

Abstract
Background and Purpose
Metabolites of the endocannabinoid, 2‐arachidonoylglycerol (2‐AG) have been postulated to act as endogenous activators of TRPV4, a Ca2+‐permeable cation channel that plays a critical role in endothelium‐dependent relaxation. However, it is unclear if TRPV4 contributes to the vascular actions of 2‐AG.
Experimental Approach
Isometric tension recording of rat small mesenteric arteries and aortae were used to assess the effect of 2‐AG and the synthetic TRPV4 activator, GSK1016790A (GSK) on vascular reactivity. Changes in intracellular Ca2+ concentration and single‐channel currents were measured in TRPV4‐expressing human coronary endothelial cells.
Key Results
In mesenteric arteries, endothelium‐dependent relaxation to both 2‐AG and GSK was attenuated by structurally distinct TRPV4 antagonists, HC067047, RN1734 and ruthenium red. The responses were inhibited by KCa inhibitors (apamin + charybdotoxin) and a gap junction inhibitor (18α‐glycyrrhetinic acid). In contrast to GSK, 2‐AG elicited considerable relaxation independently of the endothelium or TRPV4. Inhibition of 2‐AG metabolism via monoacylglycerol lipase and COX (by MAFP and indomethacin) caused potentiation, while cytochrome P450 and lipoxygenase inhibitors had no effect on 2‐AG relaxation. In coronary endothelial cells, 2‐AG (with and without MAFP) induced HC067047‐sensitive increases in intracellular Ca2+ concentration. 2‐AG also increased TRPV4 channel opening in inside‐out patches. However, in aortae, GSK induced a relaxation sensitive to HC067047 and ruthenium red, whereas 2‐AG induced contractions.
Conclusions and Implications
These data suggest that 2‐AG can directly activate endothelial TRPV4, which partly contributes to the relaxant response to 2‐AG. However, the functional role of TRPV4 is highly dependent on the vascular region.
Abbreviations
- 4α‐PDD
4α‐phorbol‐12,13‐didecanoate
- GSK
GSK1016790A
- JTE907
N‐(1,3‐benzodioxol‐5‐ylmethyl)‐1,2‐dihydro‐7‐methoxy‐2‐oxo‐8‐(pentyloxy)‐3‐quinolinecarboxamide
- SKF525A
α‐phenyl‐α‐propylbenzeneacetic acid 2‐(diethylamino)ethyl ester N,N‐diethylaminoethyl 2,2‐diphenylvalerate
Tables of Links
| TARGETS | |
|---|---|
| GPCRs a | Enzymes c |
| CB1 receptor | COX |
| CB2 receptor | Cytochrome P450 |
| Ion channels b | Lipoxygenases |
| IKCa (KCa3.1) | MGL |
| SKCa | NOS |
| TRPV1 | |
| TRPV4 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Transient receptor potential vanilloid type 4 (TRPV4) is a Ca2+‐permeable cation channel from the TRP channel family. It is widely expressed in the body, including the endothelium in conduit and resistance arteries [aorta (Watanabe et al., 2003); carotid: (Köhler et al., 2006; Hartmannsgruber et al., 2007); mesenteric: (Mendoza et al., 2010; Ma et al., 2013); and coronary; (Bubolz et al., 2012)]. There is evidence suggesting that endothelial TRPV4 channels are fundamental for the control of vascular tone and blood flow (Filosa et al., 2013). Indeed, pharmacological or genetic inhibition of TRPV4 greatly impairs the endothelium‐dependent relaxation to shear stress (Köhler et al., 2006; Mendoza et al., 2010) or ACh (Zhang et al., 2009). TRPV4 blockade also increases basal myogenic tone, governs blood flow autoregulation (Bagher et al., 2012). These effects stem from Ca2+ influx through plasmalemmal TRPV4 in the endothelium, resulting in local Ca2+ sparklets (Sonkusare et al., 2012). They, in turn, stimulate NO release and, particularly in resistance arteries, activate small‐conductance and intermediate‐conductance Ca2+‐activated K+ channels (SKCa and IKCa) (Köhler et al., 2006; Zhang et al., 2009; Mendoza et al., 2010; Sonkusare et al., 2012) leading to vasorelaxation. Activation of SKCa and IKCa is characteristic of the endothelium‐derived hyperpolarization (EDH) pathway that is independent of NO and PGs (Edwards et al., 2010). For some vessels, such as mesenteric arteries, EDH causes smooth muscle hyperpolarization at least partly via myoendothelial gap junctions (Edwards et al., 2010).
Synthetic ligands selective for TRPV4 have been developed; however, much less is known about endogenous agonists of TRPV4 (Nilius et al., 2004). Several lipid mediators have been identified as endogenous TRP agonists. More specifically, cytochrome P450 epoxygenase metabolites of arachidonic acid, epoxyeicosatrienoic acids (EETs) have been shown to directly activate TRPV4 (Watanabe et al., 2003; Zheng et al., 2013). In native endothelial cells, arachidonic acid may also activate TRPV4 (Zheng et al., 2013). Using mouse TRPV4 expressed in cultured cells, the study by Watanabe et al. (2003) demonstrated N‐arachidonyl ethanolamine (AEA), an endocannabinoid that activates cannabinoid CB receptors, is also able to activate TRPV4. This effect is dependent on AEA hydrolysis to arachidonic acid, which is then probably metabolized to EETs (Watanabe et al., 2003). It is thought that, through a similar mechanism, the other major endocannabinoid, 2‐arachidonoylglycerol (2‐AG) also acts as an endogenous activator of TRPV4. Although both AEA and 2‐AG activate G‐protein‐coupled, CB1 and CB2 receptors, there are numerous reports suggesting that other targets are involved in some of their in vitro and in vivo effects (Pertwee et al., 2010). 2‐AG is known to induce hypotension, partly through its hydrolysis to arachidonic acid (Járai et al., 2000). In animal and human isolated arteries 2‐AG often causes relaxation; an effect that is dependent on K+ channel activation induced by activating CB1 receptors or by the hydrolysis products of COX and cytochrome P450, depending on the vascular regions and species (Kagota et al., 2001; Gauthier et al., 2005; Stanley and O'Sullivan, 2014). Despite the earlier finding linking 2‐AG to TRPV4 (Watanabe et al., 2003), whether 2‐AG or its metabolites modulate vascular tone via TRPV4 activation remains to be determined.
In this study, we compared the effects of 2‐AG with the synthetic TRPV4 agonist, GSK1016790A (GSK) on vascular tone in vitro. Using a pharmacological approach, we determined the involvement of TRPV4, 2‐AG metabolism via hydrolysis, COX, cytochrome P450 and lipoxygenase, and the EDH pathway. The effects of 2‐AG on intracellular Ca2+ levels and TRPV4 single‐channel activity in endothelial cells were also investigated.
Methods
Isometric tension recording
Male Wistar rats (200–350 g; Charles River UK Ltd, Kent, UK) were killed by cervical dislocation and decapitation. All animal care and use was in accordance with the UK Animal (Scientific Procedures) Act 1986. The third‐order branches of the superior mesenteric artery were removed and cleaned of adherent tissue. Segments (2 mm in length) were mounted in a Mulvany–Halpern type wire myograph (Model 610M; Danish Myo Technology, Aarhus, Denmark) and maintained at 37°C in gassed (95% O2/5% CO2) Krebs–Henseleit solution of the following composition (mM): NaCl 118, KCl 4.7, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25, CaCl2 2, D‐glucose 10 as previously described, unless otherwise stated (Ho and Randall, 2007). Arteries were equilibrated and set to a basal tension of 2 to 2.5 mN. In a separate set of experiments, segments of thoracic aorta (3–4 mm in length) were dissected and mounted on stainless steel hooks in a 15 mL organ bath, using a basal tension of about 15 mN (Stanke‐Labesque et al., 2004). In both mesenteric arteries and aortae, the integrity of the endothelium was assessed by precontracting the vessel with 10 μM methoxamine (an α1‐adrenoceptor agonist), followed by relaxation with 10 μM carbachol (a muscarinic ACh receptor agonist); vessels showing relaxations of greater than 90% (for mesenteric arteries) or greater than 70% (for aortae) were designated as endothelium‐intact (denoted as +ec). When the endothelium was not required, it was removed by rubbing the intima with a human hair (mesenteric arteries) or a blunt needle (aortae); carbachol‐induced relaxation of less than 10% indicated successful removal (denoted as −ec). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010: McGrath et al., 2010).
To measure relaxant responses, vessels were precontracted with 10 μM methoxamine (unless otherwise stated) followed by cumulative addition of a potential TRPV4 activator (GSK, 2‐AG, noladin ether or arachidonic acid), and the maximal relaxation achieved at each concentration was determined. To investigate the mechanisms involved in the relaxation, the vessels were pre‐incubated for 30 min with various modulators, including TRPV4 antagonists, 2‐AG degradation enzyme inhibitors and KCa inhibitors, before determination of relaxant responses. Preliminary experiments indicated that continued exposure to GSK desensitized the vessels to the relaxant effect of GSK, especially at concentrations ≥300 nM and application of 1 μM GSK induced irreversible impairment of the endothelium‐dependent relaxation (data not shown). Therefore, a 4‐point concentration–response curve was obtained for GSK (mesenteric artery: 10–300 nM; aorta: 0.1–30 nM). In each arterial preparation, only a single concentration–response curve to GSK, or other potential TRPV4 activator, was constructed.
Ca2+ measurement in isolated endothelial cells
Human coronary artery endothelial cells overexpressing the human TRPV4‐GFP fusion protein were used for calcium imaging as previously described (Zheng et al., 2013). These cells allowed us to focus on the activity of TRPV4 compared with other TRP channels that might also be expressed in endothelial cells and thus provide more definitive evidence on whether 2‐AG activates endothelial TRPV4. Cells were loaded with fura‐2 AM (5 μM; Molecular Probes) for 30 min in reduced Ca2+ EGM‐2MV medium (Lonza, USA), followed by a 15 min equilibration at room temperature in a modified HBSS that contained (in mM): 123 NaCl, 5.4 KCl, 1.6 CaCl2, 0.5 MgCl2, 0.4 MgSO4, 4.2 NaHCO3, 0.3 Na2HPO4, 0.4 KH2PO4, 5.5 glucose and 20 HEPES (pH 7.4). Fluorescence images were acquired with Metafluor software (Molecular Devices, Sunnyvale, USA) for 20 to 40 min every 3 s in cells treated with 2‐AG (0.1–10 μM), noladin ether (10 μM) or the TRPV4 agonist 4α‐PDD (1 μM). For some experiments, cells were pre‐incubated with MAFP (10 μM) or SKF525A (10 μM) for 5–10 min before being assayed for Ca2+ responses to 2‐AG and 4α‐PDD respectively. The intracellular Ca2+ concentration ([Ca2+]i) was calculated according to the following formula: [Ca2+]i = Kd (Sf,2/Sb,2) (R − Rmin)/(Rmax − R) (Grynkiewicz et al., 1985), where R is the ratio of the fluorescence intensity at 340 nm (F340) over that at 380 nm (F380); Rmin and Rmax are minimal and maximal F340/F380 ratios respectively; and Sf,2/Sb,2 represents the maximal and minimal signal intensities at 380 nm respectively. K d is the apparent dissociation constant of fura‐2 (224 nM). Experiments were performed at room temperature.
Patch‐clamp recording of TRPV4 currents
Single‐channel currents were recorded in human coronary artery endothelial cells overexpressing the human TRPV4‐GFP fusion protein as previously described (Zheng et al., 2013). The pipette solution contained (in mM) 140 NaCl, 1 MgCl2 and 10 HEPES (pH 7.4 with CsOH). The bath solution for both cell‐attached and inside‐out patches was composed of (in mM) 135 KCl, 0.8 CaCl2, 1 MgCl2, 5 EGTA, 5 glucose and 10 HEPES. The pH of the bath solution was adjusted to 7.3 with CsOH (final Cs+ concentration of 4 mM) and KOH. Channel currents were recorded with an Axopatch 200B amplifier (Molecular Devices) for at least 3–6 min under control conditions and after treatment with GSK (10 nM) or 2‐AG (10 μM). All chemicals were applied by bath perfusion at a rate of 1 mL·min−1. Single‐channel currents were sampled at 5 kHz and filtered at 1 kHz and analysed using pClamp 10 software (Molecular Devices). Experiments were performed at room temperature.
Data and statistical analysis
Vascular responses are given as % relaxation (or contraction) of the tone induced by methoxamine or KCl. Data are given as mean ± SEM and n represents the number of animals used. Because it was not usually possible to fully define concentration–responses curves (to 2‐AG and GSK), maximum responses obtained at the highest concentrations used are reported, and potency is expressed as pEC40, the negative logarithm of the concentration of agent producing 40% relaxation determined directly from individual log concentration–response curves. Statistical comparisons of vascular responses were made by Student's t‐test, one‐way ANOVA or two‐way ANOVA, where appropriate (Prism 6, GraphPad Software, Inc, San Diego, CA, USA). P < 0.05 was taken as statistically significant.
All Ca2+ responses are presented as mean ± SEM and n represents the number of independent experiments, with ≥20 cells analysed in each experiment. Patch‐clamping data were obtained from at least six patches in each group. Statistical comparisons were made by Student's t‐test (Prism 6, GraphPad Software, Inc). P < 0.05 was taken as statistically significant.
Drugs
Methoxamine, carbachol, L‐NAME, SKF525A, ruthenium red, apamin (Sigma Chemical Co., Poole, UK), charybdotoxin and CGRP8‐37 (Tocris Biosciences, Bristol, UK) were dissolved in deionized water. GSK1016790A (GSK), 4α‐PDD, 18α‐glycyrrhetinic acid, ionomycin and miconazole (Sigma) were dissolved in 100% DMSO. Indomethacin, MAFP, RN1734, HC067047, capsazepine (Sigma), 2‐AG, noladin ether, arachidonic acid, ICI 192605, AM251 and JTE907 (Tocris) were dissolved in 100% ethanol.
Results
TRPV4‐mediated relaxation of mesenteric arteries
In rat small mesenteric arteries, the synthetic TRPV4 agonist GSK induced a concentration‐dependent relaxation, which was attenuated by the selective TRPV4 antagonists RN1734 (20 μM) and HC067047 (1 μM; Figure 1A and Table 1). The GSK responses were also inhibited by the non‐selective TRP channel blocker, ruthenium red (10 μM), and reduced to just below 20% relaxation after removal of the endothelium (Figure 1A and Table 1). An original trace of the GSK‐induced relaxation is shown in Figure 1B. Additional traces of GSK and its vehicle are shown in Supporting Information Figure S1.
Figure 1.
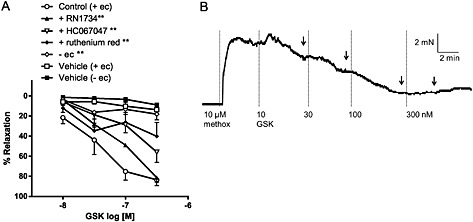
(A) Effects of TRPV4 antagonists (20 μM RN1734, 10 μM HC067047 and 10 μM ruthenium red) and endothelial removal on GSK‐induced relaxation in mesenteric arteries. The vehicle (and time) controls for GSK (0.001–0.03% vv−1 DMSO, n = 5) are also shown. Values are shown as means and vertical bars represent SEM. The data were analysed by two‐way ANOVA, followed by Bonferroni post hoc test. ** P < 0.01 in comparison with control (+ec); (B) An original trace showing the relaxant response to GSK in an endothelium‐intact mesenteric artery precontracted with 10 μM methoxamine (methox). The arrows indicate where the maximal relaxant effect was determined at each concentration.
Table 1.
Effects of TRPV4 antagonists on relaxant responses to GSK and 2‐AG in mesenteric arteries
| pEC40% | Relaxation | n | ||
|---|---|---|---|---|
| 0.3 μM (%) | 10 μM (%) | |||
| GSK (+ec) control | 7.6 ± 0.1 | 86 ± 5 | 8 | |
| + RN1734 | 7.2 ± 0.1 | 81 ± 4 | 5 ** | |
| + HC067047 | 6.9 ± 0.2 | 56 ± 10 | 4 ** | |
| + ruthenium red | — | 40 ± 14 | 4 ** | |
| GSK (−ec) | — | 18 ± 6 | 7 ** | |
| 2‐AG (+ec) control | 5.9 ± 0.1 | 90 ± 3 | 9 | |
| + RN1734 | 5.3 ± 0.1 | 57 ± 9 | 6 ** | |
| + HC067047 | 5.6 ± 0.2 | 84 ± 5 | 4 ** | |
| + Ruthenium red | 5.5 ± 0.2 | 74 ± 6 | 5 ** | |
| 2‐AG (+ec + MAFP + indomethacin) control | 6.8 ± 0.1 | 87 ± 5 | 7 ## | |
| + RN1734 | 5.8 ± 0.2 | 77 ± 7 | 5 ** | |
| 2‐AG (−ec) control | — | 43 ± 11 | 6 ## | |
| + RN1734 | — | 66 ± 7 | 5 | |
Data represent mean ± SEM.
P < 0.01 versus corresponding control;
P < 0.01 versus (+ec) control, as analysed by two‐way ANOVA of the whole data set, followed by Bonferroni post hoc tests.
Role of endothelial TRPV4 in relaxation to 2‐AG in mesenteric arteries
In rat small mesenteric arteries, 2‐AG also induced a concentration‐dependent relaxation (Figure 2A), albeit it was much less potent than GSK (Table 1). Figure 2B shows an original tracing of the 2‐AG‐induced relaxation. The TRPV4 antagonists, RN1734 (20 μM), HC067047 (1 μM) and ruthenium red (10 μM) induced rightward displacements of the concentration–response curves (Figure 2A and Table 1). There was a maximal threefold shift in EC40 values and a 30% reduction in responses to 10 μM 2‐AG (Table 1). Additional traces for effects of 2‐AG and its vehicle are shown in Supporting Information Figure S2.
Figure 2.
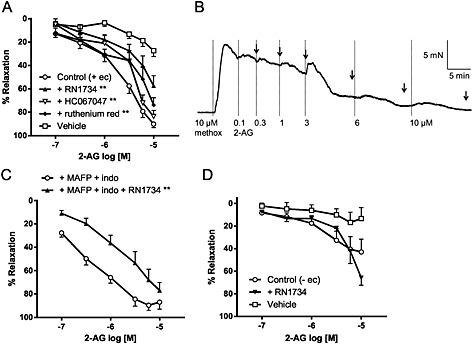
(A) Effects of TRPV4 antagonists (20 μM RN1734, 1 μM HC067047 and 10 μM ruthenium red) on 2‐AG‐induced relaxation in endothelium‐intact mesenteric arteries. The vehicle (and time) control for 2‐AG (0.001–0.1% vv−1 ethanol, n = 5) is also shown. (B) An original trace showing the relaxant response to 2‐AG in an endothelium‐intact mesenteric artery precontracted with 10 μM methoxamine (methox). The arrows indicate where the maximal relaxant effect was determined at each concentration. (C) Effects of RN1734 (20 μM), in the presence of 2‐AG metabolism inhibitors (10 μM MAFP and 10 μM indomethacin; indo), on 2‐AG‐induced relaxation in endothelium‐intact mesenteric arteries. (D) Effects of RN1734 (20 μM) on 2‐AG‐induced relaxation in endothelium‐denuded mesenteric arteries. The vehicle (and time) control for 2‐AG (0.001–0.1% vv–1 ethanol, n = 5) is also shown. In (A), (C) and (D), values are shown as means and vertical bars represent SEM. The data were analysed by two‐way ANOVA, followed by Bonferroni post hoc test. **P < 0.01 in comparison to control.
2‐AG is susceptible to hydrolysis mainly via monoacylglycerol lipase forming arachidonic acid, which is further metabolized by COX. We previously demonstrated that inhibition of monoacylglyercol lipase (by MAFP) and COX (by indomethacin) potentiated the mesenteric relaxation to 2‐AG (Ho and Randall, 2007), suggesting that the 2‐AG metabolites are not involved in this relaxant response. In this study, we confirmed that 10 μM MAFP and 10 μM indomethacin potentiated the relaxant response to 2‐AG in endothelium‐intact mesenteric arteries (Table 1). Importantly, the resultant responses were inhibited by the TRPV4 antagonist, RN1734 (Figure 2C and Table 1). In the absence of an endothelium, the 2‐AG‐induced responses were reduced to about 50% and unaffected by 20 μM RN1734 (Figure 2D and Table 1).
In contrast, the presence of CB1 and CB2 receptor antagonists did not inhibit the relaxant response to 2‐AG (control: pEC40 = 5.9 ± 0.2, relaxation at 10 μM = 93 ± 2%, n = 5; + 1 μM AM251 (CB1‐selective) + 1 μM JTE907 (CB2‐selective): pEC40 = 6.0 ± 0.1, relaxation at 10 μM = 96 ± 1%, n = 5).
Role of TRPV1/CGRP signalling in TRPV4‐mediated relaxation of mesenteric arteries
Activation of another TRPV subtype, TRPV1 channels, in perivascular sensory nerves is known to induce mesenteric relaxation through the release of the vasorelaxant CGRP (Ho et al., 2008). However, antagonism of the CGRP receptor by CGRP8‐37 (2 μM) had no significant effect on responses to GSK (control: pEC40 = 7.4 ± 0.1, relaxation at 0.3 μM = 81 ± 3%, n = 7; + CGRP8‐37: pEC40 = 7.2 ± 0.1, relaxation at 0.3 μM = 72 ± 10%, n = 6) or 2‐AG (control: pEC40 = 5.8 ± 0.2, relaxation at 10 μM = 88 ± 5%, n = 5; + CGRP8‐37: pEC40 = 5.8 ± 0.1, relaxation at 10 μM = 83 ± 3%, n = 5).
The selective TRPV1 antagonists, capsazepine (3 μM; pEC40 = 6.1 ± 0.2, relaxation at 10 μM = 98 ± 1%, n = 4) or SB366791 (2 μM; pEC40 = 5.7 ± 0.2, relaxation at 10 μM = 95 ± 4%, n = 4) also had no effect on 2‐AG‐induced relaxation.
TRPV4‐mediated Ca2+ responses in coronary endothelial cells
In isolated human coronary endothelial cells overexpressing hTRPV4, activation of TRPV4 by the synthetic phorbol derivative, 4α‐PDD (1 μM) increased intracellular Ca2+ concentration (by 2558 ± 400 nM, n = 7). Interestingly, 2‐AG (10 μM) increased intracellular Ca2+ concentration in a RN1734‐sensitive or HC067047‐sensitive manner (control: 615 ± 52 nM, n = 8; + 20 μM RN1734: 71 ± 11 nM, n = 2; + 1 μM HC067047: 29 ± 4 nM, n = 3; Figure 3A–C and F). Subsequent addition of 5 μM ionomycin (Ca2+ ionophore) induced close to maximal Ca2+ response (F340/F380 = 5.7 ± 0.3, n =6; Figure 3A and B). Inhibition of 2‐AG hydrolysis by 10 μM MAFP attenuated 2‐AG response by about 30% (439 ± 44 nM, n = 4, P > 0.05 compared with 2‐AG), but the residual Ca2+ response was reversed by HC067047 (76 ± 38 nM, n = 3; Figure 3D and F). A similar Ca2+ response (396 ± 90 nM, n = 8) was also induced by noladin ether, the non‐hydrolysable analogue of 2‐AG (Figure 3E and F).
Figure 3.
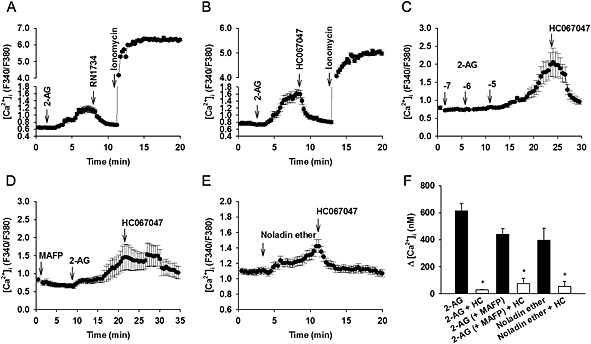
Increase in [Ca2+]i in coronary endothelial cells induced by (A–C) 2‐AG, (D) 10 μM 2‐AG in the presence of 10 μM MAFP (inhibitor of 2‐AG hydrolysis), and (E) 10 μM noladin ether (non‐hydrolysable analogue of 2‐AG). In (A) to (E), the ability of 20 μM RN1734 or 1 μM HC067047 (selective TRPV4 antagonists) to reverse the Ca2+ response, or the ability of 5 μM ionomycin (Ca2+ ionophore) to elicit [Ca2+]i increases are also shown. Traces represent mean ± SEM of an individual experiment. (F) Mean data for [Ca2+]i increases induced by 10 μM 2‐AG, 10 μM 2‐AG with MAFP and 10 μM noladin ether in the absence and presence of 1 μM HC067047. All data represent mean ± SEM (≥3 independent experiments, with ≥20 cells analysed in each experiment). *P < 0.05 in comparison with control.
Single channel recordings of TRPV4 currents in coronary endothelial cells
In cell‐attached patch clamping experiments using isolated human coronary endothelial cells overexpressing hTRPV4, single channel activity of TRPV4 was low under basal conditions, but it was markedly increased following bath perfusion of 10 nM GSK (Figure 4A and B). The currents displayed electrophysiological and pharmacological properties consistent with TRPV4 single‐channel currents previously reported in the endothelial cells (Zheng et al., 2013) and HEK‐293 cells (Nilius et al., 2004). Bath application of 2‐AG (10 μM) induced small increases in TRPV4 channel openings the in cell‐attached mode (NPo in control: 0.0008 ± 0.0005, n = 6; + 2‐AG: 0.0031 ± 0.0011, n = 6) and caused a greater activation of TRPV4 in inside‐out patches (0.0604 ± 0.0247, n =7; Figure 4C and D), suggesting 2‐AG can open the channel in a membrane‐regulated fashion. The 2‐AG activated single‐channel current had an amplitude of 2.4 ± 0.03 pA at −40 mV (equivalent to an average conductance of 60 pS) (n = 6 cells; Figure 4C), which is indistinguishable from that induced by GSK (Figure 4A).
Figure 4.
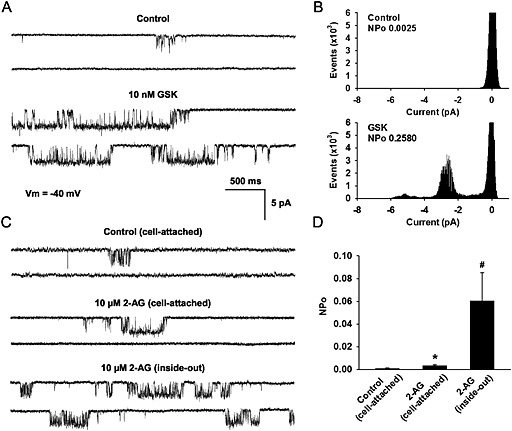
Effects of GSK and 2‐AG on single‐channel currents of TRPV4 in coronary endothelial cells. (A) Single‐channel recordings in a cell‐attached patch before and after bath perfusions of 10 nM GSK. (B) Amplitude histograms in relation to open‐state probability (NPo) corresponding to the cell recorded in panel A. (C) TRPV4 single‐channel currents recorded in a cell‐attached patch before (top) and after (middle) bath applications of 10 μM 2‐AG and in a subsequently excised inside‐out patch (bottom) in the presence of 10 μM 2‐AG. Both cell‐attached and inside‐out recordings were performed at a membrane potential (Vm) of −40 mV using a normal Na+ pipette solution (140 mM Na+, 5 mM Cs+) and a high‐K+ (145 mM K+, 5 mM Cs+) bath solution. Closed‐open transitions of the channel are shown as downward deflections. (D) Mean data for 2‐AG‐induced activation of TRPV4. All data represent mean ± SEM (≥6 patches in each group). *P < 0.05 in comparison with control; # P < 0.05 versus 2‐AG‐treated attached cell.
Role of TRPV4 in relaxation to noladin ether and arachidonic acid in mesenteric arteries
Similar to 2‐AG, noladin ether relaxed mesenteric arteries and this response was attenuated by 10 μM ruthenium red (Figure 5A and Table 2) or endothelial removal (Table 2). However, the relaxation was not significantly affected by HC067047 (1 μM) or RN1734 (20 μM) (Figure 5A and Table 2). The TRPV1 antagonist, SB366791 (2 μM) also had no effect (control: pEC40% = 6.3 ± 0.2, relaxation at 10 μM = 98 ± 1%, n = 4; + SB366791: pEC40% = 6.3 ± 0.3, relaxation at 10 μM = 92 ± 2%, n = 5). In the absence of an endothelium, ruthenium red (10 μM) significantly reduced relaxation to noladin ether (control: pEC40% = 5.5 ± 0.1, relaxation at 10 μM = 75 ± 3%, n = 7; + ruthenium red: relaxation at 10 μM = 44 ± 8%, n = 7; P < 0.001) but tended to potentiate 2‐AG responses (relaxation at 10 μM, control = 53 ± 11%, n = 6; + ruthenium red = 72 ± 12%, n = 7).
Figure 5.
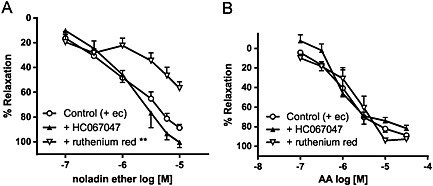
Effects of TRPV4 antagonists (1 μM HC067047 and 10 μM ruthenium red) on relaxation to (A) noladin ether and (B) arachidonic acid (AA) in endothelium‐intact mesenteric arteries. Values are shown as means and vertical bars represent SEM. The data were analysed by two‐way ANOVA, followed by Bonferroni post hoc test. **P < 0.01 in comparison with control.
Table 2.
Effects of TRPV4 antagonists on relaxant responses to noladin ether and arachidonic acid in mesenteric arteries
| pEC40% | Relaxation | n | ||
|---|---|---|---|---|
| 10 μM (%) | 30 μM (%) | |||
| Noladin ether (+ec) control | 6.2 ± 0.2 | 88 ± 4 | 10 | |
| + RN1734 | 6.1 ± 0.2 | 88 ± 4 | 5 | |
| + HC067047 | 6.1 ± 0.1 | 100 ± 4 | 4 | |
| + Ruthenium red | 5.4 ± 0.2 | 57 ± 5 | 4 ** | |
| Noladin ether (−ec) | 5.6 ± 0.1 | 61 ± 8 | 5 ** | |
| Arachidonic acid (+ec) control | 6.0 ± 0.1 | 87 ± 4 | 10 | |
| + HC067047 | 6.0 ± 0.2 | 82 ± 3 | 4 | |
| + Ruthenium red | 5.8 ± 0.1 | 93 ± 2 | 6 | |
Data represent mean ± SEM.
P < 0.01 versus corresponding control, as analysed by two‐way ANOVA of the whole data set, followed by Bonferroni post hoc tests.
In contrast, the relaxation to arachidonic acid was unaffected by both HC067047 and ruthenium red (Figure 5B and Table 2).
Effects of cytochrome P450 and lipoxygenase inhibitors on TRPV4‐mediated responses
It was previously suggested that 2‐AG indirectly activates TRPV4 through cytochrome P450 epoxygenase‐dependent metabolites (Watanabe et al., 2003). We therefore also investigated the effect of cytochrome P450 inhibitors on the mesenteric relaxation responses to GSK, 2‐AG and noladin ether. Relaxation to both 2‐AG and noladin ether was attenuated by 10 μM SKF525A, a widely used cytochrome P450 enzyme inhibitor (Figure 6A and B and Table 3). However, another cytochrome P450 inhibitor, miconazole (10 μM) had no effect (Figure 6A and B and Table 3). Interestingly, the GSK‐induced relaxation was also inhibited by SKF525A (Figure 6C and Table 3). Indeed, in human isolated coronary endothelial cells overexpressing hTRPV4, we also found that SKF525A significantly reduced the Ca2+ response to 1 μM 4α‐PDD, a synthetic TRPV4 agonist (+ SKF525A: 587 ± 493 nM, n = 3; P < 0.05).
Figure 6.
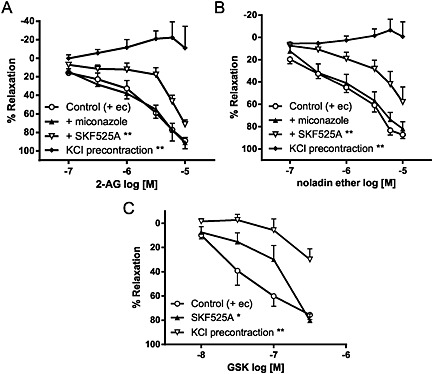
Effects of cytochrome P450 inhibitors (10 μM miconazole and 10 μM SKF525A) and KCl precontraction on relaxation to (A) 2‐AG, (B) noladin ether and (C) GSK in endothelium‐intact mesenteric arteries. Values are shown as means and vertical bars represent SEM. The data were analysed by two‐way ANOVA, followed by Bonferroni post hoc test. **P < 0.01 in comparison with control.
Table 3.
Effects of cytochrome P450 inhibitors and KCl precontraction on relaxant responses to 2‐AG, noladin ether and GSK in mesenteric arteries
| pEC40% | Relaxation | n | ||
|---|---|---|---|---|
| 10 μM (%) | 0.3 μM (%) | |||
| 2‐AG (+ec) control | 5.9 ± 0.2 | 89 ± 3 | 6 | |
| + miconazole | 5.9 ± 0.2 | 91 ± 6 | 5 | |
| + SKF525A | 5.4 ± 0.1 | 71 ± 5 | 6 ** | |
| KCl precontraction | — | −11 ± 12 | 4 ** | |
| Noladin ether (+ec) control | 6.3 ± 0.2 | 88 ± 3 | 10 | |
| + miconazole | 6.2 ± 0.3 | 82 ± 6 | 5 | |
| + SKF525A | — | 58 ± 13 | 4 ** | |
| KCl precontraction | — | −1 ± 13 | 4 ** | |
| GSK (+ec) control | 7.3 ± 0.1 | 79 ± 5 | 10 | |
| + SKF525A | 7.1 ± 0.2 | 80 ± 5 | 4 ** | |
| KCl precontraction | — | 30 ± 9 | 6 ** | |
Data represent mean ± SEM.
P < 0.01 versus corresponding control, as analysed by two‐way ANOVA of the whole data set, followed by Bonferroni post hoc tests.
Although 2‐AG also serves as a substrate for lipoxygenases (Kozak et al., 2002) and lipoxygenase products have recently been linked to TRPV4 activation (McAlexander et al., 2014), the lipoxygenase inhibitor, CDC had no significant effect on 2‐AG‐induced relaxation (control: pEC40 = 5.8 ± 0.1, relaxation at 10 μM = 90 ± 1%, n = 4; + 1 μM CDC: pEC40 = 5.7 ± 0.1, relaxation at 10 μM = 86 ± 4%, n = 4).
Relaxation mechanisms of GSK and 2‐AG in mesenteric arteries
Precontracting mesenteric arteries with high extracellular K+ (60 mM KCl), which causes smooth muscle depolarization and prevents K+ ion efflux, abolished the relaxant responses to 2‐AG and noladin ether (Figure 6A and B and Table 1), suggesting a role for K+ channel activation. The GSK‐induced relaxation was also inhibited, although a residual relaxation was seen (Figure 6C and Table 3).
Recently, endothelial TRPV4 and the subsequent increases in local [Ca2+]i have been linked to the initiation of endothelium‐derived hyperpolarization and relaxation that is independent of NO and prostanoids but is dependent on Ca2+‐activated K+ channels (Köhler et al., 2006; Sonkusare et al., 2012). Indeed, in the presence of L‐NAME (300 μM; NO synthase inhibitor) and indomethacin (10 μM; COX inhibitor), mesenteric relaxation to GSK was greatly inhibited by the KCa inhibitors, apamin and charybdotoxin (Figure 7A and Table 4). The combination of the KCa inhibitors and HC067047 had no further effect (Figure 7A and Table 4). Inhibition of gap junctions by 100 μM 18α‐glycyrrhetinic acid also attenuated the GSK responses (Figure 7A and Table 4).
Figure 7.
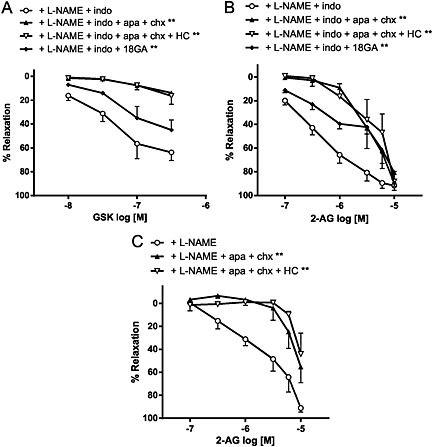
Effects of endothelium‐dependent hyperpolarization inhibitors (50 nM apamin; apa plus 50 nM charybdotoxin; chx, or 100 μM 18α‐glycyrrhetinic acid; 18GA) and selective TRPV4 antagonist (1 μM HC067047; HC) on relaxation to (A) GSK, and (B‐C) 2‐AG in endothelium‐intact mesenteric arteries. The arteries were treated with either L‐NAME (300 μM) alone, or with indomethacin (10 μM; indo), to reveal the role of endothelium‐dependent hyperpolarization. Values are shown as means and vertical bars represent SEM. The data were analysed by two‐way ANOVA, followed by Bonferroni post hoc test. **P < 0.01 in comparison with control.
Table 4.
Effects of KCa and gap junction inhibitors on relaxant responses to 2‐AG and GSK in mesenteric arteries
| pEC40% | Relaxation | n | ||
|---|---|---|---|---|
| 0.3 μM (%) | 10 μM (%) | |||
| GSK (+ec + L‐NAME + indomethacin) control | 7.2 ± 0.2 | 72 ± 4 | 4 | |
| + Apamin + charybdotoxin | — | 14 ± 1 | 4 ** | |
| + Apamin + charybdotoxin + HC067047 | — | 16 ± 7 | 4 ** | |
| +18α‐Glycyrrhetinic acid | — | 50 ± 7 | 4 ** | |
| 2‐AG (+ec + L‐NAME + indomethacin) control | 6.6 ± 0.1 | 95 ± 1 | 9 | |
| + Apamin + charybdotoxin | 5.4 ± 0.2 | 81 ± 11 | 6 ** | |
| + Apamin + charybdotoxin + HC067047 | 5.6 ± 0.3 | 85 ± 11 | 6 ** | |
| +18α‐Glycyrrhetinic acid | 5.8 ± 0.1 | 89 ± 3 | 5 ** | |
| 2‐AG (+ec + L‐NAME) control | 5.9 ± 0.2 | 92 ± 3 | 5 | |
| + Apamin + charybdotoxin | — | 55 ± 14 | 7 ** | |
| + Apamin + charybdotoxin + HC067047 | — | 62 ± 16 | 5 ** | |
Data represent mean ± SEM.
P < 0.01 versus corresponding control, as analysed by two‐way ANOVA of the whole data set, followed by Bonferroni post hoc tests.
Treatment with L‐NAME and indomethacin potentiated the relaxation to 2‐AG, the resultant relaxation was inhibited by apamin and charybdotoxin (with or without HC067047) or 18α‐glycyrrhetinic acid (Figure 7B and Table 4). Similar observations were also obtained in the presence of L‐NAME, which on its own had no effect on 2‐AG responses (Figure 7C and Table 4). It was noted that indomethacin or L‐NAME alone had no effect on the relaxation to GSK (+ec control: pEC40 = 7.6 ± 0.1, relaxation at 0.3 μM = 88 ± 5%, n = 5; + indomethacin: pEC40 = 7.7 ± 0.1, relaxation at 0.3 μM = 95 ± 2%, n = 6; + L‐NAME: pEC40 = 7.6 ± 0.1, relaxation at 0.3 μM = 85 ± 3%, n = 4).
Effects of GSK and 2‐AG in aortae
In aortae, GSK induced an endothelium‐dependent relaxation that was attenuated by HC067047 (1 μM) or abolished by ruthenium red (10 μM) (n = 4–5; Figure 8A). In contrast, 2‐AG (at 1 and 10 μM) induced small, endothelium‐dependent contractions that tended to be potentiated by MAFP and indomethacin (n = 7–9; Figure 8B). The 2‐AG‐induced contraction in the presence of MAFP and indomethacin was significantly inhibited by ruthenium red but not by HC067047 (n = 5; Figure 8B). The TxA2 receptor antagonist, ICI 192605 (1 μM) also had no effect (contractions to 10 μM 2‐AG, control: 46 ± 11%; + ICI 192605: 49 ± 19%, n = 9).
Figure 8.
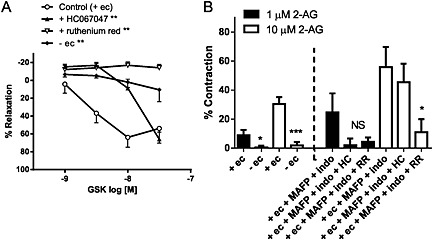
Effects of TRPV4 antagonists (1 μM HC067047; HC, or 10 μM ruthenium red; RR), or endothelium removal on (A) relaxation to GSK and (B) contraction to 2‐AG in aortae. In (B), effects of the antagonists were tested in the presence of 2‐AG metabolism inhibitors (10 μM MAFP and 10 μM indomethacin; indo). Values are shown as means and vertical bars represent SEM. The data were analysed by Student's t‐test or one‐way ANOVA, followed by Dunnett's post hoc test. *P < 0.05, ***P < 0.001 in comparison with corresponding controls.
The vehicles for GSK (up to 0.003% vv−1 DMSO) and 2‐AG (up to 0.1% vv−1 ethanol) had little effect on the precontracted tone (DMSO: 4 ± 5% relaxation, n = 4; ethanol: 2 ± 5% relaxation, n = 6).
Discussion and conclusions
The major finding of the present study is that 2‐AG activates endothelial TRPV4, which contributes to the EDH‐dependent relaxation to 2‐AG, at least in small mesenteric arteries. Overall, our data are consistent with a direct action of 2‐AG on TRPV4, and 2‐AG metabolites produced by monoacylgcerol lipase, COX, lipoxygenase or cytochrome P450 epoxygenase were shown not to be involved. However, a considerable number of vascular responses to 2‐AG are independent of TRPV4 or the endothelium. The relevance of TRPV4 to the response to 2‐AG is also highly dependent on the vascular region.
There is increasing evidence suggesting that vascular TRPV4 might act as cellular sensors that integrate diverse physical stimuli [such as shear stress: (Köhler et al., 2006; Mendoza et al., 2010); intraluminal pressure: (Bagher et al., 2012)] and chemical stimuli [EETs (Watanabe et al., 2003) and ACh: (Zhang et al., 2009)]. The TRPV4‐mediated Ca2+ entry is linked to reductions in vascular tone through NO release or activation of KCa channels. Previously, we demonstrated that TRPV4 channels are concentrated in the endothelium of mesenteric arteries and aortae (Mendoza et al., 2010), as well as coronary arteries (Bubolz et al., 2012), and TRPV4 activity contributes to endothelium‐dependent relaxation to arachidonic acid, ACh and shear stress (Zhang et al., 2009; Mendoza et al., 2010; Bubolz et al., 2012; Zheng et al., 2013). Arachidonic acid and its EET metabolites are lipid mediators that might directly activate TRPV4 (Watanabe et al., 2003; Zheng et al., 2013). Another lipid mediator, the endocannabinoid 2‐AG, which is known to induce vasorelaxation, has also been linked to TRPV4; however, the functional significance of TRPV4 in the vascular actions of 2‐AG remains speculative.
In this study, 2‐AG induced an endothelium‐dependent relaxation that was sensitive to the TRPV4 antagonists, HC067047, RN1734 and ruthenium red in rat small mesenteric arteries. A similar pharmacological profile was also observed for the potent, selective TRPV4 agonist, GSK. The potency of GSK to induce mesenteric relaxation (EC40 ~ 36 nM) is consistent with that previously reported for mouse and human TRPV4 activation (EC50 3–20 nM; Thorneloe et al., 2008; Willette et al., 2008). By comparison, 2‐AG is over 50‐fold less potent (EC40 ~ 2 μM). Nevertheless, we found that 2‐AG evoked a TRPV4 antagonist‐sensitive increase in [Ca2+]i in TRPV4‐expressing human coronary endothelial cells. This effect mimicked the responses to other TRPV4 agonists, 4α‐PDD and GSK, which we have previously shown to induce TRPV4‐mediated Ca2+ influx (Zhang et al., 2009; Zheng et al., 2013).
In the endothelium of small mesenteric arteries, TRPV4 can be found in microdomains, in which TRPV4‐mediated Ca2+ sparklets are localized with IKCa, SKCa and gap junctions between endothelial and smooth muscle cells (Saliez et al., 2008; Bagher et al., 2012; Sonkusare et al., 2012; Ma et al., 2013). As a result, TRPV4 is closely associated with EDH‐mediated relaxation that is independent of NO or prostanoid release. In this study, mesenteric relaxations to both 2‐AG and GSK were unaffected by inhibition of NO synthase but were greatly inhibited when K+ efflux was blocked by high extracellular [K+]. These findings echo the predominant role of EDH compared with NO, which is frequently reported in resistance mesenteric arteries probably due to the localization of Ca2+ microdomains near gap junctions (Edwards et al., 2010). The responses to 2‐AG and GSK, in the presence of NO and COX inhibitors, were also inhibited by apamin and charybdotoxin, which inhibit SKCa and IKCa and are commonly used to inhibit EDH (Edwards et al., 2010). This is mimicked in part by the gap junction inhibitor, 18α‐glycyrrhetinic acid, consistent with the proposal that stimulation of TRPV4 leads to activation of SKCa and IKCa in the endothelium, which in turn causes hyperpolarization of the underlying smooth muscle via myoendothelial gap junctions. In line with this, a recent study has shown that the TRPV4 agonist 4α‐PDD elicits hyperpolarization in endothelial and smooth muscle cells from rat small mesenteric arteries (Ma et al., 2013).
In addition to TRPV4, we also considered other established targets for endocannabinoids. The 2‐AG‐induced relaxation was unaffected by CB1 and CB2 receptor antagonists, excluding the involvement of these two GPCRs. Although anandamide, another major endocannabinoid, is considered to be an endogenous TRPV1 agonist (Zygmunt et al., 1999; Ho et al., 2008), it is unlikely that TRPV1 is involved in the responses to 2‐AG given that selective TRPV1 antagonists had no effect on the relaxation to 2‐AG and GSK.
On the other hand, because 2‐AG is susceptible to degradation, its metabolites could mediate some of its vascular actions. 2‐AG is hydrolysed mainly by monoacylglycerol lipase to arachidonic acid, which can be further metabolized by COX to prostanoids, or by cytochrome P450 epoxygenase to EETs (Gauthier et al., 2005). Interestingly, arachidonic acid has also been shown to activate TRPV4, either directly or indirectly via production of cytochrome P450 products EETs (Watanabe et al., 2003; Zheng et al., 2013). However, we found that TRPV4‐mediated mesenteric relaxation to 2‐AG was potentiated by monoacylglycerol lipase (MGL) and COX inhibitors, suggesting that 2‐AG responses do not require, and are in fact limited by, conversion to arachidonic acid or prostanoids. In support of this, relaxation to arachidonic acid was resistant to TRPV4 antagonists. Experiments using cytochrome P450 and lipoxygenase inhibitors also suggest that EETs or lipoxygenase metabolites of 2‐AG are unlikely to be involved in the observed relaxation. In fact, we found that a commonly used cytochrome P450 inhibitor, SKF525A also reduced GSK relaxation and Ca2+ responses, and may therefore have a direct effect on TRPV4. So, the results should be interpreted with caution if used to implicate EETs in TRPV4 activation. To explore if 2‐AG directly activates TRPV4, we further examined the effect of 2‐AG on [Ca2+]i and TRPV4 channel activity in TRPV4‐expressing human coronary endothelial cells. 2‐AG elevated [Ca2+]i even after inhibition of MGL by MAFP, suggesting a direct action on TRPV4. Nevertheless, it was noted that MAFP tended to attenuate the 2‐AG response; such differential effects of MAFP in isolated arteries and endothelial cells might be explained by inhibition of additional 2‐AG hydrolysis in vascular smooth muscle cells (Ho and Randall, 2007). In inside‐out patch‐clamping experiments, bath application of 2‐AG (and thus exposing the cytosolic side of the channels to 2‐AG) increased the opening probability of TRPV4 in the endothelial cells. Interestingly, we observed that 2‐AG was more effective in inside‐out compared with cell‐attached patches, perhaps reflecting difficulties in accessing the channels from outside the cell. The 2‐AG‐induced single‐channel currents resembled those stimulated by GSK. Our data therefore provide strong evidence that 2‐AG directly activates TRPV4, although we cannot exclude the involvement of some 2‐AG metabolites.
It is, however, clear that the functional relevance of TRPV4 in the vascular actions of 2‐AG is complex. In small mesenteric arteries, while endothelial removal almost abolished GSK responses, about 50% of 2‐AG relaxation persisted in endothelium‐denuded vessels, highlighting the presence of relaxation mechanisms unrelated to endothelial TRPV4. Similarly, inhibition of the EDH pathway largely inhibited the GSK relaxation, whereas a considerable residual relaxation (about 50%) to 2‐AG was observed. Together, these data suggest that activation of endothelial TRPV4 and subsequently the EDH pathway only partly explains the mesenteric relaxation elicited by 2‐AG, with the remaining component that appears independent of TRPV4 or the endothelium remains undefined. This is perhaps related to its lower potency/efficacy at TRPV4 compared with synthetic TRPV4 activators, such as 4α‐PDD and GSK, as evident in our Ca2+ measurements and single‐channel recordings. The presence of other relaxation pathways could also be a factor. Because blockade of K+ efflux but not the KCa inhibitors, apamin and charybdotoxin abolished the mesenteric relaxation to 2‐AG, 2‐AG might activate other K+ channel subtypes in the vascular smooth muscle. However, as discussed earlier, cannabinoid receptors, TRPV1 and 2‐AG metabolites produced by MGL, COX, lipoxygenase or cytochrome P450 epoxygenase are unlikely to be involved. Curiously, although the non‐hydrolysable analogue of 2‐AG, noladin ether also induced TRPV4‐mediated increases in [Ca2+]i in endothelial cells, TRPV4 has no significant role in its mesenteric relaxation. Ruthenium red, which inhibits TRPV4 and other TRP channels, reduced the endothelium‐independent relaxation to noladin ether but not to 2‐AG. Nonetheless, a selective TRPV1 antagonist had no effect. We therefore propose that TRP subtypes apart from TRPV4 and TRPV1 play a larger functional role in noladin ether relaxation.
The disconnection between the abilities of compounds to activate TRPV4 and induce TRPV4‐mediated relaxation is most strikingly demonstrated in the aortae. In contrast to the TRPV4‐mediated relaxation induced by GSK, 2‐AG induced further contractions of the precontracted aortae, despite inhibition of its hydrolase and COX. Several studies have suggested that the TRPV4 channel is also found in the vascular smooth muscle and might induce contractions especially at higher concentrations of GSK (Earley et al., 2005; Sukumaran et al., 2013). Recently, Saifeddine et al. (2015) reported that, in mouse aorta, endothelial TRPV4 activity can stimulate the release of contractile COX products, which activate Tx receptors, causing aortic contractions. However, in this study, the 2‐AG‐mediated contraction was not reduced by a COX inhibitor or selective antagonists for TRPV4 and the TxA2 receptor. It is possible that in the mesenteric artery and aorta the phosphorylation states of TRPV4 (Fan et al., 2009; Zheng et al., 2013) or the production of as yet‐to‐be identified 2‐AG metabolites are different, which might determine the role of TRPV4 in 2‐AG actions in different vascular regions. Indeed, initial experiments suggest that PKA activation potentiates mesenteric relaxation to 2‐AG, but not GSK, in an endothelium‐dependent fashion (Supporting Information Figure S3). We therefore surmise that, although GSK may already achieve maximal TRPV4 activation in mesenteric arteries, the contribution of TRPV4 to 2‐AG responses is more dependent on phosphorylation‐mediated regulation of the channel or 2‐AG metabolism. This aspect will be the subject of further studies.
Overall, we have provided evidence that 2‐AG directly activates TRPV4, which mediates the endothelial component of 2‐AG‐induced relaxation in rat small mesenteric arteries. Mesenteric relaxation to 2‐AG is not mediated by 2‐AG metabolites via hydrolysis, COX, lipoxygenase or cytochrome P450. Importantly, however, endothelial TRPV4 cannot fully explain the vascular actions of 2‐AG and the role of TRPV4 is highly dependent on the vascular region, inasmuch as 2‐AG does not induce TRPV4‐mediated relaxation in rat aortae. Further investigations are required to clarify the factors that determine the regional heterogeneity in TRPV4 activation by 2‐AG.
Author contributions
W. S. V. H. conceived and designed the study, performed experiments, analysed data and wrote the paper. X. Z. performed some of the experimental work and data analysis. D. X. Z. designed the study, analysed some of the data and wrote the paper.
Conflict of interest
None.
Supporting information
Figure S1 (A) Original traces showing the effects of vehicle for GSK (0.001‐0.03% vv‐1 DMSO) in endothelium‐intact and endothelium‐denuded mesenteric arteries precontracted with 10 µM methoxamine (methox). (B) Original traces showing the relaxant effects of GSK, in the absence and presence of a TRPV4 antagonist (1 µM HC067047, 20 µM RN1734 or 10 µM ruthenium red).
Figure S2 (A) Original traces showing the effects of vehicle for 2‐AG (0.001‐0.1% vv‐1 ethanol) in endothelium‐intact and endothelium‐denuded mesenteric arteries precontracted with 10 µM methoxamine (methox). (B) Original traces showing the relaxant effects of 2‐AG, in the absence and presence of a TRPV4 antagonist (1 µM HC067047, 20 µM RN1734 or 10 µM ruthenium red).
Figure S3 Effects of 8‐Br‐cAMP (1 µM; PKA activator) on relaxation to (A) 2‐AG and (B) GSK in endothelium‐intact and endothelium‐denuded mesenteric arteries. n = 4‐5. Values are shown as means and vertical bars represent s.e.mean. The data were analysed by two‐way ANOVA, followed by Bonferroni post‐hoc test. **P < 0.01 in comparison to control.
Supporting info item
Acknowledgements
We are thankful for the technical support from Nimesh Parmar at St George's University of London, and the financial support from the National Heart, Lung, and Blood Institute at the National institutes of Health, USA.
Ho, W. S. V. , Zheng, X. , and Zhang, D. X. (2015) Role of endothelial TRPV4 channels in vascular actions of the endocannabinoid, 2‐arachidonoylglycerol. British Journal of Pharmacology, 172: 5251–5264. doi: 10.1111/bph.13312.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013). The concise guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagher P, Beleznai T, Kansui Y, Mitchell R, Garland CJ, Dora KA (2012). Low intravascular pressure activates endothelial cell TRPV4 channels, local Ca2+ events, and IKCa channels, reducing arteriolar tone. Proc Natl Acad Sci U S A 109: 18174–18179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubolz AH, Mendoza SA, Zheng X, Zinkevich NS, Li R, Gutterman DD et al. (2012). Activation of endothelial TRPV4 channels mediates flow‐induced dilation in human coronary arterioles: role of Ca2+ entry and mitochondrial ROS signaling. Am J Physiol Heart Circ Physiol 302: H634–H642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards G, Félétou M, Weston AH (2010). Endothelium‐derived hyperpolarising factors and associated pathways: a synopsis. Pflugers Arch 459: 863–879. [DOI] [PubMed] [Google Scholar]
- Earley S, Heppner TJ, Nelson MT, Brayden JE (2005). TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BKCa channels. Circ Res 97: 1270–1279. [DOI] [PubMed] [Google Scholar]
- Fan HC, Zhang X, McNaughton PA (2009). Activation of the TRPV4 ion channel is enhanced by phosphorylation. J Biol Chem 284: 27884–27891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filosa JA, Yao X, Rath G (2013). TRPV4 and the regulation of vascular tone. J Cardiovasc Pharmacol 61: 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier KM, Baewer DV, Hittner S, Hillard CJ, Nithipatikom K, Reddy DS et al. (2005). Endothelium‐derived 2‐arachidonylglycerol: an intermediate in vasodilatory eicosanoid release in bovine coronary arteries. Am J Physiol Heart Circ Physiol 288: H1344–H1351. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY (1985). A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260: 3440–3450. [PubMed] [Google Scholar]
- Hartmannsgruber V, Heyken WT, Kacik M, Kaistha A, Grgic I, Harteneck C et al. (2007). Arterial response to shear stress critically depends on endothelial TRPV4 expression. PLoS One 2 e827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho WS, Barrett DA, Randall MD (2008). ‘Entourage’ effects of N‐palmitoylethanolamide and N‐oleoylethanolamide on vasorelaxation to anandamide occur through TRPV1 receptors. Br J Pharmacol 155: 837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho WS, Randall MD (2007). Endothelium‐dependent metabolism by endocannabinoid hydrolases and cyclooxygenases limits vasorelaxation to anandamide and 2‐arachidonoylglycerol. Br J Pharmacol 150: 641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Járai Z, Wagner JA, Goparaju SK, Wang L, Razdan RK, Sugiura T et al. (2000). Cardiovascular effects of 2‐arachidonoyl glycerol in anesthetized mice. Hypertension 35: 679–684. [DOI] [PubMed] [Google Scholar]
- Kagota S, Yamaguchi Y, Nakamura K, Sugiura T, Waku K, Kunitomo M (2001). 2‐Arachidonoylglycerol, a candidate of endothelium‐derived hyperpolarizing factor. Eur J Pharmacol 415: 233–238. [DOI] [PubMed] [Google Scholar]
- Kozak KR, Gupta RA, Moody JS, Ji C, Boeglin WE, DuBois RN et al. (2002). 15‐Lipoxygenase metabolism of 2‐arachidonylglycerol. Generation of a peroxisome proliferator‐activated receptor alpha agonist. J Biol Chem 277: 23278–23286. [DOI] [PubMed] [Google Scholar]
- Köhler R, Heyken WT, Heinau P, Schubert R, Si H, Kacik M et al. (2006). Evidence for a functional role of endothelial transient receptor potential V4 in shear stress‐induced vasodilatation. Arterioscler Thromb Vasc Biol 26: 1495–1502. [DOI] [PubMed] [Google Scholar]
- Ma X, Du J, Zhang P, Deng J, Liu J, Lam FF et al. (2013). Functional role of TRPV4‐KCa2.3 signaling in vascular endothelial cells in normal and streptozotocin‐induced diabetic rats. Hypertension 62: 134–139. [DOI] [PubMed] [Google Scholar]
- McAlexander MA, Luttmann MA, Hunsberger GE, Undem BJ (2014). Transient receptor potential vanilloid 4 activation constricts the human bronchus via the release of cysteinyl leukotrienes. J Pharmacol Exp Ther 349: 118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza SA, Fang J, Gutterman DD, Wilcox DA, Bubolz AH, Li R et al. (2010). TRPV4‐mediated endothelial Ca2+ influx and vasodilation in response to shear stress. Am J Physiol Heart Circ Physiol 298: H466–H476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B, Vriens J, Prenen J, Droogmans G, Voets T (2004). TRPV4 calcium entry channel: a paradigm for gating diversity. Am J Physiol Cell Physiol 286: C195–C205. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al. (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledgebase of drug targets and their ligands. Nucl Acids Res 42 (Database Issue): D1098–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR et al. (2010). International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB₁ and CB2 . Pharmacol Rev 62: 588–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saifeddine M, El‐Daly M, Mihara K, Bunnett NW, McIntyre P, Altier C et al. (2015). GPCR‐mediated EGF receptor transactivation regulates TRPV4 action in the vasculature. Br J Pharmacol 172: 2493–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliez J, Bouzin C, Rath G, Ghisdal P, Desjardins F, Rezzani R et al. (2008). Role of caveolar compartmentation in endothelium‐derived hyperpolarizing factor‐mediated relaxation: Ca2+ signals and gap junction function are regulated by caveolin in endothelial cells. Circulation 117: 1065–1074. [DOI] [PubMed] [Google Scholar]
- Stanke‐Labesque F, Mallaret M, Lefebvre B, Hardy G, Caron F, Bessard G (2004). 2‐Arachidonoyl glycerol induces contraction of isolated rat aorta: role of cyclooxygenase‐derived products. Cardiovasc Res 63: 155–160. [DOI] [PubMed] [Google Scholar]
- Stanley CP, O'Sullivan SE (2014). Cyclooxygenase metabolism mediates vasorelaxation to 2‐arachidonoylglycerol (2‐AG) in human mesenteric arteries. Pharmacol Res 81: 74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumaran SV, Singh TU, Parida S, Narasimha Reddy CE, Thangamalai R, Kandasamy K et al. (2013). TRPV4 channel activation leads to endothelium‐dependent relaxation mediated by nitric oxide and endothelium‐derived hyperpolarizing factor in rat pulmonary artery. Pharmacol Res 78: 18–27. [DOI] [PubMed] [Google Scholar]
- Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ et al. (2012). Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 336: 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorneloe KS, Sulpizio AC, Lin Z, Figueroa DJ, Clouse AK, McCafferty GP et al. (2008). N‐((1S)‐1‐{[4‐((2S)‐2‐{[(2,4‐dichlorophenyl)sulfonyl]amino}‐3‐hydroxypropanoyl)‐1‐piperazinyl]carbonyl}‐3‐methylbutyl)‐1‐benzothiophene‐2‐carboxamide GSK1016790A, a novel and potent transient receptor potential vanilloid 4 channel agonist induces urinary bladder contraction and hyperactivitiy: part 1. J Pharmacol Exp Ther 326: 432–442. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B (2003). Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature 424: 434–438. [DOI] [PubMed] [Google Scholar]
- Willette RN, Bao W, Nerurkar S, Yue TL, Doe CP, Stankus G et al. (2008). Systemic activation of the transient receptor potential vanilloid subtype 4 channel causes endothelial failure and circulatory collapse: part 2. J Pharmacol Exp Ther 326: 443–452. [DOI] [PubMed] [Google Scholar]
- Zhang DX, Mendoza SA, Bubolz AH, Mizuno A, Ge ZD, Li R et al. (2009). Transient receptor potential vanilloid type 4‐deficient mice exhibit impaired endothelium‐dependent relaxation induced by acetylcholine in vitro and in vivo. Hypertension 53: 532–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Zinkevich NS, Gebremedhin D, Gauthier KM, Nishijima Y, Fang J et al. (2013). Arachidonic acid‐induced dilation in human coronary arterioles: convergence of signaling mechanisms on endothelial TRPV4‐mediated Ca2+ entry. J Am Heart Assoc 2 e000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sørgård M, Di Marzo V et al. (1999). Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 400: 452–457. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (A) Original traces showing the effects of vehicle for GSK (0.001‐0.03% vv‐1 DMSO) in endothelium‐intact and endothelium‐denuded mesenteric arteries precontracted with 10 µM methoxamine (methox). (B) Original traces showing the relaxant effects of GSK, in the absence and presence of a TRPV4 antagonist (1 µM HC067047, 20 µM RN1734 or 10 µM ruthenium red).
Figure S2 (A) Original traces showing the effects of vehicle for 2‐AG (0.001‐0.1% vv‐1 ethanol) in endothelium‐intact and endothelium‐denuded mesenteric arteries precontracted with 10 µM methoxamine (methox). (B) Original traces showing the relaxant effects of 2‐AG, in the absence and presence of a TRPV4 antagonist (1 µM HC067047, 20 µM RN1734 or 10 µM ruthenium red).
Figure S3 Effects of 8‐Br‐cAMP (1 µM; PKA activator) on relaxation to (A) 2‐AG and (B) GSK in endothelium‐intact and endothelium‐denuded mesenteric arteries. n = 4‐5. Values are shown as means and vertical bars represent s.e.mean. The data were analysed by two‐way ANOVA, followed by Bonferroni post‐hoc test. **P < 0.01 in comparison to control.
Supporting info item