

Abstract
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal neoplasms of gastrointestinal tract. They feature heterogeneous triggering mechanisms, implying relevant clinical differences. The vast majority of GISTs are sporadic tumors. Rarely, however, GIST-prone syndromes occur, mostly depending on heritable GIST predisposing molecular defects involving the entire organism. These conditions need to be properly identified in order to plan appropriate diagnostic, prognostic and therapeutic procedures.
Clinically, GIST-prone syndromes must be thought of whenever GISTs are multiple and/or associated with accompanying signs peculiar to the background tumorigenic trigger, either in single individuals or in kindreds. Moreover, syndromic GISTs, individually considered, tend to show distinctive features depending on the underlying condition. When applicable, genotyping is usually confirmatory.
In GIST-prone conditions, the prognostic features of each GIST, defined according to the criteria routinely applied to sporadic GISTs, combine with the characters proper to the background syndromes, defining peculiar clinical settings which challenge physicians to undertake complex decisions. The latter concern preventive therapy and single tumor therapy, implying possible surgical and molecularly targeted options.
In the absence of specific comprehensive guidelines, this review will highlight the traits characteristic of GIST-predisposing syndromes, with particular emphasis on diagnostic, prognostic and therapeutic implications, which can help the clinical management of these rare diseases.
Keywords: Syndromic GIST, Hereditary GIST, Familial GIST, PDGFRA-mutant syndrome, Carney’s triad, Carney-Stratakis syndrome, Succinate dehydrogenase, germline mutations
Background
Gastrointestinal (GI) stromal tumors (GISTs) are the most common GI mesenchymal neoplasms [1, 2]. They generally express KIT (CD117) and DOG1, similar to interstitial cells of Cajal (ICC) [3, 4].
GISTs have been a paradigm of molecular targeted therapy since they revealed activating KIT mutations [5], leading to the successful employment of the tyrosin kinase (TK) inhibitor (TKI) imatinib [6]. Since then, besides KIT mutations (~¾ of cases), GISTs have revealed other possible triggers: platelet-derived growth factor (PDGF) receptor α (PDGFRA) mutations (~7 %), succinate dehydrogenase (SDH) complex deficiency (~5 %, half of which depending on mutations of SDH subunits), and mutations of BRAF (2 %) or neurofibromatosis type 1 (NF1) (1, 5 %). This heterogeneity implies different pathogenic, diagnostic and prognostic features characterizing distinct GIST subgroups, entailing diverse therapeutic approaches [1].
All of these pathogenic mechanisms but BRAF mutations have been rarely described to involve the entire organism, causing syndromes featuring multiple GISTs and peculiar associated signs. The resulting repertoire of syndromic GISTs constitute ~3-4 % of GISTs. Within them, NF1-associated GISTs prevail [1]. Much rarer are GISTs hinging upon germline KIT or PDGFRA mutations, with <50 kindreds/individuals described [7–48].
An overall favorable prognosis has been attributed to GISTs when multiple (including syndromic ones), no matter their number/phenotype [27]. However, despite the relatively high fraction of indolent GISTs due to NF1 or SDH-deficiency, “multiple GISTs” is a crucible where heterogeneous conditions merge in, differing in pathogenesis, prognosis and therapy. The approach to syndromic GISTs must therefore be personalized, considering the characters of both individual tumors, influencing their own natural history, and of the background syndrome, defining peculiar clinical settings.
GIST-predisposing syndromes will be herein reviewed, emphasizing inherent diagnostic, prognostic and therapeutic implications. Additionally, pertinent fundamentals of GIST pathogenesis will be recalled.
Features of GIST-predisposing syndromes
KIT mutant syndrome
KIT is a transmembrane type III TK receptor (TKR), whose gene is mapped to 4q12. Physiologically, KIT activation follows homodimerization upon stem cell factor (SCF) binding. Mutated KIT homodimerizes in a ligand-independent way. Activated KIT initiates RAS/MAPK, PI3K/AKT/mTOR and JAK/STAT3 signaling [49–51] (Fig. 1).
Fig. 1.
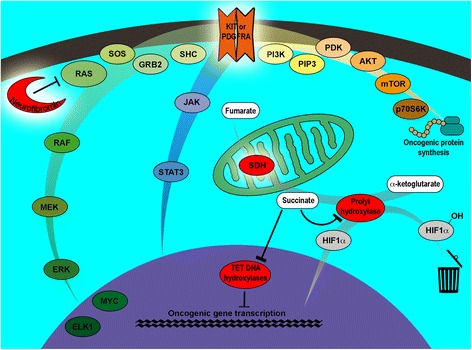
Molecular triggers and intracellular pathways involved in syndromic GIST arousal. Syndromic GISTs reported so far hinge upon alterations of one of the following (evidenced with a halo): KIT, PDGFRA, neurofibromin or SDH. KIT and PDGFRA activation initiates a downstream signaling involving multiple pathways: RAS/RAF/MEK/ERK (MAPK) (left, green hue); JAK/STAT3 (centre, blue hue); and PI3K/AKT/mTOR (top right, yellow/brown hue), stimulating oncogenic gene transcription or protein synthesis. In NF1-associated GISTs, tumoral inactivation of the WT neurofibromin impairs its RAS inhibiting effect, resulting in the activation of MAPK cascade downstream to KIT and PDGFRA. Impairment of the SDH enzymatic complex prevents succinate conversion to fumarate. Accumulated succinate inhibits prolyl-hydroxylase; the missed hydroxylation of HIF1-α prevents the degradation of this molecule which, consequently, heterodimerizes with HIF1-β and translocates into the nucleus acting as an oncogenic transcription factor. Furthermore, succinate accumulation inhibits TET DNA hydroxylases resulting in impaired conversion of 5-methylcytosine to 5-hydroxymethylcytosine, required for DNA demethylation, thereby influencing gene expression
At the best of my knowledge, germline KIT-mutant GISTs have been described in 31 kindreds and 6 individuals without familial history [7–44]. Additionally, ungenotyped kindreds with signs coherent with KIT-dependent familial GISTs have been described prior to the discovery of KIT role in GISTs [52–55].
Average age-at-diagnosis of germline KIT-dependent GISTs anticipates that of sporadic cases of ~10 years: late forties/early fifties versus early/middle sixties [20, 29, 56–58]. This is true also if, in case of KIT-mutant kindreds, only the first individuals diagnosed with GIST (or GIST-compatible tumors in “pre-KIT era”) are considered, resulting in a 48-year mean, not influenced by familial screening. This anticipation likely parallels the early GIST trigger intrinsic to the connatal KIT mutation. The probability of GIST diagnosis in germline KIT-mutants increases with age, raising from 0.077 before the age of 40 to 0.462 by the age of 50 [29]. The age-at-diagnosis decrease reported in successive generations [17] is possibly due to subclinical GISTs diagnosed at screening.
KIT-mutant syndrome features no sex predilection, as expected given its autosomal dominant inheritance. Penetrance for GISTs is high [13, 20], unlike that for altered skin pigmentation [29].
Familial KIT-mutant GISTs occur along the whole GI tract (especially in small bowel/stomach), featuring a spindle, epithelioid or spindle-and-epithelioid citology (with the former prevailing), commonly expressing CD117 and DOG1 (Table 1), similar to their sporadic counterparts.
Table 1.
Kindreds and individuals affected by gastrointestinal stromal tumors associated with germline KIT mutations
| Reference | Type of report (family vs. single individual) | Mutant exon (mutation) | GIST | Other manifestations | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sitea | Histologyb | Mc | ICCHd | Altered skin pigmentation | Mast cell disorders | GI motility disorders | Diverticula | Others | |||
| Nishida et al. [7] | family | 11 (p.V560del) | SIe | S, Mf | Perineal hyperpigmentation | ||||||
| O’Brien et al. [8]; Hirota et al. [9]; Chen et al. [10] | family | 11 (p.W557R) | SI | S, M | yes | yes | |||||
| Isozaki et al. [11]; Handra-Luca et al. [12]; Bachet et al. [13] | family | 13 (p.K642E) | ST, SI | S, M | yes | yes | Lentigines on trunk, limbs, palms and soles | Dysphagia | |||
| Maeyama et al. [14] | family | 11 (p.V559A) | ST, SI | S | Hyperpigmentation and nevi | ||||||
| Beghini et al. [15] | family | 11 (p.V559A) | ST, SI | S, E, M | yes | yes | Hyperpigmentation of face, trunk, extremities and mucous membranes | Urticaria pigmentosa | |||
| Hirota et al. [16] | family | 17 (p.D820Y) | ST, SI | Mf | yes | Dysphagia | |||||
| Robson et al. [17] | family | 11 (p.W557R) | ST, SI | S, M | yes | Hyperpigmentation of hands, knees, perineum and circumoral areas | Dysphagia | Small bowel | |||
| Antonescu et al. [18] | individual | 11 (p.W557R) | ST, SI | S | yes | ||||||
| Carballo et al. [19] | family | 11 (p.L756_P577InsQL) | ST, SI | S | yes | Hyperpigmentation of neck, hands, feet and circumoral area | |||||
| Li et al. [20] | family | 11 (p.V559A) | ST, SI | Sg | yes | Hyperpigmentation, lentigines, café-au-lait maculesh, nevi (all these lesions variably involved neck, perioral area, scrotal region/pelvic/genital/inguinal area, axillae, buttocks); vitiligo | Urticaria pigmentosa | 1 Melanoma, 1 angioleiomyoma of ankle skin | |||
| Tarn et al. [21] | family | 11 (p.D579del) | ST | M | yes | ||||||
| Hartmann et al. [22] | family | 8 (p.D419del) | SI | M | yes | Mastocytosis | Dysphagia | ||||
| Kim et al. [23] | individual | 11 (p.V559A) | SI | S, M | yes | ||||||
| O’Riain et al. [24] | family | 17 (p.D820Y) | ST, SI, ICV | Mf, S | yes | yes | Dysphagia | Small bowel | |||
| Miettinen et al. [25]; Lasota and Miettinen [26] | family | 11 (p.D579del) | SI, A, C | NS | |||||||
| Kang et al. [27] | family | 11 (p.V560G) | SI | NS | yes | Hyperpigmentation | |||||
| Kang et al. [27] | individual | 11 (p.V559A) | SI | NS | yes | ||||||
| Graham et al. [28] | individual | 13 (p.K642E) | E, ST, SI, R | NS | yes | Vitiligo | |||||
| Kleinbaum et al. [29] | family | 11 (c.D579del) | ST, SI, C | S | yes | yes | Hyperpigmentation, nevi | ||||
| Woźniak et al. [30] | family | 11 (p.Q575_577delinsH) | R | S, M | yes | Constipation | |||||
| Thalheimer et al. [31] | family | 17 (p. N822Y) | ST, SI, A, R | S | yes | yes | |||||
| Campbell et al. [32] | family | 11 (NSi) | ST, SI, C | NS | Dysplastic nevi, lentigines, darkening of labia minora pudendi | ||||||
| Veiga et al. [33] | family | 17 (p.D820Y) | ST, SI, R | NS | yes | 1 endometrial stromal sarcoma | |||||
| Kuroda et al. [34] | family | 11 (p. V559A) | ST, SI, C | S | yes | Hyperpigmentation of external genitalia and axilla | |||||
| Vilain et al. [35] | family | 13 (p. K642E) | ST, SI | Sf | yes | Hyperpigmentation (multiple nevi in the axillae and trunk and spontaneously resolving childhood facial hyperpigmentation) and hypopigmentation consistent with WSj type 2 | Dysphagia | Oesophagus | |||
| Nakai et al. [36] | family | 11 (p. Y553K) | ST, SI, C | NS | yes | ||||||
| Wadt et al. [37] | family | 13 (p.K642E) | ST, SI | NS | yes | yes | 1 breast cancer | ||||
| Speight et al. [38] | individual | 9 (p.K509I) | SI | S | Mastocytosis | ||||||
| Bachet et al. [13] | family | 13 (p.K642E) | ST, SI, C, R | S | yes | yes | Lentigines and nevi | Multiple cutaneous angiolypomas in one individual | |||
| Bachet et al. [13] | family | 13 (p.K642E) | ST, SI | S | yes | ||||||
| Neuhann et al. [39] | family | 11 (p.L576P) | ST, SI, C | S | yes | Multiple lentigines on face, neck, chest, back, axillae, legs | Achalasia, dysphagia | ||||
| Yamanoi et al. [40] | individual | 13 (p.K642T) | ST, SI | S | yes | Dyshpagia | |||||
| Adela Avila et al. [41] | family | 11 (p.V559A) | SI | S | Diffuse melanosis, generalized lentiginosis, palmar crease hyperpigmentation | Dysphagia | |||||
| Jones et al. [42] | family | 11 (p.D579del) | ST, SI | S | |||||||
| Jones et al. [42] | family | 11 (p.D579del) | ST, SI | S | |||||||
| Bamba et al. [43] | family | 11 (p.V560del) | ST, SI | S | |||||||
| Forde et al. [44] | family | 11 (p.D579del) | ST, SI | NS | Skin hyperpigmentation | Dysphagia | |||||
a E esophagus, ST stomach, SI small intestine, ICV ileocecal valve, A appendix, C colon, R rectum, NS not specified
b S spindle cell, E epithelioid, M mixed spindle cell and epithelioid, NS not specified
c M, GIST metastases
d Diffuse Intersitial cell of Cajal hyperplasia
e inferred from the mention of intestinal obstruction
f inferred from published microphotographs
g not specified in the referred paper; inferred from the diagnosis of “neurofibromatosis” previously made in several of the family members
h café-au-lait macules were reported in one of the individuals originally thought to have neurofibromatosis; this could have influenced the term used
i NS, not specified
j Waardenburg syndrome
As shown in Table 1, other features of germline KIT mutants include: diffuse ICC hyperplasia (ICCH) (with related peristalsis disturbances), skin pigmentation alterations and mast-cell disorders. Sporadically, melanoma, non-GIST stromal tumors and breast cancer have been signaled. KIT mutations could be implied in the arousal of the former [49]; the latter two are likely incidental. Sometimes, non-GIST signs are the first reason for patients to seek medical help [15, 32, 35, 41, 44].
Skin pigmentation defects mostly occur in excess; nevertheless, hypopigmentation has been reported [28, 35]. Of note, lentigines and vitiligo can coexist [20]. Finally, pigmentation alterations consistent with Waardenburg syndrome type 2 have also been reported in the absence of pathognomonic mutations, candidating the detected KIT variant among the possible causes of this disease [35].
The role of KIT in ICC development and gut motility regulation, and in the development and neoplastic transformation of melanocytes and mast cells [49], explains some of the germline KIT-mutants’ features. Conversely, the variable manifestations of these signs have not been satisfactorily justified. In particular, the suggested association between dysphagia and KIT TK II domain mutations and the inability of KIT TK I domain mutations to affect skin pigmentation [11, 16] proved wrong [13, 28, 35]. Acute myeloid leukemia, seminoma/dysgerminoma and sinonasal NK/T-cell lymphoma, neoplasms related to KIT mutations (often in exon 17) [49], at the best of my knowledge have never been described in germline KIT-mutants (curiously, some familial germ-cell tumors revealed somatic KIT mutations [59]). K559I and D816V KIT mutations, found in GISTs, can cause familial mastocytosis without detectable GISTs [38, 60–63]. Thus, genotype-phenotype correlation appears loose [39].
KIT mutations in sporadic GISTs cluster in exons 11, 9, 13, 17 and 8, with a frequency of ~65 %, ~8 %, ~1 %, ~1 % and < <1 %, respectively [2, 64]. In germline mutants, however, the KIT mutational spectrum differs (Table 1). In fact, focusing each single GIST oncogenic mutation arousal (counting each KIT-mutant kindred as one): 1) mutations involve exon 9 in 1/37 (3 %) and exon 17 in 4/37 (11 %) cases; 2) among exon-11 mutations, substitutions (61 %) prevail over deletions and insertion/deletions (29 %) (reversing the 31 %/60 % proportion found in sporadic GISTs); 3) KIT exon-11 mutations appear enriched in p.V559A, p.W557R and p.L576P substitutions (48 %, versus 10 % of sporadic GISTs) [58]. These differences suggest a selection of favorable genotypes in germline mutants, possibly less life-threatening, because: 1) exon-9 mutations proved aggressive in imatinib-naive GISTs; 2) exon-17 mutations appear more favorable than exon-11 ones [65]; 3) 5’-end exon-11 mutations, especially deletions, are likely biologically severe [30]; 4) sporadic GISTs with exon-11 substitutions or duplications revealed smaller than those with exon-11 deletions, and the former featured lower mitotic rates, supporting a lower biological impact of exon-11 substitutions; 5) GISTs bearing p.V559A, p.W557R and p.L576P mutations tend to feature better relapse-free survivals [58].
The first step of GIST tumoral progression in germline KIT-mutants is ICCH. “ICCH” and “micro-GIST” are terms applied to a variety of microscopic/tiny CD117+ cell lesions. Despite gross dimensional criteria have been proposed for separating them [66], a distinction between diffuse (ICCH) and nodular/focal (micro-GISTs) lesions prevails [2, 50]. Thus defined, ICCH is a non-neoplastic polyclonal lesion [10], constituting the only known GIST precursor. Subsequent events leading to overt tumors follow those found in sporadic GISTs: chromosomes 14 and 22 deletions [20], and loss of heterozigosity (LOH) involving the KIT wild-type (WT) allele at progression to malignancy [29]. The oncological impact of individual syndromic KIT-mutant GISTs is estimated using the parameters adopted for their sporadic counterparts [67]. Besides, germline KIT-mutant subjects may suffer frequent/severe gut occlusion/hemorrage, due to the tumor numerousness.
KIT mutations alter KIT structure simulating SCF-binding induced activation (if in exons 8 and 9, coding for the extracellular, ligand-binding domain), or allowing the kinase activation loop to switch to activation (exon 11, juxtamembrane regulatory domain), or directly imparting an active conformation to TK domains (exons 13 and 17, intracellular ATP-binding region and activation loop, respectively). This explains the differences in imatinib sensitivity depending on the KIT-mutant exon [50], either germline or not. Relatively better results are achieved when mutations occur “upstream” to the imatinib targeted site (i.e. in KIT exons 8, 9 and 11; for the rare exon 8 mutations evidences are limited [22, 64]), while the highest resistance rates are found in exon-13 and 17 mutations [50].
Table 1 details the features of the published germline KIT mutant kindreds/individuals manifesting GISTs.
PDGFRA-mutant syndrome
PDGFRA is a type III TKR whose gene is mapped to 4q12 [51], probably sharing with KIT a common ancestor [68]. PDGFRA is physiologically activated through binding to all PDGFs except PDGF-DD [69], triggering the same pathways elicited by KIT [50] (Fig. 1), albeit differentially activating PI3K/AKT/mTOR over RAS/MAPK [70]. Coherently, activating mutations of PDGFRA and KIT can raise similar tumors and are usually mutually exclusive.
At the best of my knowledge, germline PDGFRA-mutant GISTs have been signaled in two kindreds and in an individual without familial history [45–48]. A PDGFRA-mutant family bearing intestinal tumors defined as GISTs, but lacking GIST hallmarks except PDGFRA status, has also been reported [71]; these tumors are probably fibrous tumors, possible variants of another GI PDGFRA-driven tumor: inflammatory fibroid polyp (IFP) [48]. Moreover, a germline PDGFRA-mutant individual bearing multiple IFPs (and a likely PDGFRA-unrelated GIST hosting a somatic KIT mutation) has been reported very recently [72]; this individual shares both germline defect and geographical origin with one of the above mentioned kindreds [48], and is therefore probably related to it. Finally, three families featuring multiple IFPs have been described without PDGFRA genotyping [73–76]. PDGFRA-mutant syndrome is the term proposed for defining this clinical spectrum [48], formerly termed “intestinal neurofibromatosis/neurofibromatosis 3b” [71, 77, 78].
First diagnoses of IFP (including tumors misdiagnosed as neurofibromas) tend to precede those of GIST (means 40.6 and 48.1 years, respectively, p = 0.12 -Mann–Whitney U Test-) in PDGFRA-mutant syndrome, suggesting a faster IFP tumorigenesis (of note, IFP and GIST are unrelated lesions) [45, 47, 48, 71, 72, 78].
Although PDGFRA-mutant syndrome features an autosomal dominant pattern of inheritance with high penetrance [48], the sex distribution of GI tumors is apparently unbalanced, with a 4:11 male-to-female ratio (7/15 considering also pathologically undiagnosed GI tumors). Affected females outnumber males (11:1) also in ungenotyped kindreds featuring familial IFPs or “intestinal neurofibromatosis/NF3b” (12:2 including suspected GI tumors) [45–48, 71–78]. However, females prevail (14:5) also among the 19 ascertained PDGFRA-mutants, compensating the corresponding 10:4 female-to-male tumor distribution (coherently, Fisher exact test, one-tailed, proved not significant: p = 0.60).
PDGFRA-mutant GISTs (germline or not) are mostly at-least-in-part epithelioid, and gastric [48, 50]. The latter location appears so far exclusive in germline-mutant examples, as the only extra-gastric GIST reported in this genetic setting bore a concomitant somatic KIT mutation, and likely hinged exclusively on it [72]. PDGFRA-mutant GISTs express CD117 (less frequently than KIT-mutant GISTs and often weakly/patchy) and DOG1.
PDGFRA-mutant syndrome may also feature IFPs (including GI fibrous tumors), GI lipomas or large hands (Table 2). Diffuse ICCH has never been described in germline PDGFRA mutants; the reported “focal ICCs” [45] rather fits micro-GIST. These findings support PDGFRA-mutant and KIT-mutant GISTs as distinct entities. Accordingly, GI motility disturbance, frequently accompanying ICCH, is not typical of PDGFRA-mutant syndrome. Although the PDGFRA role in GIST and IFP pathogenesis [79] justifies the presence of these two tumor types in PDGFRA-mutant syndrome, the occurrence of GI lipomas (sporadic GI lipomas revealed PDGFRA WT [80]) and large hands, and the variability of the observed phenotypic assortment are presently unexplained.
Table 2.
Kindreds and individuals affected by PDGFRA-mutant syndrome
| Reference | Type of report (family vs. single individual) | Mutant exon (mutation) | GIST | Associated signs | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sitea | Histologyb | IFPc | GI fibrous tumorsd | GI lipomas | Large hands | Other manifestations | ||||
| Chompret et al. [45] | family | 18 (p.D846Y) | yes | ST | M (mainly E) | yes | ||||
| de Raedt et al. [71]; Heimann et al. [78] | family | 12 (p.Y555C) | yese | yes | Broad wrists, glaucoma | |||||
| Pasini et al.[46]; Carney and Stratakis [47] | individual | 12 (p.V561D) | yes | ST | M (mainly E) | yes | yes | |||
| Ricci et al. [48] | family | 14 (p.P653L) | yes | ST | E, M (mainly E) | yes | yes | yes | ||
| Ricci et al. [72] | Individual (mother and grandmother suffered a gut occlusion) (likely related to the above mentioned kindred [48]) | 14 (p.P653L) | yes (bearing a concomitant somatic KIT mutation) | SI | S | yes | ||||
a: SI small intestine, ST stomach
b: E epithelioid, M mixed spindle cell and epithelioid; S spindle cell
c: Inflammatory fibroid polyp
d: Fibrous tumor is likely a variant of IFP [48]
e: When a germline PDGFRA mutation was found in the referred kindred, these tumors were considered GISTs since GISTs were the only PDGFRA-mutant GI tumors known at that time
GIST PDGFRA mutations cluster in exons 12 (juxtamembrane regulatory domain), 14 and 18 (TK domains -ATP binding region and activation loop, respectively-). Mutations in all of these exons are represented in PDGFRA-mutant syndrome (Table 2). Two of them (involving exon 12) are relatively indolent, one at intermediate risk (exon 14) and another probably at high risk (exon 18-non-p.D842V) [65]. Besides, PDGFRA exon-14 mutations proved prognostically favorable in another work [81]. Similarly to KIT-mutant GISTs (counting each kindred as one, and considering the recently reported p.P653L individual [72] as a member of a previously published PDGFRA-mutant family [48] given the identity of both genetic defect and geographical origin), with the caveats of the limited sample, PDGFRA mutation distribution differs in germline mutations with respect to sporadic GISTs, with exon 18 involved in 25 % (Table 2) and 82 % [65] of cases, respectively. Germline-mutants appear enriched in presumably low-biological-impact mutations [65], with low molecular risk exon-12 ones found in 50 % of cases.
Germline PDGFRA-mutant GISTs feature 14q LOH [46], similar to sporadic, germline KIT-mutant and NF1-associated ones.
In PDGFRA-mutant kindreds, small bowel occlusions due to IFP can be life-threatening. Of note, only one individual died of malignancy possibly related to GIST described as “gastric cancer” in the absence of pathological records [48], coherently with the relative indolence of PDGFRA-mutant GISTs [82]. In need of treatment, however, TKI use must be pondered, evaluating the specific mutation present. In fact, although never reported in germline-mutants, p.D842V, the most common PDGFRA mutation of sporadic GISTs, confers resistance to both imatinib and the second-line TKI sunitinib, justifying alternative approaches using dasatinib and crenolanib [83–85].
Table 2 details the features of published PDGFRA-mutants syndrome cases.
NF1-associated GISTs
Neurofibromin, encoded by NF1 gene on 17q11.2 [51], accelerates the conversion from active GTP-bound to inactive GDP-bound RAS. NF1 inactivation stimulates MAPK cascade through increasing RAS activity [50], promoting tumorigenesis (Fig. 1).
NF1 is relatively common, with a ~1:3,000 birth incidence and a 1:4–5,000 prevalence [86]. NF1 transmission is autosomal dominant with complete penetrance and variable expression. ~50 % of NF1 lack familial history due to the high human NF1 mutation rate. In NF1, one NF1 allele is germline-mutant, the other displays tumoral somatic inactivation.
National Institutes of Health NF1 diagnostic criteria consist of ≥2 among: ≥6 café-au-lait macules >5 mm or >15 mm in pre-pubertal or post-pubertal subjects, respectively; ≥2 neurofibromas (one if plexiform); axillary/inguinal freckles; optic glioma; ≥2 iris hamartomas; bony dysplasia; and a NF1-affected first-degree relative [87]. 0.1-6 % of NF1 feature paragangliomas [88].
Clues of a possible association between NF1 and GIST have been known for decades [89]. As differential diagnosis between neurofibroma (NF) and GIST was made reliable [90], this association became evident. GISTs are the most frequent GI NF1 manifestation with a 7 % prevalence in NF1 patients, increasing to 25 % at autopsy. NF1 is >45-fold overrepresented among GIST patients. Coherently, NF1-mutated GISTs account for ~1,5 % of all GISTs. Average age-at-diagnosis of NF1-associated GISTs is ~49 years [1, 86, 91, 92].
NF1-associated GISTs are often multiple and small intestinal, with possible gastric exceptions (NF1 frequency is 6 %, 4 % and 0.06 % among patients with duodenal, jejunal/ileal and gastric GISTs, respectively); singly considered, they do not differ from sporadic intestinal GISTs, mostly featuring spindle cells, collagen globules (skeinoid fibers) and CD117 positivity [91]. Other NF1 GI lesions include: ICCH, with related GI motility disorders, and neuroendocrine tumors (especially periampullary somatostatinomas) [86]. The awareness of these GI signs can help to avoid diagnostic omissions caused by the variable clinical presentation and frequent non-familial form of NF1 (the lack of NF1 mutational hotspots makes genotyping an unpractical diagnostic tool) [86].
The discovery of NF1 second-hit in NF1 GISTs, already revealed peculiar because of their usually WT KIT/PDGFRA [91, 93], disclosed their pathogenesis and molecular link with NF1 [94]. KIT/PDGFRA mutations in NF1-associated GISTs are nevertheless possible, although probably incidental [95], globally accounting for ~8 % of cases [96].
NF1 GIST tumoral progression resembles that of germline KIT mutants, featuring preneoplastic ICCH followed by 14q and 22q LOH [97].
Although mostly indolent, ≤15-20 % of NF1 GISTs behave aggressively [91, 93]. NF1 GISTs are poorly responsive to imatinib, since NF1 trigger occurs downstream to imatinib targets KIT/PDGFRA; coherently, NF1 GISTs should not be treated with adjuvant imatinib, whatever their risk [98–100].
Syndromic SDH-deficient GISTs
SDH is a 4-subunit (A/B/C/D) Krebs cycle enzymatic complex, located in the inner mitochondrial membrane, but encoded by chromosomal DNA. SDHA/B/C/D (collectively, SDHx) are mapped to 5p15.33, 1p36.13, 1q23.3 and 11q23.1, respectively [51]. Whatever subunit is damaged, the entire complex is hampered, impairing succinate-to-fumarate conversion; succinate accumulation inhibits prolyl-hydroxylase, decreasing the hydroxylation of hypoxia-inducible factor (HIF)-1α (HIF-1α) and its consequent degradation; HIF heterodimers can thus translocate into the nucleus initiating tumorigenic transcription. Furthermore, succinate accumulation inhibits TET DNA-hydroxylases, compromising the 5-methylcytosine conversion to 5-hydroxymethylcytosine, required for DNA demethylation; coherently, SDHx-deficient GISTs feature pervasive DNA hypermethylation, likely implied in oncogenesis [101–105] (Fig. 1).
SDH-deficiency characterizes the largest KIT/PDGFRA-WT GIST subgroup (accounting for ~5 % of GISTs) [1]. SDH deficiency is also a feature of paragangliomas, renal cell carcinomas and pituitary adenomas [106, 107]. SDH-deficient GISTs arise either in germline SDHx-mutant and/or manifest syndromic settings or not [105, 108]. Thus, not surprisingly, in case of constitutive predisposition to SHD-deficiency, GISTs can associate with paragangliomas producing two syndromes: Carney’s triad (CT) and Carney-Stratakis Syndrome (CSS), both affecting young people (mean ages-at-diagnosis 22 and 19 years −22 and 24 years concerning GIST only-, respectively) [109–111]. CT and CSS are separated by the presence of pulmonary chondromas and of a striking female predilection in the former, and of an autosomal-dominant inheritance pattern in the latter, with incomplete penetrance [111]. Additionally, CT may feature esophageal leiomyoma and adrenal cortical adenoma [110]. SDH-deficient GISTs are restricted to stomach (especially antrum) and show a multinodular pattern (referred to as “plexiform” by pathologists) [103, 110].
≤50 % and ≤10 % of SDH-deficient GISTs manifest lymph-vascular invasion and lymph node metastases, respectively [112], explaining the frequent relapses despite apparently radical surgery.
SDH-deficient GIST are mostly at-least-in-part epithelioid, and express DOG1, CD34 (≥75 % of cases) and CD117 (strongly/diffusely, unlike PDGFRA-driven GISTs) [103, 110]. They are peculiarly SDHB-, whatever the damaged SDH subunit (unlike SDHA positivity, lost only in SDHA-mutations), and overexpress insulin-like growth factor 1 receptor (IGF1R) [103, 113–115].
Only micro GISTs have been found to precede overt SDH-deficient syndromic GIST [116]. ICCH is not a feature of CT [110], nor has ever been described in SDH deficiencies.
The basis of SDH impairment in CSS is mutational, with a typical second-hit mechanism involving SDHB/C/D [117, 118]. Significantly, germline SDHA mutations, described in patients bearing GISTs or paragangliomas and in paraganglioma/pituitary adenoma familial associations, are unreported in CSS, possibly due to their low penetrance [106, 108, 115, 118–125]. Of note, at the best of my knowledge, the only SDHD-mutant CSS with a reported pedigree hinged on a paternally inherited defective allele [126], coherently with the parent-of-origin effect of SDHD-depending hereditary paraganglioma syndrome PGL1, simulating maternal imprinting. However, SDHD lacks physical imprinting and PGL1 is exceptionally maternally transmitted, invoking the involvement of a second, paternally imprinted, tumor suppressor gene (TSG) (possibly H19) located on 11p15, i.e. the same chromosome of SDHD. Both WT SDHD and functional 11p15 TSG can thus be inactivated by non-disjunctional loss of the maternal chromosome 11, justifying the paternal disease transmission. More complex events, such as mitotic recombination of 11p15 TSG followed by loss of the paternal chromosome, would explain the exceptional maternal transmission [127–129].
Unlike CSS, CT tumorigenesis depends on epigenetic tumoral SDHC inactivation through SDHC hypermethylation [130]. This phenomenon, distinct from the global DNA hypermethylation of SDH-deficient GISTs as a whole, is common to most SDH-deficient, SDHx-WT GISTs, irrespective of CT presence; however, “non-CT” cases could be formes frustes of CT, as suggested by the mosaic constitutional SDHC promoter hypermethylation, implying a risk for metachronous paraganglioma/pulmonary chondroma [105].
Another oncogenic stimulus of SDH deficient GISTs is the hyperactivation of the IGF1R pathway, physiologically involved in cell survival/proliferation [113, 131].
≥50 % CT GISTs do not bear chromosomal imbalances; their rare LOHs preferentially involve 1p; 14q or 22q losses occur infrequently [132].
SDH-deficient GISTs often behave indolently, even in case of metastases, probably due to the metabolic disadvantage caused by SDH deficiency. They can nevertheless be aggressive (the SDH-deficient GIST overall mortality approaches 15 %). Of note, current risk classifications do not fit SDH-deficient GISTs [110, 112].
TKI imatinib and sunitinib proved ineffective or only partially effective, respectively, in SDH-deficient GISTs, coherently with SHD deficiency constituting a pathway independent of KIT/PDGFRA. Therefore, in the absence of guidelines specific for these tumors, TKI treatment is suggested for progressive disease but not after complete surgical resection [98–100, 112]. Inhibitors of IGF1R are being evaluated: testing of linsitinib in adult and pediatric WT GISTs (enriched in SDH-deficient GISTs), although without evidence of RECIST response, indicates a 45 % clinical benefit/52 % progression-free survival at 9 months [133]. When compared to KIT-mutant GISTs, a higher fraction of KIT/PDGFRA-WT (and PDGFRA-mutant) GISTs displayed activation of the mTOR pathway [70]; Therefore, CT/CSS GISTs can potentially benefit of PI3K/AKT/mTOR inhibitors [131]. Regorafenib, nilotinib, sorafenib and heat-shock protein inhibitors are other drugs potentially usable; finally, targeting of HIF-1α, α-ketoglutarate, and demethylating agents such as decitabine are theoretically attractive approaches [104, 105, 134–136].
CSS should be readily distinguishable from CT based on the presence or not of pulmonary chondromas, germline SDHB/C/D mutations and inheritance; female predilection of CT is another helping feature [137]. However, the arousal of tumors in SDH-deficient syndromes, including CT and CSS, can span over dozens of years [112]. Thus, if the “missing” tumor of a CT is pulmonary chondroma, CT/CSS morphologic differential diagnosis is impossible. Under these circumstances, in the absence of familial history, genotyping becomes pivotal. But recent findings have eroded also this cornerstone, evidencing germline SDHA/B/C variants, at least part of which pathogenic, in 6/63 (9.5 %) CT patients “certified” by pulmonary chondromas. On top of that, unlike typical CT, these patients were equally distributed between sexes [138]. Additionally, three males diagnosed with CT (one with a lesion “consistent with pulmonary chondroma”) were subsequently found to bear germline SDHx mutations [105, 139]. Noticeably, CT diagnosis in these cases appears questionable, with genotype and sex distribution rather supporting CSS.
GISTs could be also considered an infrequent component of SDH-deficient paraganglioma syndromes PGL1/3/4/5 [140], depending on SDHD/C/B/A germline mutations, respectively [141]. A risk of WT SDHx allele loss lower in GIST precursor cells than in paraganglial ones could explain the relative GIST rarity [112]. Noticeably, associations between GIST and renal cell carcinoma, another SDH-deficient tumor reported in paraganglioma syndromes, has been found in germline SDHA/B/C mutants [141–144].
Perhaps CT, CSS and PGL1-5 will be reconsidered in the future as different aspects of a single SDH-deficient disease.
Practical approach to GIST-predisposing syndromes
GIST-prone syndromes must be suspected in the presence of GISTs either multiple or associated with the peculiar manifestations previously described (which can raise suspicions even by themselves) (Table 3). Genotyping, if applicable, is confirmatory. NCCN guidelines suggest to investigate every KIT/PDGFRA-WT GIST patient for germline SDHx mutations [84]. Multiple GISTs sharing a phenotype not found in the germline favor a metastatic condition.
Table 3.
Main features of GIST-predisposing syndromes
| Syndrome | Trigger | Inheritance | Sex predilection | Average age at diagnosis (years) | GIST features | Other manifestations | |||
|---|---|---|---|---|---|---|---|---|---|
| Sitea | Morphologyb | Immunohistochemistry | |||||||
| KIT-mutant | germline KIT mutation | Autosomal dominant, high penetrance | None | 48 | SI, ST > C, R > E | S > M> > E | CD117+ DOG1+ | Skin hyperpigmentation, mast cell disorders, ICCHc, dysphagia | |
| PDGFRA-mutant | germline PDGFRA mutation | Autosomal dominant, high penetrance | None | 48 (GIST), 41 (inflammatory fibroid polyp) | ST | E, M | CD117+/− DOG1+/− | Inflammatory fibroid polyps (including GId “fibrous tumors”), GI lipomas, large hands | |
| Neurofibromatosis type 1 | Germline NF1 mutation + tumor 2nd hit in WT allele | Autosomal dominant, complete penetrance and variable expression | None | 49 | SI > ST | S > M | CD117+ DOG1+ | Neurofibromas and other signs of Neurofibromatosis type 1, ICCH, dysphagia | |
| SDH-deficient syndromes | CTe | epigenetic SDHC promoter hypermethylation in tumors | Nonef | F> > M | 22 (either whatever tumor type or GIST) | ST | E > M, S; plexiform | CD117 + DOG1+ | Paragangliomas/pheocromocytomas, pulmonary chondromas, esophageal leiomyoma, adrenal cortical adenoma |
| CSSg | Germline SDHB, C or D mutation + tumor 2nd hit in WT allele | Autosomal dominant, incomplete penetrance. Parent-of-origin if SDHD-mutant | None | 19 (whatever tumor type), 24 (GIST) | ST | E > M, S; plexiform | CD117 + DOG1+ | Paragangliomas/pheocromocytomas | |
a E esophagus, ST stomach, SI small intestine, C colon, R rectum
b S spindle cell, E epithelioid, M mixed spindle cell and epithelioid
c Diffuse Intersitial cell of Cajal hyperplasia
d GI, gastrointestinal
e Carney’s triad
f Recently reported 6 germline SDHx-mutant cases, one of which with inherited paragangliomas
g Carney-Stratakis syndrome
Comprehensive guidelines specific for the management of GIST-prone syndromes are lacking. Existing recommendations, dealing with adjuvant therapy of NF1-associated and SDH-deficient GISTs [99], have been treated in the pertaining chapters of this review.
Once a patient is diagnosed with a GIST-predisposing condition depending on a germline DNA defect, predictive genetic testing in family members should be considered. The latter is indicated for the highly penetrant germline KIT/PDGFRA/SDHB/SDHD mutations; conversely, the opportunity of genetic evaluation is controversial for the low penetrance SDHC and, especially, SDHA variants [141]. It is worth recalling the parent-of-origin effect of familial SDHD mutations, herein previously discussed.
Periodic computed-tomography-scan or 18F-FDG PET-computed-tomography have been suggested for the surveillance of syndromic GISTs [145]. Endoscopic ultrasound joined to fine needle tissue acquisition allowing histological assessment [146, 147] appear valid complements. Colonscopy is expectedly of limited utility in familial KIT-dependent GISTs [29] (mostly gastric/small intestinal) and useless in SDH-deficient ones (strictly stomach restricted), while can detect colonic/ileal IFPs in PDGFRA-mutant syndrome [48]. In case of SDH-deficient GISTs, chest X-ray is indicated to look for pulmonary chondromas (especially if SDHx-WT), and plasma/urine determination of metanephrines/catecholamines and PET-tracers 68Ga-DOTATATE, 18F-DOPA and 18F-FDA can be used for investigating paragangliomas [141].
Intracellular signaling machineries and gene expressions are common to homologous syndromic and sporadic GISTs [20]. Coherently, no differences in imatinib sensitivity exist between GISTs sharing the same genotype, no matters whether somatic or germline [98]. Thus, the approach to individual syndromic GISTs in need of treatment does not differ from that to their sporadic counterparts.
There is no evidence that GIST hereditary predispositions constitute an independent prognostic factor warranting a differential GIST treatment [145]. Nevertheless, the frequent multiplicity of GI tumors constitutes a problem peculiar to GIST-syndromes deserving a special attention, commonly causing acute complications such as hemorrage and, especially in germline KIT/PDGFRA mutations and NF1 (where the small bowel can be involved), occlusion/perforation which can be life-threatening [13, 43, 48]. Although surgery is frequently necessary as affected patients are often symptomatic [13], no agreement exists as to whether preventive therapy, either surgical or molecularly targeted, is indicated in asymptomatic subjects [145]. It has been suggested to postpone surgery based on the frequent indolence of germline-mutant GISTs [148]; however, the latter’s possible aggressiveness recommends to remove GISTs at presumable significant risk, based on size/site/genotype. Similarly, lesions at risk of occlusion/hemorrhage should be removed too. Surgery should focus on resecting the outstanding tumor(s) disregarding possible tiny nodules if numerous and involving extended areas, since their removal would sacrifice substantial portions of functional GI tissue without proven benefit in terms of tumor recurrence risk; this is particularly true in CT and CSS patients, given their frequently young age [109]. Preventive molecular therapy poses several problems. In fact, although theoretically attractive, it is presently not evidence-based. Prospective clinical trials will be hardly achieved, given the rarity of syndromic GISTs. Moreover, the risks of lifelong exposures to GIST-targeted drugs are presently unknown. Thus, active surveillance has been adopted after surgery [44]. Alternatively, a long term preventive treatment with TKI in patients bearing a sensitive mutation has been proposed [13]. A compromise has been adopted by employing half-dose imatinib in a KIT exon-11 germline mutant with multiple GISTs, obtaining marked tumor reductions after one-year [43]. Interestingly, imatinib therapy has been reported to reduce cutaneous melanosis in germline KIT-mutants [32, 41].
Exceptionally, GISTs in syndromic contexts can reveal superimposed triggers [96, 149]. Anomalous GIST site and/or morphology with respect to a given hosting syndrome can help in suspecting these “divergent” GISTs, whose pathogenesis can hinge mainly or even exclusively upon the “extra-syndromic” molecular defect, with relevant clinical implications [72]. In any event, to be on the safe side it is advisable to fully genotype whatever apparently syndromic GIST to be molecularly treated, independently of its morphology and site.
Conclusions
The correct approach to syndromic GISTs results from the integration between congruent diagnostic strategies, including familial screening, and treatment of individual tumors and background syndromic manifestations. Protean signs prompt to undertake complex choices involving the treatment of symptomatic lesions and the prevention of future complications. Specific comprehensive guidelines are lacking, primarily due to the rarity of syndromic GISTs. However, these diseases have been the subject of an increasing number of publications, resulting in a conspicuous amount of data with relevant clinical implications. The latter allowed the draft of the present review, which hopefully will help physicians facing GIST-prone syndromes, waiting for the development of dedicated guidelines.
Abbreviations
CSS, Carney-Stratakis syndrome; CT, Carney’s triad; GIST, gastrointestinal stromal tumor; ICCH, interstitial cell of Cajal hyperplasia; IFP, inflammatory fibroid polyp; NF1, neurofibromatosis type 1; PDGF, platelet-derived growth factor; SCF, stem cell factor; SDH, succinate dehydrogenase
Acknowledgements
Funding
This work was supported by the Università Cattolica del Sacro Cuore (Linea D1 grants number 70200367).
Competing interests
The author declares speaker’s honoraria from Novartis.
Author's note
During the review process and after acceptance of this manuscript, data relevant with respect to SDH-deficient GISTs, one of the main topics of the present review, have been published: 1) Ben-Ami et al. (Ben-Ami E, Barysauskas CM, von Mehren M, Heinrich MC, Corless CL, Butrynski JE, et al. Long-term follow-up of the multicenter phase II trial of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of standard tyrosin kinase inhibitor therapy. Ann Oncol. 2016; doi: 10.1093/annonc/mdw228) reported that regorafenib induced objective responses and durable benefit in SDH-deficient GISTs; 2) Boikos et al. (Boikos SA, Pappo AS, Killian JK, LaQuaglia MP, Weldon CB, George S, et al. Molecular subtypes of KIT/PDGFRA wild-type gastrointestinal stromal tumors. A report from the National Institutes of Health Gastrointestinal Stromal Tumor Clinic. JAMA Oncol. 2016; doi: 10.1093/annonc/mdw228) propose genotype rather than presence or absence of chondromas as the discriminating factor between CT and CSS.
References
- 1.Ricci R, Dei Tos AP, Rindi G. GISTogram: a graphic presentation of the growing GIST complexity. Virchows Arch. 2013;463:481–7. doi: 10.1007/s00428-013-1467-4. [DOI] [PubMed] [Google Scholar]
- 2.Corless CL. Gastrointestinal stromal tumors: what do we know now? Mod Pathol. 2014;27(Suppl 1):S1–16. doi: 10.1038/modpathol.2013.173. [DOI] [PubMed] [Google Scholar]
- 3.Thomsen L, Robinson TL, Lee JC, Farraway LA, Hughes MJ, Andrews DW, et al. Interstitial cells of Cajal generate a rhythmic pacemaker current. Nat Med. 1998;4:848–51. doi: 10.1038/nm0798-848. [DOI] [PubMed] [Google Scholar]
- 4.Kindblom LG, Remotti HE, Aldenborg F, Meis-Kindblom JM. Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol. 1998;152:1259–69. [PMC free article] [PubMed] [Google Scholar]
- 5.Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–80. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- 6.Joensuu H, Roberts PJ, Sarlomo-Rikala M, Andersson LC, Tervahartiala P, Tuveson D, et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med. 2001;344:1052–6. doi: 10.1056/NEJM200104053441404. [DOI] [PubMed] [Google Scholar]
- 7.Nishida T, Hirota S, Taniguchi M, Hashimoto K, Isozaki K, Nakamura H, et al. Familial gastrointestinal stromal tumours with germline mutation of the KIT gene. Nat Genet. 1998;19:323–4. doi: 10.1038/1209. [DOI] [PubMed] [Google Scholar]
- 8.O’Brien P, Kapusta L, Dardick I, Axler J, Gnidec A. Multiple familial gastrointestinal autonomic nerve tumors and small intestinal neuronal dysplasia. Am J Surg Pathol. 1999;23:198–204. doi: 10.1097/00000478-199902000-00009. [DOI] [PubMed] [Google Scholar]
- 9.Hirota S, Okazaki T, Kitamura Y, O’Brien P, Kapusta L, Dardick I. Cause of familial and multiple gastrointestinal autonomic nerve tumors with hyperplasia of interstitial cells of Cajal is germline mutation of the c-kit gene. Am J Surg Pathol. 2000;24:326–7. doi: 10.1097/00000478-200002000-00045. [DOI] [PubMed] [Google Scholar]
- 10.Chen H, Hirota S, Isozaki K, Sun H, Ohashi A, Kinoshita K, et al. Polyclonal nature of diffuse proliferation of interstitial cells of Cajal in patients with familial and multiple gastrointestinal stromal tumours. Gut. 2002;51:793–6. doi: 10.1136/gut.51.6.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Isozaki K, Terris B, Belghiti J, Schiffmann S, Hirota S, Vanderwinden JM. Germline-activating mutation in the kinase domain of KIT gene in familial gastrointestinal stromal tumors. Am J Surg Pathol. 2000;157:1581–5. doi: 10.1016/S0002-9440(10)64795-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Handra-Luca A, Flejou JF, Molas G, Sauvanet A, Belghiti J, Degott C, et al. Familial multiple gastrointestinal stromal tumours with associated abnormalities of the myenteric plexus layer and skeinoid fibres. Histopathology. 2001;39:359–63. doi: 10.1046/j.1365-2559.2001.01214.x. [DOI] [PubMed] [Google Scholar]
- 13.Bachet JB, Landi B, Laurent-Puig P, Italiano A, Le Cesne A, Levy P, et al. Diagnosis, prognosis and treatment of patients with gastrointestinal stromal tumour (GIST) and germline mutation of KIT exon 13. Eur J Cancer. 2013;49:2531–41. doi: 10.1016/j.ejca.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 14.Maeyama H, Hidaka E, Ota H, Minami S, Kajiyama M, Kuraishi A, et al. Familial gastrointestinal stromal tumor with hyperpigmentation: association with a germline mutation of the c-kit gene. Gastroenterology. 2001;120:210–5. doi: 10.1053/gast.2001.20880. [DOI] [PubMed] [Google Scholar]
- 15.Beghini A, Tibiletti MG, Roversi G, Chiaravalli AM, Serio G, Capella C, et al. Germline mutation in the juxtamembrane domain of the kit gene in a family with gastrointestinal stromal tumors and urticaria pigmentosa. Cancer. 2001;92:657–62. doi: 10.1002/1097-0142(20010801)92:3<657::AID-CNCR1367>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 16.Hirota S, Nishida T, Isozaki K, Taniguchi M, Nishikawa K, Ohashi A, et al. Familial gastrointestinal stromal tumors associated with dysphagia and novel type germline mutation of KIT gene. Gastroenterology. 2002;122:1493–9. doi: 10.1053/gast.2002.33024. [DOI] [PubMed] [Google Scholar]
- 17.Robson ME, Glogowski E, Sommer G, Antonescu CR, Nafa K, Maki RG, et al. Pleomorphic characteristics of a germ-line KIT mutation in a large kindred with gastrointestinal stromal tumors, hyperpigmentation, and dysphagia. Clin Cancer Res. 2004;10:1250–4. doi: 10.1158/1078-0432.CCR-03-0110. [DOI] [PubMed] [Google Scholar]
- 18.Antonescu CR, Viale A, Sarran L, Tschernyavsky SJ, Gonen M, Segal NH, et al. Gene expression in gastrointestinal stromal tumors is distinguished by KIT genotype and anatomic site. Clin Cancer Res. 2004;10:3282–90. doi: 10.1158/1078-0432.CCR-03-0715. [DOI] [PubMed] [Google Scholar]
- 19.Carballo M, Roig I, Aguilar F, Pol MA, Gamundi MJ, Hernan I, et al. Novel c-KIT germline mutation in a family with gastrointestinal stromal tumors and cutaneous hyperpigmentation. Am J Med Genet A. 2005;132A:361–4. doi: 10.1002/ajmg.a.30388. [DOI] [PubMed] [Google Scholar]
- 20.Li FP, Fletcher JA, Heinrich MC, Garber JE, Sallan SE, Curiel-Lewandrowski C, et al. Familial gastrointestinal stromal tumor syndrome: phenotypic and molecular features in a kindred. J Clin Oncol. 2005;23:2735–43. doi: 10.1200/JCO.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 21.Tarn C, Merkel E, Canutescu AA, Shen W, Skorobogatko Y, Heslin MJ, et al. Analysis of KIT mutations in sporadic and familial gastrointestinal stromal tumors: therapeutic implications through protein modeling. Clin Cancer Res. 2005;11:3668–77. doi: 10.1158/1078-0432.CCR-04-2515. [DOI] [PubMed] [Google Scholar]
- 22.Hartmann K, Wardelmann E, Ma Y, Merkelbach-Bruse S, Preussner LM, Woolery C, et al. Novel germline mutation of KIT associated with familial gastrointestinal stromal tumors and mastocytosis. Gastroenterology. 2005;129:1042–6. doi: 10.1053/j.gastro.2005.06.060. [DOI] [PubMed] [Google Scholar]
- 23.Kim HJ, Lim SJ, Park K, Yuh YJ, Jang SJ, Choi J. Multiple gastrointestinal stromal tumors with a germline c-kit mutation. Pathol Int. 2005;55:655–9. doi: 10.1111/j.1440-1827.2005.01885.x. [DOI] [PubMed] [Google Scholar]
- 24.O’Riain C, Corless CL, Heinrich MC, Keegan D, Vioreanu M, Maguire D, et al. Gastrointestinal stromal tumors: insights from a new familial GIST kindred with unusual genetic and pathologic features. Am J Surg Pathol. 2005;29:1680–3. doi: 10.1097/01.pas.0000173024.79852.08. [DOI] [PubMed] [Google Scholar]
- 25.Miettinen M, Makhlouf H, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the jejunum and ileum: a clinicopathologic, immunohistochemical, and molecular genetic study of 906 cases before imatinib with long-term follow-up. Am J Surg Pathol. 2006;30:477–89. doi: 10.1097/00000478-200604000-00008. [DOI] [PubMed] [Google Scholar]
- 26.Lasota J, Miettinen M. A new familial GIST identified. Am J Surg Pathol. 2006;30:1342. doi: 10.1097/01.pas.0000213364.56498.3b. [DOI] [PubMed] [Google Scholar]
- 27.Kang DY, Park CK, Choi JS, Jin SY, Kim HJ, Joo M, et al. Multiple gastrointestinal stromal tumors: clinicopathologic and genetic analysis of 12 patients. Am J Surg Pathol. 2007;31:224–32. doi: 10.1097/01.pas.0000213318.66800.94. [DOI] [PubMed] [Google Scholar]
- 28.Graham J, Debiec-Rychter M, Corless CL, Reid R, Davidson R, White JD. Imatinib in the management of multiple gastrointestinal stromal tumors associated with a germline KIT K642E mutation. Arch Pathol Lab Med. 2007;131:1393–6. doi: 10.5858/2007-131-1393-IITMOM. [DOI] [PubMed] [Google Scholar]
- 29.Kleinbaum EP, Lazar AJ, Tamborini E, McAuliffe JC, Sylvestre PB, Sunnenberg TD, et al. Clinical, histopathologic, molecular and therapeutic findings in a large kindred with gastrointestinal stromal tumor. Int J Cancer. 2008;122:711–8. doi: 10.1002/ijc.23137. [DOI] [PubMed] [Google Scholar]
- 30.Wozniak A, Rutkowski P, Sciot R, Ruka W, Michej W, Debiec-Rychter M. Rectal gastrointestinal stromal tumors associated with a novel germline KIT mutation. Int J Cancer. 2008;122:2160–4. doi: 10.1002/ijc.23338. [DOI] [PubMed] [Google Scholar]
- 31.Thalheimer A, Schlemmer M, Bueter M, Merkelbach-Bruse S, Schildhaus HU, Buettner R, et al. Familial gastrointestinal stromal tumors caused by the novel KIT exon 17 germline mutation N822Y. Am J Surg Pathol. 2008;32:1560–5. doi: 10.1097/PAS.0b013e318172ce6f. [DOI] [PubMed] [Google Scholar]
- 32.Campbell T, Felsten L, Moore J. Disappearance of lentigines in a patient receiving imatinib treatment for familial gastrointestinal stromal tumor syndrome. Arch Dermatol. 2009;145:1313–6. doi: 10.1001/archdermatol.2009.263. [DOI] [PubMed] [Google Scholar]
- 33.Veiga I, Silva M, Vieira J, Pinto C, Pinheiro M, Torres L, et al. Hereditary gastrointestinal stromal tumors sharing the KIT Exon 17 germline mutation p.Asp820Tyr develop through different cytogenetic progression pathways. Genes Chromosomes Cancer. 2010;49:91–8. doi: 10.1002/gcc.20720. [DOI] [PubMed] [Google Scholar]
- 34.Kuroda N, Tanida N, Hirota S, Daum O, Hes O, Michal M, et al. Familial gastrointestinal stromal tumor with germ line mutation of the juxtamembrane domain of the KIT gene observed in relatively young women. Ann Diagn Pathol. 2011;15:358–61. doi: 10.1016/j.anndiagpath.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 35.Vilain RE, Dudding T, Braye SG, Groombridge C, Meldrum C, Spigelman AD, et al. Can a familial gastrointestinal tumour syndrome be allelic with Waardenburg syndrome? Clin Genet. 2011;79:554–60. doi: 10.1111/j.1399-0004.2010.01489.x. [DOI] [PubMed] [Google Scholar]
- 36.Nakai M, Hashikura Y, Ohkouchi M, Yamamura M, Akiyama T, Shiba K, et al. Characterization of novel germline c-kit gene mutation, KIT-Tyr553Cys, observed in a family with multiple gastrointestinal stromal tumors. Lab Invest. 2012;92:451–7. doi: 10.1038/labinvest.2011.165. [DOI] [PubMed] [Google Scholar]
- 37.Wadt K, Andersen MK, Hansen TV, Gerdes AM. A new genetic diagnosis of familiar gastrointestinal stromal tumour. Ugeskr Laeger. 2012;174:1462–4. [PubMed] [Google Scholar]
- 38.Speight RA, Nicolle A, Needham SJ, Verrill MW, Bryon J, Panter S. Rare, germline mutation of KIT with imatinib-resistant multiple GI stromal tumors and mastocytosis. J Clin Oncol. 2013;31:e245–7. doi: 10.1200/JCO.2012.42.0133. [DOI] [PubMed] [Google Scholar]
- 39.Neuhann TM, Mansmann V, Merkelbach-Bruse S, Klink B, Hellinger A, Hoffkes HG, et al. A novel germline KIT mutation (p.L576P) in a family presenting with juvenile onset of multiple gastrointestinal stromal tumors, skin hyperpigmentations, and esophageal stenosis. Am J Surg Patghol. 2013;37:898–905. doi: 10.1097/PAS.0b013e31827bc071. [DOI] [PubMed] [Google Scholar]
- 40.Yamanoi K, Higuchi K, Kishimoto H, Nishida Y, Nakamura M, Sudoh M, et al. Multiple gastrointestinal stromal tumors with novel germline c-kit gene mutation, K642T, at exon 13. Hum Pathol. 2014;45:884–8. doi: 10.1016/j.humpath.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 41.Adela Avila S, Penaloza J, Gonzalez F, Abdo I, Rainville I, Root E, et al. Dysphagia, melanosis, gastrointestinal stromal tumors and a germinal mutation of the KIT gene in an Argentine family. Acta Gastroenterol Latinoam. 2014;44:9–15. [PubMed] [Google Scholar]
- 42.Jones DH, Caracciolo JT, Hodul PJ, Strosberg JR, Coppola D, Bui MM. Familial gastrointestinal stromal tumor syndrome: report of 2 cases with KIT exon 11 mutation. Cancer Control. 2015;22:102–8. doi: 10.1177/107327481502200113. [DOI] [PubMed] [Google Scholar]
- 43.Bamba S, Hirota S, Inatomi O, Ban H, Nishimura T, Shioya M, et al. Familial and multiple gastrointestinal stromal tumors with fair response to a half-dose of imatinib. Intern Med. 2015;54:759–64. doi: 10.2169/internalmedicine.54.3585. [DOI] [PubMed] [Google Scholar]
- 44.Forde PM, Cochran RL, Boikos SA, Zabransky DJ, Beaver JA, Meyer CF et al. Familial GI Stromal Tumor With Loss of Heterozygosity and Amplification of Mutant KIT. J Clin Oncol. 2014; doi:10.1200/JCO.2013.51.6633. [DOI] [PubMed]
- 45.Chompret A, Kannengiesser C, Barrois M, Terrier P, Dahan P, Tursz T, et al. PDGFRA germline mutation in a family with multiple cases of gastrointestinal stromal tumor. Gastroenterology. 2004;126:318–21. doi: 10.1053/j.gastro.2003.10.079. [DOI] [PubMed] [Google Scholar]
- 46.Pasini B, Matyakhina L, Bei T, Muchow M, Boikos S, Ferrando B, et al. Multiple gastrointestinal stromal and other tumors caused by platelet-derived growth factor receptor alpha gene mutations: a case associated with a germline V561D defect. J Clin Endocrinol Metab. 2007;92:3728–32. doi: 10.1210/jc.2007-0894. [DOI] [PubMed] [Google Scholar]
- 47.Carney JA, Stratakis CA. Stromal, fibrous, and fatty gastrointestinal tumors in a patient with a PDGFRA gene mutation. Am J Surg Pathol. 2008;32:1412–20. doi: 10.1097/PAS.0b013e31816250ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ricci R, Martini M, Cenci T, Carbone A, Lanza P, Biondi A, et al. PDGFRA-mutant syndrome. Mod Pathol. 2015;28:954–64. doi: 10.1038/modpathol.2015.56. [DOI] [PubMed] [Google Scholar]
- 49.Roberts R, Govender D. Gene of the month: KIT. J Clin Pathol. 2015;68:671–4. doi: 10.1136/jclinpath-2015-203207. [DOI] [PubMed] [Google Scholar]
- 50.Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11:865–78. doi: 10.1038/nrc3143. [DOI] [PubMed] [Google Scholar]
- 51.Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD). 1985. [database on the Internet]. http://www.ncbi.nlm.nih.gov/omim. Accessed: 20 Mar 2016.
- 52.Babin RW, Ceilley RI, DeSanto LW. Oral hyperpigmentation and occult malignancy--report of a case. J Otolaryngol. 1978;7:389–94. [PubMed] [Google Scholar]
- 53.Suda M, Ishii H, Kashiwazaki K, Tsuchiya M. Hyperpigmentation of skin and nails in a patient with intestinal leiomyosarcoma. Dig Dis Sci. 1985;30:1108–11. doi: 10.1007/BF01315610. [DOI] [PubMed] [Google Scholar]
- 54.Marshall JB, Diaz-Arias AA, Bochna GS, Vogele KA. Achalasia due to diffuse esophageal leiomyomatosis and inherited as an autosomal dominant disorder. Report of a family study. Gastroenterology. 1990;98:1358–65. doi: 10.1016/0016-5085(90)90357-7. [DOI] [PubMed] [Google Scholar]
- 55.el-Omar M, Davies J, Gupta S, Ross H, Thompson R. Leiomyosarcoma in leiomyomatosis of the small intestine. Postgrad Med J. 1994;70:661–4. doi: 10.1136/pgmj.70.827.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rossi S, Miceli R, Messerini L, Bearzi I, Mazzoleni G, Capella C, et al. Natural history of imatinib-naive GISTs: a retrospective analysis of 929 cases with long-term follow-up and development of a survival nomogram based on mitotic index and size as continuous variables. Am J Surg Pathol. 2011;35:1646–56. doi: 10.1097/PAS.0b013e31822d63a7. [DOI] [PubMed] [Google Scholar]
- 57.Joensuu H, Hohenberger P, Corless CL. Gastrointestinal stromal tumour. Lancet. 2013;382:973–83. doi: 10.1016/S0140-6736(13)60106-3. [DOI] [PubMed] [Google Scholar]
- 58.Joensuu H, Rutkowski P, Nishida T, Steigen SE, Brabec P, Plank L, et al. KIT and PDGFRA Mutations and the Risk of GI Stromal Tumor Recurrence. J Clin Oncol. 2015;33:634–42. doi: 10.1200/JCO.2014.57.4970. [DOI] [PubMed] [Google Scholar]
- 59.Rapley EA, Hockley S, Warren W, Johnson L, Huddart R, Crockford G, et al. Somatic mutations of KIT in familial testicular germ cell tumours. Br J Cancer. 2004;90:2397–401. doi: 10.1038/sj.bjc.6601880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chan EC, Bai Y, Kirshenbaum AS, Fischer ER, Simakova O, Bandara G, et al. Mastocytosis associated with a rare germline KIT K509I mutation displays a well-differentiated mast cell phenotype. J Allergy Clin Immunol. 2014;134:178–87. doi: 10.1016/j.jaci.2013.12.1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bodemer C, Hermine O, Palmerini F, Yang Y, Grandpeix-Guyodo C, Leventhal PS, et al. Pediatric mastocytosis is a clonal disease associated with D816V and other activating c-KIT mutations. J Invest Dermatol. 2010;130:804–15. doi: 10.1038/jid.2009.281. [DOI] [PubMed] [Google Scholar]
- 62.Rossi S, Gasparotto D, Toffolatti L, Pastrello C, Gallina G, Marzotto A, et al. Molecular and clinicopathologic characterization of gastrointestinal stromal tumors (GISTs) of small size. Am J Surg Pathol. 2010;34:1480–91. doi: 10.1097/PAS.0b013e3181ef7431. [DOI] [PubMed] [Google Scholar]
- 63.Calabuig-Farinas S, Lopez-Guerrero JA, Navarro S, Machado I, Poveda A, Pellin A, et al. Evaluation of prognostic factors and their capacity to predict biological behavior in gastrointestinal stromal tumors. Int J Surg Pathol. 2011;19:448–61. doi: 10.1177/1066896911402327. [DOI] [PubMed] [Google Scholar]
- 64.Huss S, Kunstlinger H, Wardelmann E, Kleine MA, Binot E, Merkelbach-Bruse S, et al. A subset of gastrointestinal stromal tumors previously regarded as wild-type tumors carries somatic activating mutations in KIT exon 8 (p.D419del) Mod Pathol. 2013;26:1004–12. doi: 10.1038/modpathol.2013.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rossi S, Gasparotto D, Miceli R, Toffolatti L, Gallina G, Scaramel E, et al. KIT, PDGFRA, and BRAF mutational spectrum impacts on the natural history of imatinib-naive localized GIST: a population-based study. Am J Surg Pathol. 2015;39:922–30. doi: 10.1097/PAS.0000000000000418. [DOI] [PubMed] [Google Scholar]
- 66.Agaimy A, Markl B, Arnholdt H, Hartmann A, Schneider-Stock R, Chetty R. Sporadic segmental Interstitial cell of cajal hyperplasia (microscopic GIST) with unusual diffuse longitudinal growth replacing the muscularis propria: differential diagnosis to hereditary GIST syndromes. Int J Clin Exp Pathol. 2010;3:549–56. [PMC free article] [PubMed] [Google Scholar]
- 67.Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol. 2006;23(2):70–83. doi: 10.1053/j.semdp.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 68.Giebel LB, Strunk KM, Holmes SA, Spritz RA. Organization and nucleotide sequence of the human KIT (mast/stem cell growth factor receptor) proto-oncogene. Oncogene. 1992;7:2207–17. [PubMed] [Google Scholar]
- 69.Demoulin JB, Essaghir A. PDGF receptor signaling networks in normal and cancer cells. Cytokine Growth Factor Rev. 2014;25:273–83. doi: 10.1016/j.cytogfr.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 70.Sapi Z, Fule T, Hajdu M, Matolcsy A, Moskovszky L, Mark A, et al. The activated targets of mTOR signaling pathway are characteristic for PDGFRA mutant and wild-type rather than KIT mutant GISTs. Diagn Mol Pathol. 2011;20:22–33. doi: 10.1097/PDM.0b013e3181eb931b. [DOI] [PubMed] [Google Scholar]
- 71.de Raedt T, Cools J, Debiec-Rychter M, Brems H, Mentens N, Sciot R, et al. Intestinal neurofibromatosis is a subtype of familial GIST and results from a dominant activating mutation in PDGFRA. Gastroenterology. 2006;131:1907–12. doi: 10.1053/j.gastro.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 72.Ricci R, Martini M, Cenci T, Riccioni ME, Maria G, Cassano A et al. Divergent gastrointestinal stromal tumors in syndromic settings. Cancer Genet. 2016; doi: http://dx.doi.org/10.1016/j.cancergen.2016.05.073 [DOI] [PubMed]
- 73.Spencer D. Recurrent familial inflammatory fibroid polyps of the small intestine. J Clin Pathol. 1969;22:743. doi: 10.1136/jcp.22.6.743-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Anthony PP, Morris DS, Vowles KD. Multiple and recurrent inflammatory fibroid polyps in three generations of a Devon family: a new syndrome. Gut. 1984;25:854–62. doi: 10.1136/gut.25.8.854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Allibone RO, Nanson JK, Anthony PP. Multiple and recurrent inflammatory fibroid polyps in a Devon family (‘Devon polyposis syndrome’): an update. Gut. 1992;33:1004–5. doi: 10.1136/gut.33.7.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bayle S, Rossi P, Bagneres D, Demoux AL, Ashero A, Dales JP, et al. Ileum inflammatory fibroid polyp revealed by intussusception. About one familial case. Rev Med Interne. 2005;26:233–7. doi: 10.1016/j.revmed.2004.10.030. [DOI] [PubMed] [Google Scholar]
- 77.Lipton S, Zuckerbrod M. Familial enteric neurofibromatosis. Med Times. 1966;94:544–8. [PubMed] [Google Scholar]
- 78.Heimann R, Verhest A, Verschraegen J, Grosjean W, Draps JP, Hecht F. Hereditary intestinal neurofibromatosis. I. A distinctive genetic disease. Neurofibromatosis. 1988;1:26–32. [PubMed] [Google Scholar]
- 79.Schildhaus HU, Cavlar T, Binot E, Buttner R, Wardelmann E, Merkelbach-Bruse S. Inflammatory fibroid polyps harbour mutations in the platelet-derived growth factor receptor alpha (PDGFRA) gene. J Pathol. 2008;216:176–82. doi: 10.1002/path.2393. [DOI] [PubMed] [Google Scholar]
- 80.Dubova M, Sedivcova M, Saskova B, Hadravska S, Daum O. Nonsyndromic Intestinal Lipomas are Probably not Associated With Mutations of PDGFRA. Appl Immunohistochem Mol Morphol. 2016; doi:10.1097/PAI.0000000000000356. [DOI] [PubMed]
- 81.Lasota J, Stachura J, Miettinen M. GISTs with PDGFRA exon 14 mutations represent subset of clinically favorable gastric tumors with epithelioid morphology. Lab Invest. 2006;86:94–100. doi: 10.1038/labinvest.3700360. [DOI] [PubMed] [Google Scholar]
- 82.Lasota J, Dansonka-Mieszkowska A, Sobin LH, Miettinen M. A great majority of GISTs with PDGFRA mutations represent gastric tumors of low or no malignant potential. Lab Invest. 2004;84:874–83. doi: 10.1038/labinvest.3700122. [DOI] [PubMed] [Google Scholar]
- 83.Heinrich MC, Griffith D, McKinley A, Patterson J, Presnell A, Ramachandran A, et al. Crenolanib inhibits the drug-resistant PDGFRA D842V mutation associated with imatinib-resistant gastrointestinal stromal tumors. Clin Cancer Res. 2012;18:4375–84. doi: 10.1158/1078-0432.CCR-12-0625. [DOI] [PubMed] [Google Scholar]
- 84.von Mehren M, Randall RL, Benjamin RS, Boles S, Bui MM, Casper ES, et al. Gastrointestinal stromal tumors, version 2.2014. J Natl Compr Canc Netw. 2014;12:853–62. doi: 10.6004/jnccn.2014.0080. [DOI] [PubMed] [Google Scholar]
- 85.Liegl-Atzwanger B, Fletcher JA, Fletcher CD. Gastrointestinal stromal tumors. Virchows Arch. 2010;456(2):111–27. doi: 10.1007/s00428-010-0891-y. [DOI] [PubMed] [Google Scholar]
- 86.Agaimy A, Vassos N, Croner RS. Gastrointestinal manifestations of neurofibromatosis type 1 (Recklinghausen’s disease): clinicopathological spectrum with pathogenetic considerations. Int J Clin Exp Pathol. 2012;5:852–62. [PMC free article] [PubMed] [Google Scholar]
- 87.National Institutes of Health National Institutes of Health Consensus Development Conference Statement: neurofibromatosis. Bethesda, Md., USA, July 13–15, 1987. Neurofibromatosis. 1988;1:172–8. [PubMed] [Google Scholar]
- 88.Welander J, Soderkvist P, Gimm O. Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr Relat Cancer. 2011;18:R253–76. doi: 10.1530/ERC-11-0170. [DOI] [PubMed] [Google Scholar]
- 89.Lukash WM, Morgan RI, Sennett CO, Nielson OF. Gastrointestinal neoplasms in von Recklinghausen’s disease. Arch Surg. 1966;92:905–8. doi: 10.1001/archsurg.1966.01320240093020. [DOI] [PubMed] [Google Scholar]
- 90.Sarlomo-Rikala M, Kovatich AJ, Barusevicius A, Miettinen M. CD117: a sensitive marker for gastrointestinal stromal tumors that is more specific than CD34. Mod Pathol. 1998;11:728–34. [PubMed] [Google Scholar]
- 91.Miettinen M, Fetsch JF, Sobin LH, Lasota J. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: a clinicopathologic and molecular genetic study of 45 cases. Am J Surg Pathol. 2006;30:90–6. doi: 10.1097/01.pas.0000176433.81079.bd. [DOI] [PubMed] [Google Scholar]
- 92.Zoller ME, Rembeck B, Oden A, Samuelsson M, Angervall L. Malignant and benign tumors in patients with neurofibromatosis type 1 in a defined Swedish population. Cancer. 1997;79:2125–31. doi: 10.1002/(SICI)1097-0142(19970601)79:11<2125::AID-CNCR9>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 93.Kinoshita K, Hirota S, Isozaki K, Ohashi A, Nishida T, Kitamura Y, et al. Absence of c-kit gene mutations in gastrointestinal stromal tumours from neurofibromatosis type 1 patients. J Pathol. 2004;202:80–5. doi: 10.1002/path.1487. [DOI] [PubMed] [Google Scholar]
- 94.Maertens O, Prenen H, Debiec-Rychter M, Wozniak A, Sciot R, Pauwels P, et al. Molecular pathogenesis of multiple gastrointestinal stromal tumors in NF1 patients. Hum Mol Genet. 2006;15:1015–23. doi: 10.1093/hmg/ddl016. [DOI] [PubMed] [Google Scholar]
- 95.Lasota J, Miettinen M. KIT and PDGFRA mutations in gastrointestinal stromal tumors (GISTs) Semin Diagn Pathol. 2006;23:91–102. doi: 10.1053/j.semdp.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 96.Mussi C, Schildhaus HU, Gronchi A, Wardelmann E, Hohenberger P. Therapeutic consequences from molecular biology for gastrointestinal stromal tumor patients affected by neurofibromatosis type 1. Clin Cancer Res. 2008;14:4550–5. doi: 10.1158/1078-0432.CCR-08-0086. [DOI] [PubMed] [Google Scholar]
- 97.Yamamoto H, Tobo T, Nakamori M, Imamura M, Kojima A, Oda Y, et al. Neurofibromatosis type 1-related gastrointestinal stromal tumors: a special reference to loss of heterozygosity at 14q and 22q. J Cancer Res Clin Oncol. 2009;135:791–8. doi: 10.1007/s00432-008-0514-z. [DOI] [PubMed] [Google Scholar]
- 98.Gronchi A. Risk stratification models and mutational analysis: keys to optimising adjuvant therapy in patients with gastrointestinal stromal tumour. Eur J Cancer. 2013;49:884–92. doi: 10.1016/j.ejca.2012.10.025. [DOI] [PubMed] [Google Scholar]
- 99.Group ESESNW Gastrointestinal stromal tumours: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014;25(Suppl 3):iii21–6. doi: 10.1093/annonc/mdu255. [DOI] [PubMed] [Google Scholar]
- 100.Maki RG, Blay JY, Demetri GD, Fletcher JA, Joensuu H, Martin-Broto J, et al. Key issues in the clinical management of gastrointestinal stromal tumors: an expert discussion. Oncologist. 2015;20:823–30. doi: 10.1634/theoncologist.2014-0471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Killian JK, Kim SY, Miettinen M, Smith C, Merino M, Tsokos M, et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 2013;3:648–57. doi: 10.1158/2159-8290.CD-13-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Raimundo N, Baysal BE, Shadel GS. Revisiting the TCA cycle: signaling to tumor formation. Trends Mol Med. 2011;17:641–9. doi: 10.1016/j.molmed.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Miettinen M, Wang ZF, Sarlomo-Rikala M, Osuch C, Rutkowski P, Lasota J. Succinate dehydrogenase-deficient GISTs: a clinicopathologic, immunohistochemical, and molecular genetic study of 66 gastric GISTs with predilection to young age. Am J Surg Pathol. 2011;35:1712–21. doi: 10.1097/PAS.0b013e3182260752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mason EF, Hornick JL. Succinate dehydrogenase deficiency is associated with decreased 5-hydroxymethylcytosine production in gastrointestinal stromal tumors: implications for mechanisms of tumorigenesis. Mod Pathol. 2013;26:1492–7. doi: 10.1038/modpathol.2013.86. [DOI] [PubMed] [Google Scholar]
- 105.Killian JK, Miettinen M, Walker RL, Wang Y, Zhu YJ, Waterfall JJ, et al. Recurrent epimutation of SDHC in gastrointestinal stromal tumors. Sci Transl Med. 2014;6:268ra177. [DOI] [PMC free article] [PubMed]
- 106.Dwight T, Mann K, Benn DE, Robinson BG, McKelvie P, Gill AJ, et al. Familial SDHA mutation associated with pituitary adenoma and pheochromocytoma/paraganglioma. J Clin Endocrinol Metab. 2013;98:E1103–8. [DOI] [PubMed]
- 107.Ricketts CJ, Forman JR, Rattenberry E, Bradshaw N, Lalloo F, Izatt L, et al. Tumor risks and genotype-phenotype-proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Hum Mutat. 2010;31:41–51. doi: 10.1002/humu.21136. [DOI] [PubMed] [Google Scholar]
- 108.Oudijk L, Gaal J, Korpershoek E, van Nederveen FH, Kelly L, Schiavon G, et al. SDHA mutations in adult and pediatric wild-type gastrointestinal stromal tumors. Mod Pathol. 2013;26:456–63. doi: 10.1038/modpathol.2012.186. [DOI] [PubMed] [Google Scholar]
- 109.Pappo AS, Janeway K, Laquaglia M, Kim SY. Special considerations in pediatric gastrointestinal tumors. J Surg Oncol. 2011;104:928–32. doi: 10.1002/jso.21868. [DOI] [PubMed] [Google Scholar]
- 110.Zhang L, Smyrk TC, Young WF, Jr, Stratakis CA, Carney JA. Gastric stromal tumors in Carney triad are different clinically, pathologically, and behaviorally from sporadic gastric gastrointestinal stromal tumors: findings in 104 cases. Am J Surg Pathol. 2010;34:53–64. doi: 10.1097/PAS.0b013e3181c20f4f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Carney JA, Stratakis CA. Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet. 2002;108:132–9. doi: 10.1002/ajmg.10235. [DOI] [PubMed] [Google Scholar]
- 112.Miettinen M, Lasota J. Succinate dehydrogenase deficient gastrointestinal stromal tumors (GISTs) - a review. Int J Biochem Cell Biol. 2014;53:514–9. doi: 10.1016/j.biocel.2014.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chou A, Chen J, Clarkson A, Samra JS, Clifton-Bligh RJ, Hugh TJ, et al. Succinate dehydrogenase-deficient GISTs are characterized by IGF1R overexpression. Mod Pathol. 2012;25(9):1307–13. doi: 10.1038/modpathol.2012.77. [DOI] [PubMed] [Google Scholar]
- 114.Dwight T, Benn DE, Clarkson A, Vilain R, Lipton L, Robinson BG, et al. Loss of SDHA expression identifies SDHA mutations in succinate dehydrogenase-deficient gastrointestinal stromal tumors. Am J Surg Pathol. 2013;37:226–33. doi: 10.1097/PAS.0b013e3182671155. [DOI] [PubMed] [Google Scholar]
- 115.Miettinen M, Killian JK, Wang ZF, Lasota J, Lau C, Jones L, et al. Immunohistochemical loss of succinate dehydrogenase subunit A (SDHA) in gastrointestinal stromal tumors (GISTs) signals SDHA germline mutation. Am J Surg Pathol. 2013;37:234–40. doi: 10.1097/PAS.0b013e3182671178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Perez-Atayde AR, Shamberger RC, Kozakewich HW. Neuroectodermal differentiation of the gastrointestinal tumors in the Carney triad. An ultrastructural and immunohistochemical study. Am J Surg Pathol. 1993;17:706–14. doi: 10.1097/00000478-199307000-00008. [DOI] [PubMed] [Google Scholar]
- 117.McWhinney SR, Pasini B, Stratakis CA, International Carney T, Carney-Stratakis SC. Familial gastrointestinal stromal tumors and germ-line mutations. N Engl J Med. 2007;357:1054–6. doi: 10.1056/NEJMc071191. [DOI] [PubMed] [Google Scholar]
- 118.Pasini B, McWhinney SR, Bei T, Matyakhina L, Stergiopoulos S, Muchow M, et al. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet. 2008;16:79–88. doi: 10.1038/sj.ejhg.5201904. [DOI] [PubMed] [Google Scholar]
- 119.Korpershoek E, Favier J, Gaal J, Burnichon N, van Gessel B, Oudijk L, et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J Clin Endocrinol Metab. 2011;96:E1472–6. doi: 10.1210/jc.2011-1043. [DOI] [PubMed] [Google Scholar]
- 120.Wagner AJ, Remillard SP, Zhang YX, Doyle LA, George S, Hornick JL. Loss of expression of SDHA predicts SDHA mutations in gastrointestinal stromal tumors. Mod Pathol. 2013;26:289–94. doi: 10.1038/modpathol.2012.153. [DOI] [PubMed] [Google Scholar]
- 121.Burnichon N, Briere JJ, Libe R, Vescovo L, Riviere J, Tissier F, et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet. 2010;19:3011–20. doi: 10.1093/hmg/ddq206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pantaleo MA, Astolfi A, Indio V, Moore R, Thiessen N, Heinrich MC, et al. SDHA loss-of-function mutations in KIT-PDGFRA wild-type gastrointestinal stromal tumors identified by massively parallel sequencing. J Natl Cancer Inst. 2011;103:983–7. doi: 10.1093/jnci/djr130. [DOI] [PubMed] [Google Scholar]
- 123.Italiano A, Chen CL, Sung YS, Singer S, DeMatteo RP, LaQuaglia MP, et al. SDHA loss of function mutations in a subset of young adult wild-type gastrointestinal stromal tumors. BMC Cancer. 2012;12:408. doi: 10.1186/1471-2407-12-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pantaleo MA, Astolfi A, Urbini M, Nannini M, Paterini P, Indio V, et al. Analysis of all subunits, SDHA, SDHB, SDHC, SDHD, of the succinate dehydrogenase complex in KIT/PDGFRA wild-type GIST. Eur J Hum Genet. 2014;22:32–9. doi: 10.1038/ejhg.2013.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nannini M, Biasco G, Astolfi A, Pantaleo MA. An overview on molecular biology of KIT/PDGFRA wild type (WT) gastrointestinal stromal tumours (GIST) J Med Genet. 2013;50:653–61. doi: 10.1136/jmedgenet-2013-101695. [DOI] [PubMed] [Google Scholar]
- 126.Tenorio Jimenez C, Izatt L, Chang F, Moonim MT, Carroll PV, McGowan BM. Carney Stratakis syndrome in a patient with SDHD mutation. Endocr Pathol. 2012;23:181–6. doi: 10.1007/s12022-012-9213-z. [DOI] [PubMed] [Google Scholar]
- 127.Hensen EF, Jordanova ES, van Minderhout IJ, Hogendoorn PC, Taschner PE, van der Mey AG, et al. Somatic loss of maternal chromosome 11 causes parent-of-origin-dependent inheritance in SDHD-linked paraganglioma and phaeochromocytoma families. Oncogene. 2004;23:4076–83. doi: 10.1038/sj.onc.1207591. [DOI] [PubMed] [Google Scholar]
- 128.Yeap PM, Tobias ES, Mavraki E, Fletcher A, Bradshaw N, Freel EM, et al. Molecular analysis of pheochromocytoma after maternal transmission of SDHD mutation elucidates mechanism of parent-of-origin effect. J Clin Endocrinol Metab. 2011;96:E2009–13. doi: 10.1210/jc.2011-1244. [DOI] [PubMed] [Google Scholar]
- 129.Bayley JP, Oldenburg RA, Nuk J, Hoekstra AS, van der Meer CA, Korpershoek E, et al. Paraganglioma and pheochromocytoma upon maternal transmission of SDHD mutations. BMC Med Genet. 2014;15:111. doi: 10.1186/s12881-014-0111-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Haller F, Moskalev EA, Faucz FR, Barthelmess S, Wiemann S, Bieg M, et al. Aberrant DNA hypermethylation of SDHC: a novel mechanism of tumor development in Carney triad. Endocr Relat Cancer. 2014;21:567–77. doi: 10.1530/ERC-14-0254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Songdej N, von Mehren M. GIST treatment options after tyrosine kinase inhibitors. Curr Treat Options Oncol. 2014;15:493–506. doi: 10.1007/s11864-014-0295-3. [DOI] [PubMed] [Google Scholar]
- 132.Matyakhina L, Bei TA, McWhinney SR, Pasini B, Cameron S, Gunawan B, et al. Genetics of carney triad: recurrent losses at chromosome 1 but lack of germline mutations in genes associated with paragangliomas and gastrointestinal stromal tumors. J Clin Endocrinol Metab. 2007;92:2938–43. doi: 10.1210/jc.2007-0797. [DOI] [PubMed] [Google Scholar]
- 133.Huss S, Elges S, Trautmann M, Sperveslage J, Hartmann W, Wardelmann E. Classification of KIT/PDGFRA wild-type gastrointestinal stromal tumors: implications for therapy. Expert Rev Anticancer Ther. 2015;15:623–8. doi: 10.1586/14737140.2015.1032941. [DOI] [PubMed] [Google Scholar]
- 134.Celestino R, Lima J, Faustino A, Maximo V, Gouveia A, Vinagre J, et al. A novel germline SDHB mutation in a gastrointestinal stromal tumor patient without bona fide features of the Carney-Stratakis dyad. Fam Cancer. 2012;11:189–94. doi: 10.1007/s10689-011-9499-x. [DOI] [PubMed] [Google Scholar]
- 135.Kim SY, Janeway K, Pappo A. Pediatric and wild-type gastrointestinal stromal tumor: new therapeutic approaches. Curr Opin Oncol. 2010;22:347–50. doi: 10.1097/CCO.0b013e32833aaae7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Vadakara J, von Mehren M. Gastrointestinal stromal tumors: management of metastatic disease and emerging therapies. Hematol Oncol Clin North Am. 2013;27:905–20. doi: 10.1016/j.hoc.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Stratakis CA, Carney JA. The triad of paragangliomas, gastric stromal tumours and pulmonary chondromas (Carney triad), and the dyad of paragangliomas and gastric stromal sarcomas (Carney-Stratakis syndrome): molecular genetics and clinical implications. J Intern Med. 2009;266:43–52. doi: 10.1111/j.1365-2796.2009.02110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Boikos SA, Xekouki P, Fumagalli E, Faucz FR, Raygada M, Szarek E et al. Carney triad can be (rarely) associated with germline succinate dehydrogenase defects. Eur J Hum Genet. 2015. [DOI] [PMC free article] [PubMed]
- 139.Alrashdi I, Bano G, Maher ER, Hodgson SV. Carney triad versus Carney Stratakis syndrome: two cases which illustrate the difficulty in distinguishing between these conditions in individual patients. Fam Cancer. 2010;9:443–7. doi: 10.1007/s10689-010-9323-z. [DOI] [PubMed] [Google Scholar]
- 140.Ayala-Ramirez M, Callender GG, Kupferman ME, Rich TA, Chuang HH, Trent J, et al. Paraganglioma syndrome type 1 in a patient with Carney-Stratakis syndrome. Nat Rev Endocrinol. 2010;6:110–5. doi: 10.1038/nrendo.2009.250. [DOI] [PubMed] [Google Scholar]
- 141.Benn DE, Robinson BG, Clifton-Bligh RJ. 15 YEARS OF PARAGANGLIOMA: clinical manifestations of paraganglioma syndromes types 1–5. Endocr Relat Cancer. 2015;22:T91–103. doi: 10.1530/ERC-15-0268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Jiang Q, Zhang Y, Zhou YH, Hou YY, Wang JY, Li JL, et al. A novel germline mutation in SDHA identified in a rare case of gastrointestinal stromal tumor complicated with renal cell carcinoma. Int J Clin Exp Pathol. 2015;8:12188–97. [PMC free article] [PubMed] [Google Scholar]
- 143.Gill AJ, Pachter NS, Chou A, Young B, Clarkson A, Tucker KM, et al. Renal tumors associated with germline SDHB mutation show distinctive morphology. Am J Surg Pathol. 2011;35:1578–85. doi: 10.1097/PAS.0b013e318227e7f4. [DOI] [PubMed] [Google Scholar]
- 144.Gill AJ, Lipton L, Taylor J, Benn DE, Richardson AL, Frydenberg M, et al. Germline SDHC mutation presenting as recurrent SDH deficient GIST and renal carcinoma. Pathology. 2013;45:689–91. doi: 10.1097/PAT.0000000000000018. [DOI] [PubMed] [Google Scholar]
- 145.Agarwal R, Robson M. Inherited predisposition to gastrointestinal stromal tumor. Hematol Oncol Clin North Am. 2009;23:1–13. doi: 10.1016/j.hoc.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 146.Larghi A, Fuccio L, Chiarello G, Attili F, Vanella G, Paliani GB, et al. Fine-needle tissue acquisition from subepithelial lesions using a forward-viewing linear echoendoscope. Endoscopy. 2014;46:39–45. doi: 10.1055/s-0033-1344895. [DOI] [PubMed] [Google Scholar]
- 147.Ricci R, Chiarello G, Attili F, Fuccio L, Alfieri S, Persiani R, et al. Endoscopic ultrasound-guided fine needle tissue acquisition biopsy samples do not allow a reliable proliferation assessment of gastrointestinal stromal tumours. Dig Liver Dis. 2015;47:291–5. doi: 10.1016/j.dld.2014.12.011. [DOI] [PubMed] [Google Scholar]
- 148.Antonescu CR. Gastrointestinal stromal tumor (GIST) pathogenesis, familial GIST, and animal models. Semin Diagn Pathol. 2006;23:63–9. doi: 10.1053/j.semdp.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 149.Gasparotto D, Rossi S, Campagna D, Scavina P, Tiziano FD, Marzotto A et al. Imatinib-Sensitizing KIT Mutation in a Carney-Stratakis-Associated GI Stromal Tumor. J Clin Oncol. 2014; doi:10.1200/JCO.2012.44.7300. [DOI] [PubMed]