophagy also was preferentially recruited to mitochondria after T3 treatment (Fig. 2a). Additionally, increased mitochondrial protein ubiquitination was observed in T3-treated cells (Fig. 2a) consistent with the notion that mitochondrial ubiquitination precedes mitophagy. Confocal imaging of mt-RFP-EGFP in conjunction with the mitochondrial marker, TOMM20, showed that T3 increased mitophagy. However, treatment with ULK1 siRNA decreased mitophagy induced by T3 back to baseline level. Thus, mitophagy critically depends upon ULK1 and suggests that the latter is a necessary component for forming the nascent autophagosome that engulfs mitochondria (Fig. 2b, c) [16]. Interestingly, siRNA knockdown of ULK1 did not abrogate general autophagy suggesting that, unlike mitophagy, this process might be complemented by another isoform of ULK, ULK2 [16].
Fig. 2.
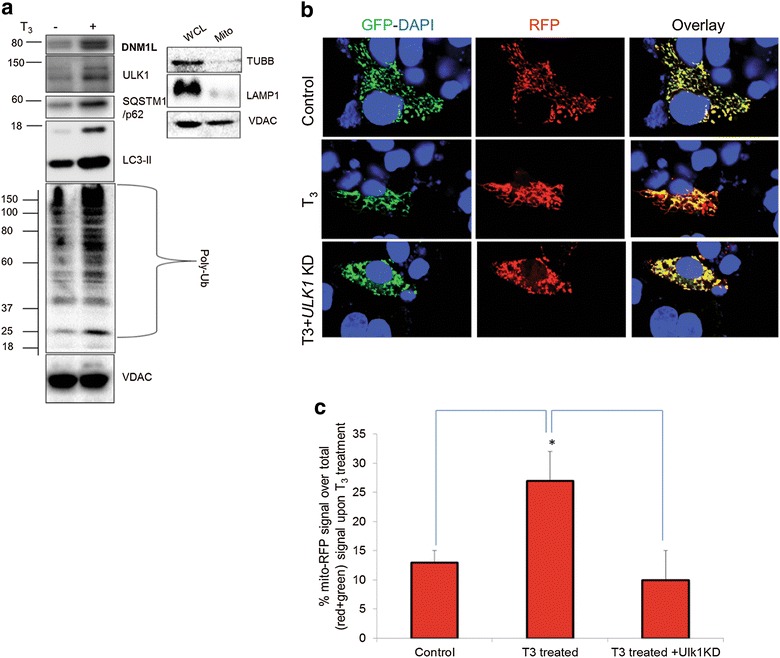
Mitophagy proteins translocate to mitochondria and are necessary for T3 stimulation of mitophagy. a Immunoblot showing mitochondrial protein ubiquitination and localization of ULK1, p62, LC3-II, and Drp1 proteins in isolated mitochondrial fraction from T3 (100 nM/48 h)-treated TRβ1-HepG2 cells. Purity/enrichment of the mitochondrial fraction (Mito) was verified by the absence of β-Tubulin (cytosolic) and LAMP-1 (lysosomal) relative to its level in the whole cell lysate (WCL) for the same amount of VDAC levels. b TRβ-HepG2 cells transiently expressing Mito-mRFP-EGFP were treated with 100 nM T3 for 48 h with or without ULK1 KD followed by visualization using confocal microscopy (40× magnification). Nuclei were stained with DAPI (blue). In the images, fluorescence signals indicate the expression of Mito-mRFP-EGFP targeting mitochondria: yellow color no mitophagy, red color mitophagy. c Quantitative analysis of the RFP (red) fluorescence to denote % mitophagy. Quantification of images (at least 10 transfected cells per each sample in 3 different fields) was conducted with ImageJ software. Bars represent the mean of the respective individual ratios ± SD (*p < 0.05).
Adapted from Ref. [16], Fig. 9
We observed induction of hepatic mitochondria biogenesis by T3-mediated stimulation of PGC1a and mitochondrial protein expression. The latter proteins increased their accumulation when autophagy was blocked suggesting that there was increased mitochondrial turnover involving both mitophagy and mitochondrial synthesis. The transcriptional expression of several genes involved in mitophagy, Bnip, Nix, ULK1, p62, and LC3 mRNAs also were induced by T3. Additionally, the master regulator of autophagy and lysosomal genes, Transcription factor EB (TFEB) as well as PGC1a, Tfam, and Cox 4 mRNAs were induced by T3.
Tissue-specific hypothyroidism in NAFLD
We examined livers from mice fed a methionine and choline deficient (MCD) diet for 12 weeks and found that the highly sensitive TH-responsive gene, Deiodinase 1 (DIO1), a deiodinase enzyme that converts T4 to T3, was significantly reduced in livers of MCD-fed rats compared to livers from control animals fed normal chow diet. Moreover, the MCD fed group exhibited grade 2 steatohepatitis on histology. We then measured intrahepatic T3, T4, and rT3 concentrations in livers from MCD-fed rats vs. rats fed normal chow diet. For the MCD fed rats, hepatic T3 concentration was significantly decreased, rT3 whereas hepatic T4, and rT3 concentrations were not changed (Sinha and Yen, unpublished data). In pilot studies in these rats, we found that DIO1 as well as OATP1 and MCT8 (thyroid hormone transporters) mRNA expression were decreased suggesting that intrahepatic hypothyroidism may be a feature of, as well as a contributor towards, the development of NASH in these rats.
Consistent with our data, two previous studies showed that T3 or TH analogs decreased hepatosteatosis in mouse and rat models [5, 6]. In order to assess whether T3 decreased lipotoxicity, a common feature of steatohepatitis, we examined the effects of TH on palmitate-induced cell death. Preliminary results showed palmitate markedly increased cleaved caspase 3 in TRβ-HepG2 cells, and this was attenuated by co-treatment with T3. Palmitate itself induced an increase in autophagy. However, the improvement in cell survival provided by T3 also was accompanied by a further increase in autophagy. Although palmitate increased oxidative phosphorylation as measured by the Seahorse XF Analyzer available in our laboratory, T3 further increased oxidative phosphorylation (Sinha and Yen, unpublished results.). These results suggest that induction of lipophagy and β-oxidation by T3 may protect against lipotoxicity due to toxic lipids derived from excessive intracellular palmitate such as such as ceramides or diacylglycerol. On the basis of the foregoing pre-clinical data, we recently have initiated a pilot clinical study on the effects of low dose levothyroxine therapy in reducing hepatic fat content and improving glucose control in diabetic Chinese male patients with hepatosteatosis. Patients are treated with low dose levothyroxine for 4 months, with their liver fat content measured both before and after treatment by MRI spectroscopy. Currently, we are in the middle of the enrollment of patients for our study.
Role of SIRT1 on T3-mediated autophagy
We also have shown that a subset of forkhead box protein O1 (FOXO1) target genes, including those involved in gluconeogenesis, are co-regulated by T3 through its metabolic activation of SIRT1 and its regulation of downstream deacetylation and dephosphorylation of FOXO1 [19, 20]. SIRT1 is a deactylase that can be activated by increased NAD+ concentration, and thus can act as an intracellular energy sensor to modulate the transcriptional activity by both TH and FOXO1 [21]. Besides its critical role in gluconeogenesis, we also have found that SIRT1 is required for TH-mediated autophagy by virtue of its ability to stimulate the expression of target genes involved in autophagy as well as deacetylate ATG proteins. Thus, SIRT1 plays vital roles in in initiating and maintaining the autophagy that is required for β-oxidation of fatty acids as well as mitophagy by TH. In this connection, mitochondria degeneration has been associated with metabolic disorders and aging. Thus, the maintenance of normal mitophagy may be a crucial aspect in preventing cell death in tissues such as the pancreas and liver in diabetes. Furthermore, it is possible that maintaining tissue-specific euthyroidism may promote normal lipid metabolism as well as preserve mitochondrial function in the liver. Indeed, the expression of target genes involved in lipid metabolism that are regulated by TH are decreased in liver samples from patients undergoing bariatric surgery [7].
Conclusions
In summary, we believe that our studies to elucidate the mechanisms of hepatic mitochondrial turnover by TH and SIRT1 and the role of autophagy in NAFLD, will lead to better understanding of the role of hormones and their potential dysregulation in the pathogenesis and progression of this condition. This information could lead to better diagnosis and treatments for NAFLD as well as other metabolic disorders.
Authors’ contributions
RAS searched the related articles, and prepared figures. PMY drafted and revised the manuscript and approved for publication. Both authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Availability of data and material
Not applicable.
Funding
This work was supported by NMRC/CSA/0054/2013 (PMY), NMRC/CIRG/1340/2012 (PMY), and from Singapore Ministry of Health NMRC/BNIG/2025/2014 (RAS). The following fundings helped in the design and completion of this work.
Abbreviations
- Drp1
dynamin 1-like protein
- FOXO1
forkhead box protein O1
- MCD
methionine and choline deficient
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- TH
thyroid hormones
- ULK1
ubiquitin-like protein 1
Contributor Information
Rohit Anthony Sinha, Email: paul.yen@duke-nus.edu.sg.
Paul M. Yen, Phone: (+65) 6516 6719, Email: paul.yen@duke-nus.edu.sg
References
- 1.Farrell GC, Wong VW, Chitturi S. NAFLD in Asia–as common and important as in the West. Nat Rev Gastroenterol Hepatol. 2013;10:307–318. doi: 10.1038/nrgastro.2013.34. [DOI] [PubMed] [Google Scholar]
- 2.Federico A, Zulli C, de Sio I, Del Prete A, Dallio M, Masarone M, Loguercio C. Focus on emerging drugs for the treatment of patients with non-alcoholic fatty liver disease. World J Gastroenterol. 2014;20:16841–16857. doi: 10.3748/wjg.v20.i45.16841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salgado AL, Carvalho L, Oliveira AC, Santos VN, Vieira JG, Parise ER. Insulin resistance index (HOMA-IR) in the differentiation of patients with non-alcoholic fatty liver disease and healthy individuals. Arq Gastroenterol. 2010;47:165–169. doi: 10.1590/S0004-28032010000200009. [DOI] [PubMed] [Google Scholar]
- 4.Liangpunsakul S, Chalasani N. Is hypothyroidism a risk factor for non-alcoholic steatohepatitis? J Clin Gastroenterol. 2003;37:340–343. doi: 10.1097/00004836-200310000-00014. [DOI] [PubMed] [Google Scholar]
- 5.Perra A, Simbula G, Simbula M, Pibiri M, Kowalik MA, Sulas P, Cocco MT, Ledda-Columbano GM, Columbano A. Thyroid hormone (T3) and TRbeta agonist GC-1 inhibit/reverse nonalcoholic fatty liver in rats. FASEB J. 2008;22:2981–2989. doi: 10.1096/fj.08-108464. [DOI] [PubMed] [Google Scholar]
- 6.Cable EE, Finn PD, Stebbins JW, Hou J, Ito BR, van Poelje PD, Linemeyer DL, Erion MD. Reduction of hepatic steatosis in rats and mice after treatment with a liver-targeted thyroid hormone receptor agonist. Hepatology. 2009;49:407–417. doi: 10.1002/hep.22572. [DOI] [PubMed] [Google Scholar]
- 7.Pihlajamaki J, Boes T, Kim EY, Dearie F, Kim BW, Schroeder J, Mun E, Nasser I, Park PJ, Bianco AC, et al. Thyroid hormone-related regulation of gene expression in human fatty liver. J Clin Endocrinol Metab. 2009;94:3521–3529. doi: 10.1210/jc.2009-0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carulli L, Ballestri S, Lonardo A, Lami F, Violi E, Losi L, Bonilauri L, Verrone AM, Odoardi MR, Scaglioni F, et al. Is nonalcoholic steatohepatitis associated with a high-though-normal thyroid stimulating hormone level and lower cholesterol levels? Intern Emerg Med. 2013;8:297–305. doi: 10.1007/s11739-011-0609-4. [DOI] [PubMed] [Google Scholar]
- 9.Li M, Xu Y, Xu M, Ma L, Wang T, Liu Y, Dai M, Chen Y, Lu J, Liu J, et al. Association between nonalcoholic fatty liver disease (NAFLD) and osteoporotic fracture in middle-aged and elderly Chinese. J Clin Endocrinol Metab. 2012;97:2033–2038. doi: 10.1210/jc.2011-3010. [DOI] [PubMed] [Google Scholar]
- 10.Nozaki S, Shimomura I, Funahashi T, Menju M, Kubo M, Matsuzawa Y. Stimulation of the activity and mRNA level of hepatic triacylglycerol lipase by triiodothyronine in HepG2 cells. Biochim Biophys Acta. 1992;1127:298–302. doi: 10.1016/0005-2760(92)90235-N. [DOI] [PubMed] [Google Scholar]
- 11.Choi SE, Lee SM, Lee YJ, Li LJ, Lee SJ, Lee JH, Kim Y, Jun HS, Lee KW, Kang Y. Protective role of autophagy in palmitate-induced INS-1 beta-cell death. Endocrinology. 2009;150:126–134. doi: 10.1210/en.2008-0483. [DOI] [PubMed] [Google Scholar]
- 12.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–478. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sinha RA, Singh BK, Yen PM. Thyroid hormone regulation of hepatic lipid and carbohydrate metabolism. Trends Endocrinol Metab. 2014;25:538–545. doi: 10.1016/j.tem.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 15.Sinha RA, You SH, Zhou J, Siddique MM, Bay BH, Zhu X, Privalsky ML, Cheng SY, Stevens RD, Summers SA, et al. Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. J Clin Invest. 2012;122:2428–2443. doi: 10.1172/JCI60580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sinha RA, Singh BK, Zhou J, Wu Y, Farah BL, Ohba K, Lesmana R, Gooding J, Bay BH, Yen PM. Thyroid hormone induction of mitochondrial activity is coupled to mitophagy via ROS-AMPK-ULK1 signaling. Autophagy. 2015;11:1341–1357. doi: 10.1080/15548627.2015.1061849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh BK, Sinha RA, Zhou J, Xie SY, You SH, Gauthier K, Yen PM. FoxO1 deacetylation regulates thyroid hormone-induced transcription of key hepatic gluconeogenic genes. J Biol Chem. 2013;288(30365–30372):17. doi: 10.1074/jbc.M113.504845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Held NM, Houtkooper RH. Mitochondrial quality control pathways as determinants of metabolic health. BioEssays. 2015;37(8):867–876. doi: 10.1002/bies.201500013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shitara H, Kaneda H, Sato A, Iwasaki K, Hayashi J, Taya C, Yonekawa H. Non-invasive visualization of sperm mitochondria behavior in transgenic mice with introduced green fluorescent protein (GFP) FEBS Lett. 2015;500:7–11. doi: 10.1016/S0014-5793(01)02574-1. [DOI] [PubMed] [Google Scholar]
- 20.Singh BK, Sinha RA, Zhou J, Tripathi M, Ohba K, Wang ME, Astapova I, Ghosh S, Hollenberg AN, Gauthier K, et al. Hepatic FOXO1 target genes are co-regulated by thyroid hormone via RICTOR deacetylation and MTORC2-AKT inhibition. J Biol Chem. 2015;291:198. doi: 10.1074/jbc.M115.668673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, Finkel T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci USA. 2008;105:3374–3379. doi: 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]