

Abstract
The amiloride-sensitive epithelial Na+ channel (ENaC) is a key player in the regulation of Na+ homeostasis. Its functional activity is under continuous control by a variety of signaling molecules including bioactive peptides of endothelin family. Since ENaC dysfunction is causative for disturbances in total body Na+ levels associated with abnormal regulation of blood volume, blood pressure, and lung fluid balance, the uncovering the molecular mechanisms of inhibitory modulation or inappropriate activation of ENaC is crucial for the successful treatment of a variety of human diseases including hypertension. The precise regulation of ENaC is particularly important for normal Na+ and fluid homeostasis in organs where endothelins are known to act: kidneys, lung and colon. Inhibition of ENaC by endothelin-1 (ET-1) has been established in renal cells and several molecular mechanisms of inhibition of ENaC by ET-1 are proposed and will be reviewed in this chapter.
Keywords: sodium reabsorption, ET-1, blood pressure, collecting duct, signal transduction, ETRA, ETRB, βPix
1. Introduction
The amiloride-sensitive epithelial sodium channel (ENaC) is expressed in the apical membrane of polarized epithelial cells in the distal part of kidney tubule, the lung and the distal colon. These cells are in constant contact with other cells, which is achieved by specific reception and secretion of a number of regulatory molecules, including potent bioactive peptides endothelins. The family of endothelin proteins consists of 3 members: endothelin-1, endothelin-2 and endothelin-3 (ET-1, ET-2 and ET-3, respectively) known to play important roles in renal physiology and pathophysiology due to their involvement into the physiological control of blood pressure and salt homeostasis (Kohan, Inscho, Wesson, & Pollock, 2011; Kohan, Rossi, Inscho, & Pollock, 2011; Simonson & Dunn, 1993; Sorokin & Kohan, 2003). ET-1 is the main isoform which is secreted by renal cells and which also operates on a number of renal targets, including its effects in different parts of nephron on top of action as multifunctional bioactive peptides in renal mesangium (Sorokin, 2011). Its production is largely controlled at the level of gene expression (Foschi et al., 2001; Sorokin & Kohan, 2003). ET-1 appears to be an important negative regulator of ENaC in vivo and endothelin-dependent regulation of ENaC activity, critical aspect of the functional role of endothelins, will be the subject of this review.
2. Regulation of sodium reabsorption – role of ENaC
Hypertension represents one of the most prevalent chronic health problems of Western nations but the mechanisms responsible for this disease remain poorly understood. Long term control of blood pressure involves Na+ homeostasis through the precise regulation of the epithelial Na+ channels (ENaC) in the aldosterone-sensitive distal nephron (ASDN). Although only a small percent of the glomerular filtrate (less than 5–10%) reaches the connecting tubules (CNT) and collecting ducts (CD), these segments are critical for water and electrolyte homeostasis since fine tuning of electrolyte and fluid balance is mediated in these nephron segments through reabsorption and secretion and these processes are under tight control of hormones and non-hormonal factors (Staruschenko, 2012). Na+ reabsorption in the CNT and CDs is transcellular and is mediated by the connecting tubule cells and the principal cells of CDs. Shown on Figure 1 is a structure of the nephron with segments expressing ENaC. At the basolateral membrane of the principal cells Na+ extrusion is mediated by the Na+/K+-ATPase, which also provides the electrochemical driving force for the apical entry of Na+ (Feraille & Doucet, 2001). ENaC, which appears to be a trimeric channel comprised of 1 α-, 1 β-, and 1 γ-subunits (Jasti, Furukawa, Gonzales, & Gouaux, 2007; Staruschenko, Adams, Booth, & Stockand, 2005) is responsible for sodium reabsorption in these segments. ENaC dysfunction is causative for disturbances in total body Na+ levels associated with abnormal regulation of blood volume and blood pressure, as well as alterations in lung fluid balance (Bhalla & Hallows, 2008; Pearce et al., 2014; Soundararajan, Pearce, Hughey, & Kleyman, 2010; Rossier, 2014; Alvarez, Navarro-Gonzalez, & Giraldez, 2013; Warnock et al., 2014). Mutations in genes encoding the ENaC subunits corroborate critical role of this channel in the control of blood pressure. For instance, Liddle Syndrome is an autosomal dominant form of hypertension that results from the C-terminal truncation mutations in the β- or γ-ENaC subunits, which prevents the channel’s retrieval from the apical membrane and subsequent degradation, thus leading to increased basal ENaC expression and activity at the apical membrane (Hansson et al., 1995a; Hansson et al., 1995b; Shimkets et al., 1994; Lifton, Gharavi, & Geller, 2001). Loss-of-function mutations in any of the three different ENaC subunits also cause the autosomal recessive form of Pseudohypoaldosteronism type I (PHAI) (Chang et al., 1996; Lifton et al., 2001; Grunder et al., 1997). ENaC-mediated electrogenic sodium entry also provides the driving force for luminal potassium exit via potassium (renal outer medullary K channel (ROMK) and large conductance calcium-activated Maxi-K (BK)) channels (Staruschenko, 2012; Wen, Cornelius, & Sansom, 2014; Welling, 2013). Mutations in ROMK channel result in the Type II Bartter syndrome (Simon et al., 1996; Welling & Ho, 2009; Srivastava et al., 2013).
Figure 1.
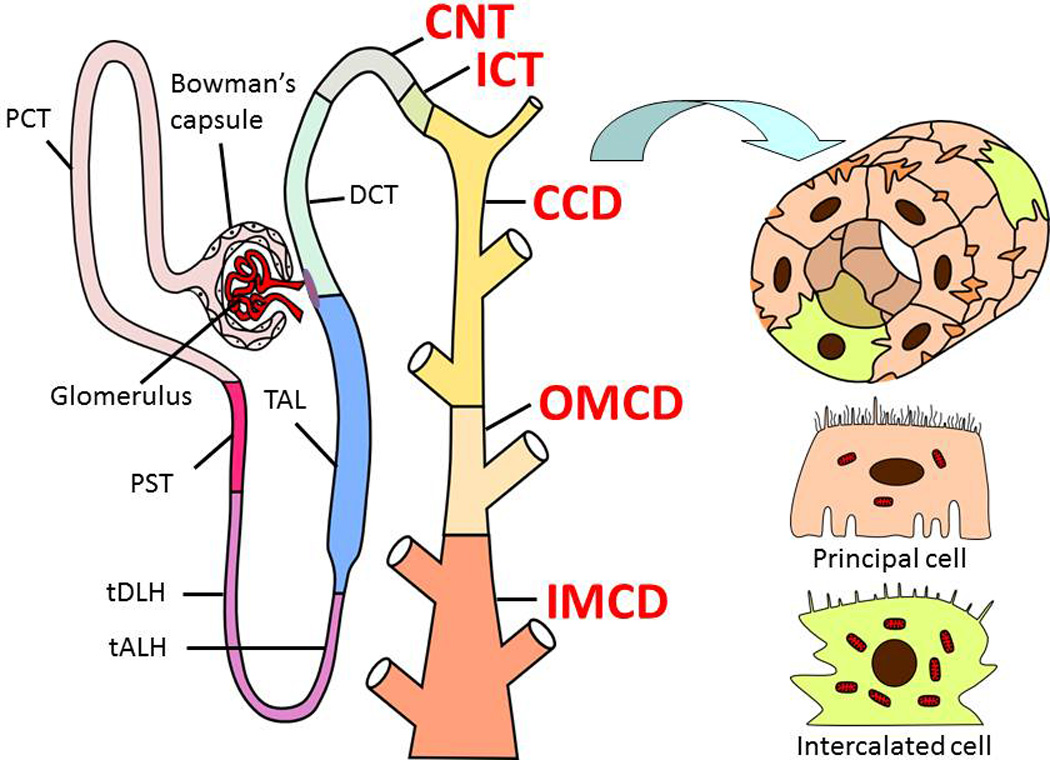
Structure of the nephron and specific segments involved in effects of ET-1 on ENaC. Every nephron contains a renal corpuscle (glomerulus and Bowman's capsule), a proximal tubule (proximal convoluted and straight tubules; PCT and PST, respectively), a loop of Henle (thin descending limb of Henle’s loop (tDLH), thin ascending limb of Henle’s loop (tALH), and thick ascending limb of Henle’s loop (TAL)), a distal convoluted tubule (DCT), connecting tubule (CNT), and the collecting duct system, which includes the initial collecting tubule (ICT), the cortical collecting duct (CCD), the outer medullary collecting duct (OMCD), and the inner medullary collecting duct (IMCD). Structure of the CCD as a cross-section and schematic presentation of principal and intercalated cells that comprise these segments are also shown. Modified from (Staruschenko, 2012).
The expression and activity of ENaCs are regulated by specific hormones and different extra- and intracellular regulatory mechanisms. Considering that ENaC is responsible for the fine-tuning of sodium reabsorption in the last nephron segment, the role of this channel in sodium reabsorption in the kidney in critical and unique. The tight regulation of transcellular Na+ concentrations is so important that multiple mechanisms work in concert to control them. One of the main mechanisms controlling ENaC activity is activation of the RAAS (Pearce et al., 2014; Rossier, 2014; Quinn, Harvey, & Thomas, 2014; Alvarez et al., 2013). Activation of the RAAS is well known to enhance activity of ENaC. Aldosterone, as well as other corticosteroid hormones, binds to the mineralocorticoid receptor and triggers coordinated changes in the expression of multiple aldosterone-induced genes in a ligand-dependent manner (Stockand, 2002; Kone, 2013). Hyperaldosteronismis results in enhanced sodium reabsorption in the distal nephron, whereas aldosterone insufficiency leads to reduced ENaC activity and urinary sodium wasting. However, substantial experimental data suggest a role for aldosterone-independent mechanisms in regulation of ENaC (Zaika, Mamenko, Staruschenko, & Pochynyuk, 2013). For instance, mice lacking mineralocorticoid receptors still possess ENaC-mediated Na+ reabsorption in the ASDN (Berger et al., 1998). Besides, it was reported that mice with surgically removed adrenal glands have no detectable circulating aldosterone, but possess robust ENaC activity in the distal nephron (Mironova, Bugaj, Roos, Kohan, & Stockand, 2012). Recent studies further revealed that ENaC activity remained above control levels during maximal mineralocorticoid receptors inhibition with spironolactone (Mamenko et al., 2013). Therefore, other signaling pathways, which modulate ENaC either acutely or at transcription level, work in parallel with the RAAS.
Current chapter is focused on the control of ENaC by ET-1. However, as noticed above, multiple mechanisms are involved in the tight control of ENaC expression and activity in the ASDN. Hormones are key regulators of sodium transport in the kidney and in ASDN specifically. In addition to the RAAS, it was shown that arginine vasopressin (AVP) (Ecelbarger et al., 2000; Mironova et al., 2012; Stockand, 2012; Bankir, Bouby, & Ritz, 2013; Bankir et al., 2013; Sanghi et al., 2014), atrial natriuretic peptide (ANP) (Kudo & Baird, 1984; Wang et al., 2006; Guo, Alli, Eaton, & Bao, 2013) and insulin (alongside with Insulin-like growth factor 1, IGF-1) (Blazer-Yost, Liu, & Helman, 1998; Li et al., 2013; Ilatovskaya, Pavlov, Levchenko, & Staruschenko, 2013; Pavlov et al., 2013a) are critical modulators of ENaC activity. The kallikrein-kinin systems through its peptide bradykinin also play a specific role in blunting ENaC activity, particularly under conditions of elevated sodium intake (Zaika, Mamenko, O'Neil, & Pochynyuk, 2011; Mamenko, Zaika, Doris, & Pochynyuk, 2012; Mamenko, Zaika, & Pochynyuk, 2014). In addition to hormonal regulation of ENaC-mediated sodium transport in the kidney, a number of local autocrine and paracrine factors play critical role in the modulation of ENaC. For instance, recent review article by Stockand and colleagues highlight regulation of ENaC-mediated sodium excretion and blood pressure by purinergic signaling (Mironova, Boiko, Bugaj, Kucher, & Stockand, 2014). Multiple evidence reveal that a robust inhibitory purinergic signaling system intrinsic to the ASDN dynamically regulates ENaC through paracrine ATP signaling via the metabotropic P2Y2 purinergic receptor to properly match urinary Na+ excretion to dietary Na+ intake (Pochynyuk et al., 2008; Pochynyuk et al., 2010; Rieg et al., 2007; Rieg, Gerasimova, Boyer, Insel, & Vallon, 2011; Birch, Schwiebert, Peppiatt-Wildman, & Wildman, 2013; Stockand et al., 2010). This enables blood pressure to be maintained within a normal range despite broad changes in dietary Na+ consumption. We and others also identified that members of the epidermal growth factors (EGF) are involved in the control of ENaC ((reviewed in (Staruschenko, Palygin, Ilatovskaya, & Pavlov, 2013)). Using the Dahl salt-sensitive rat model, we evaluated the role of EGF and identified that deficiency of renal cortical EGF increases ENaC activity and contributes to salt-sensitive hypertension (Pavlov et al., 2013b). Prostaglandins, cytochrome P450 metabolites, nitric oxide, peroxisome proliferator-activated receptor agonists and other molecules are also among important paracrine and autocrine factors modulating ENaC activity.
3. Endothelin signaling and control of blood pressure
3.1. Endothelin Receptors
Endothelins are multifunctional 21 amino acid vasoactive peptides secreted by numerous cell types (Simonson & Dunn, 1993). All of endothelin’s effects are elicited by binding to specific G-protein coupled receptors Endothelin Receptor A (ETRA) (Arai, Hori, Aramori, Ohkubo, & Nakanishi, 1990) and Endothelin Receptor B (ETRB) (Sakurai et al., 1990). ETRA and ETRB demonstrate considerable variability in the distribution between different tissues and among species (Pollock, 1998). Human and rat renal tubular cells, vascular smooth muscle cells, glomerular mesangial cells and many other cell types express substantial levels of both ETRA and ETRB (Orth et al., 2000; Kohan et al., 2011; Bouallegue, Daou, & Srivastava, 2007; Sorokin, 2011). A progressive renal injury like inflammation and fibrosis are mediated via both ETRA and ETRB receptors, while constrictor effects are primarily transduced by ETRA receptors (Neuhofer & Pittrow, 2006). Thus, actions of ET-1 depends on the cell type and physiological situation (Vignon-Zellweger, Heiden, & Emoto, 2011)
3.2. Endothelin Signaling
ET-1 receptors couple to members of the Gαi, Gαq, Gαs and Gα12/13 G protein families (Sorokin & Kohan, 2003). Accordingly, when ET-1 binds to its receptor the multiple intracellular pathways are triggered (Sorokin, Foschi, & Dunn, 2002; Sorokin, 2011). Among most important cellular events are activation of phospholipases A, C and D, activation of serine-threonine kinases, increased protein tyrosine phosphorylation and a stimulated influx of Ca2+ from outside the cell as well as mobilization of intracellular Ca2+ from internal sources.
ET-1 acts via ETRA to induce GTP loading of Gαq and Gαs to stimulate phospholipase C (PLC) and adenylate cyclase respectively. Correspondingly, ETRB stimulates PLC via Gαq, and causes the inhibition of adenylate cyclase through Gαi. The ET receptors co-localize and interact with caveolin-1 in caveolae and since caveolae integrates different receptor and signaling proteins it adds to the complexity of ET-1 signaling. It is of note, that activation of endothelin receptors leads to the compartmentalization and the binding of Gαq to guanine exchange factor βPix (p21-activated kinase (PAK)-interacting exchange factor β) in caveolae (Chahdi & Sorokin, 2010b). Another important feature of ET-1 signaling is transactivation of growth factor receptors, such as EGF receptor (Chahdi & Sorokin, 2010a; Hua, Munk, & Whiteside, 2003; Vacca, Bagnato, Catt, & Tecce, 2000; Iwasaki, Eguchi, Ueno, Marumo, & Hirata, 1999; Kodama et al., 2002) and PDGF receptor (Harada et al., 2014). This ability ET-1 shares with some other ligands of G-protein coupled receptors (Little, 2013; Daub, Weiss, Wallasch, & Ullrich, 1996).
The contraction of smooth muscle and glomerular mesangial cells induced by ET-1 depends upon stimulation of inositol triphosphate (IP3) generation and elevation of cytosolic free Ca2+ concentration ([Ca2+]i) (Badr et al., 1989; Bouallegue et al., 2007). The calcium influx and mobilization pathways activated by ET-1 vary immensely and are cell-type-specific with regard to which ET receptor subtypes are involved (Tykocki & Watts, 2010). Initial transient increase in ([Ca2+]i) after ET-1 treatment is followed by a sustained lesser increase. It has been shown that early transient peak reflects IP3-mediated release of Ca2+ from intracellular sources, which is associated with cell alkalinization through augmented Na+/H+ exchange (Simonson et al., 1989; Simonson & Dunn, 1991; Simonson & Dunn, 1993). The continued plateau of [Ca2+]i which over time decreases towards the initial level is due to the increased influx of Ca2+ across the plasma membrane. This pattern of initial increase followed by sustained influx is induced by ET-1 in both vascular and non-vascular cells (Tykocki & Watts, 2010). Vascular smooth muscle cells and glomerular mesangial cells express L-type Ca2+ channels (McDermott, Hurst, & Whiteside, 1993; Sonkusare et al., 2006) as well as transient receptor potential canonical (TRPC) proteins, a family of nonselective Ca2+-permeable cation channels (Du et al., 2007; Gonzalez-Cobos & Trebak, 2010). Members of TRPC family which were shown to be activated by stimulation of endothelin receptors in various cells or tissues include TRPC1, TRPC3, TRPC6 and TRPC7 (Horinouchi, Terada, Higashi, & Miwa, 2013). Low concentrations of ET-1 added to mesangial cells induced slow sustained increase in [Ca2+]i through a voltage-channel-independent mechanism, whereas higher than 100 pM concentrations caused a transient increase of [Ca2+]i, which was dependent on Ca2+ release from intracellular stores and required activation of PLC and Protein kinase C (PKC) (Simonson et al., 1989).
Endothelin-mediated increase of [Ca2+]i causes the activation of calcium-dependent intracellular tyrosine kinase Pyk2 (Sorokin, Kozlowski, Graves, & Philip, 2001; Park, Bose, Leszyk, & Czech, 2001; Kawanabe, Hashimoto, & Masaki, 2003) providing therefore the link between G-protein coupled endothelin receptors and the induction of tyrosine phosphorylation via mobilization of intracellular calcium. Activation of Pyk2 in ET-1-treated cells requires not only Ca2+ mobilization, but also an increased activity of Src kinases (Sorokin et al., 2001). ET-1 was shown to activate Src kinases (Simonson & Herman, 1993) and it appears that at least some members of Src family are recruited to the complex of β-arrestin 1 with ETRA in an agonist-dependent manner (Imamura et al., 2001). Pyk2/p130Cas/BCAR3/Rap1 signaling cascade activated via ETRA receptors is involved in regulation of adhesion and spreading of mesangial cells (Rufanova, Alexanian, Wakatsuki, Lerner, & Sorokin, 2009). Pyk2 also mediates ET-1 signaling to GLUT4 glucose transporters in 3T3-L1 adipocytes (Park et al., 2001).
The ability of ET-1 to act as a mitogen is well established (Bouallegue et al., 2007; Fukuda et al., 1996; Sorokin, 2011). Since GTP-loading of the small GTPase Ras, which is followed by activation of the Raf-MEK-ERK signaling pathway, is indispensable for cell proliferation, it is not surprising that ET-1 is not only a potent inducer of Ras activation, but also is an effective stimulator of ERK signaling cascade (Wang, Simonson, Pouyssegur, & Dunn, 1992; Foschi, Chari, Dunn, & Sorokin, 1997). Remarkably, other mitogen-activated protein kinases (MAPK) cascades: JNK/SAPK (Araki, Haneda, Togawa, & Kikkawa, 1997), p38 MAP kinase (Sorokin et al., 2001) and ERK5 (Dorado et al., 2008) are also induced by ET-1. The significance of activation of these MAPKs by ET-1 has been investigated in different cellular systems and is partially responsible for multifunctional activity of endothelins.
3.3. Targeting Endothelins
The increase of ET-1 production accompanies the wide variety of renal diseases and since the most efficient way to interfere with excessive ET-1 action is to block its specific reception by ETR receptors, the pharmacological blockade of ETR received unprecedented attention from multiple pharmaceutical companies. Quite a few nonselective (targeting both ETRA and ETRB) and selective (targeting either ETRA or ETRB) antagonists were created and evaluated for their efficiency (Motte, McEntee, & Naeije, 2005). This class of compounds showed noteworthy promise in different animal models of renal and cardiovascular diseases and sufficient rationale for testing them for treatment of human pathologies was obtained (Benigni et al., 1993; Kohan & Pollock, 2013; Boffa, Tharaux, Dussaule, & Chatziantoniou, 2001; Dhein et al., 2000; Fukuda et al., 1996; Gomez-Garre et al., 1996; Neuhofer & Pittrow, 2006). Numerous clinical trials addressing the benefits of blockade of ETR receptors in the treatment of cardiovascular and renal diseases has been initiated. It was reported that ETR antagonists can reverse proteinuric renal disease and glomerulosclerosis, and their remarkable antiproteinuric effects are additive to those of standard antiproteinuric therapy (Barton, 2008). There are two ETRA antagonists approved in the United States for the treatment of pulmonary hypertension: Ambrisentan and Bosentan. Sitaxsentan, the most selective ETRA antagonist clinically available, has been approved for use in Europe, Canada, and Australia but not in the USA. It was however recently voluntarily withdrawn by Pfizer driven by a review of evolving safety information from clinical trials regarding concerns about idiosyncratic, fatal hepatic failure (Raja & Raja, 2011). Nevertheless, experimental data and recent clinical studies suggest that, despite limitations connected with side effects and relatively poor antagonist selectivity, the antagonists of ETRA, hold promise in the treatment of hypertension and diabetic nephropathy (Meyers & Sethna, 2013). Antagonists of the ETRB are not usually beneficial since they might cause vasoconstriction along with salt and water retention. The blockade of ET-1 actions reduces the progression of chronic kidney disease and might even be beneficial in dialysis patients, even though the potential therapeutic effect of ETR antagonists for treatment of end-stage kidney disease still requires the verification by clinical trials (Kohan & Pollock, 2013).
3.4. Endothelin-mediated control of blood pressure
Regulation of blood pressure by endothelin is a result of complex interweaving of endothelin’s effects upon vasculature, humoral systems, nervous system, heart function and specialized kidney cells (Kohan et al., 2011). It is generally accepted that effects of endothelin upon blood pressure and Na+ homeostasis are the consequences of vasodilation caused by stimulated nitric oxide (NO) production by endothelial cells, attenuation of water reabsorption in the CD, decrease of Na+ transport in several segments of nephron, impairment of afferent nerve inputs and inhibition of renin release (Kohan et al., 2011).
ET-1 is among the most potent vasoconstrictors. In terms of contractility, the net effect of ETRA activation is contraction, whereas the net effect of activation of ETRB tends to be vasorelaxation (Sorokin & Kohan, 2003). Renal microvascular smooth muscle cells which express both ETRA and ETRB react to ET-1 by vasoconstriction, whereas ETRB microvascular endothelial cells promote vasodilator responses (Guan & Inscho, 2011; Pollock, Keith, & Highsmith, 1995). The vasorelaxant effect of ET-1 is due to stimulated secretion of endothelium-derived relaxing factor NO (Levin, 1996). The ability of ETRB to induce NO release is linked to nitric oxide synthase (NOS) activation which is markedly increased when intracellular Ca2+ rises (Forstermann & Sessa, 2012). In turn, NO can modulate ET-1 activity via inhibiting ET-1 formation/secretion (Bourque, Davidge, & Adams, 2011). Since NO and ET-1 continuously interfere with each other actions, the pressure regulation by ET-1 and NO is more complex than simply their individual pressor and depressor action (Rapoport, 2014). Nonetheless, it is widely recognized that endothelial NOS keeps all types of blood vessels dilated, making NO/ET-1 interferences an important factor of endothelin-mediated control of blood pressure (Forstermann & Sessa, 2012).
The role of ET-1 in regulation of water reabsorption became evident from studies utilizing either in vivo administration of ET-1 or analysis of mice with CD specific knockouts of ET-1. Administration of ET-1 inhibits vasopressin-stimulated osmotic water permeability in CDs (Oishi, Nonoguchi, Tomita, & Marumo, 1991; Nadler, Zimpelmann, & Hebert, 1992). Mice lacking ET-1 production in CD, the major renal site of ET-1 production, have impaired excretion of an acute, but not a chronic, water load suggesting that ET-1 functions as a physiological autocrine regulator of vasopressin-mediated water permeability in the CDs (Ge et al., 2005; Kohan, 2013). These effects seem to be mediated by ETRB, since blockade of ETRA was inefficient, while agonists of ETRB had inhibitory effect (Kohan, 2013). The data obtained with ETRB CD specific knockout indicates however that ETRB of CD only partially mediates the antihypertensive and natriuretic effects of ET-1 (Ge et al., 2006). It is likely, that ET-1 effects are associated with via inhibition of vasopressin-stimulated cAMP accumulation, which is achieved by ET-1 triggered elevation of intracellular Ca2+ and PLC-mediated activation of PKC (Teitelbaum & Berl, 1994; Kohan, 2013).
The overall action of ET-1 to increase blood pressure and vascular tone is partially due to ET-1-mediated stimulation of the sympathetic nervous system (Agapitov & Haynes, 2002; Kohan et al., 2011). The renal tissues are densely innervated by sympathetic nerves and increases in renal sympathetic activity, to which ET-1 contributes importantly, decreases urinary sodium excretion (Kopp, 2011). Endothelins are significantly involved in modulation of the activation of the afferent renal nerves and can induce the release of neurotransmitters, promote the development of postganglionic neurons and influence the generation of action potentials (Kopp, 2011; Kohan et al., 2011). Even though the mechanisms responsible for effects of ET-1 in ganglia, peripheral nerves and central nervous system resulting in blood regulation are not sufficiently studied, ET-1 and its receptors have been implicated in the enhanced sympathetic excitability associated with salt-sensitive hypertension, regulation of baroreflex activity and systemic hemodynamics (Kohan et al., 2011).
ET-1 was shown to inhibit renin release from rat kidney cortical slices and isolated juxtaglomerular cells (Moe, Tejedor, Campbell, Alpern, & Henrich, 1991; Kramer et al., 1994). Studies utilizing administration of ET-1 intravenously suggest that endothelin inhibits renin release in vivo (Otsuka et al., 1989). Even though inhibition of renin release by ET-1 in vitro and in vivo has been documented a long time ago, there is no consensus on what is the physiological significance of this effect. It has been hypothesized that high salt diet induces ET-1 production which promotes Na excretion by attenuating the renal baroreceptor responsible for renin release (Kohan et al., 2011). It has been reported that ET-1 generation is increased by Ang II and that vasoconstrictor effects of Ang II are partially mediated by ET-1 (Imai et al., 1992). Furthermore, synergistic effect of ET-1 and Ang II on blood pressure has been observed in rats and enhancement of ET-1-induced vasoconstriction by Ang II was shown through upregulation of ETRA (Lin et al., 2014). The crosstalk between the angiotensin and endothelin system is complex and is observed in a number of cellular systems including cells of cerebrovasculature, hepatic stellate cells, renal mesangial and endothelial cells (Lopez-Ongil, Diez-Marques, Griera, Rodriguez-Puyol, & Rodriguez-Puyol, 2005; Konczalla et al., 2013; He, Miao, Li, & Qi, 2013) .
4. Collecting duct: ET-1 and ENaC
4.1. ET receptors expression and endothelin production in CDs
Renal ET-1 production is facilitated by shear stress, inflammation, oxidative stress, the Renin-Angiotensin-Aldosterone System (RAAS) and other systems and mechanisms (for review see (Kohan et al., 2011)). Changes in extracellular fluid volume also increase ET-1 levels. Importantly, most of these mechanisms result in activation of ENaC in ASDN. Thus, endothelin system might represent feedback mechanism, which plays protective role in the maintenance of ENaC-mediated sodium absorption in the kidney and blood pressure, respectively. As summarized in excellent reviews by Kohan and colleagues (Kohan et al., 2011; Kohan et al., 2011)), the CDs are the major source of ET-1 in the kidney and may produce and release more ET-1 than any other cell type in the body. Consistent with this conclusion is that mice with the CD specific ET-1 knock out have markedly reduced urinary ET-1 excretion in response to salt loading (Ahn et al., 2004). A numerous in vitro and ex vivo studies suggested that ET-1 inhibits Na+ and water reabsorption in the CDs (Takemoto, Uchida, Ogata, & Kurokawa, 1993; Tomita, Nonoguchi, Terada, & Marumo, 1993; Kohan, Padilla, & Hughes, 1993; Edwards, Stack, Pullen, & Nambi, 1993; Nadler, Zimpelmann, & Hebert, 1992). However, central evidence of ET-1 in CDs was published only ten years ago when Ahn et al. demonstrated that the absence of CD principal cell ET-1 causes hypertension and sodium retention (Ahn et al., 2004). Mice with CD-specific knockout of ET-1 (under control of the AQP2 promoter) were hypertensive and had reduced sodium excretion in response to sodium loading. Systolic blood pressure was increased by approximately 15 and 35 mmHg during normal and high Na+ intake, respectively (Ahn et al., 2004).
As discussed above, effects of endothelins are mediated by activation of ETRA and ETRB receptors. Both receptors are expressed in the CNT and entire CDs, where ENaC is involved in sodium transport (see Figure 1). Importantly, ET receptors are localized at the basolateral membranes with no evidence of their expression at the luminal side. A numerous studies identified expression of the ETRA receptor in these nephron segments (Kohan, Hughes, & Perkins, 1992; Wendel, Knels, Kummer, & Koch, 2006; Yukimura et al., 1996; Yamamoto, Suzuki, Kubo, Matsumoto, & Uemura, 2008). While some studies suggest that ETRA receptor is expressed in the distal tubules, majority of work identified that the greatest levels of these receptors are localized in the inner medullary collecting ducts (IMCD). Functional studies using genetic deletion of ETRA receptor in the principal cell of CDs (Ge, Stricklett, Hughes, Yanagisawa, & Kohan, 2005) or entire nephron (Stuart et al., 2012) revealed that renal ETRA receptor, at least under normal physiological conditions, does not play a major role in the regulation of blood pressure, sodium excretion or systemic hemodynamics. In following study using tissue specific promoters to target ETRA receptors in heart, smooth muscles, nephron and collecting duct, the authors demonstrated that fluid retention is due to a direct effect on the CDs and partially to a vascular effect but not related to cardiac function (Stuart, Chapman, Rees, Woodward, & Kohan, 2013). Recent studies tested the effects of ET-1 on ENaC-mediated (benzamil-sensitive) Na+ transport by measuring lumen-to-bath isotopic 22Na reabsorption (JNa) using in vitro microperfusion and identified that ET-1 regulation of Na+ reabsorption in the CDs involves both ETRA and ETRB receptors (Lynch, Welch, Kohan, Cain, & Wingo, 2013). However, despite the data demonstrating expression ETRA receptor and some evidence of its functional role, it appears that the ETRB receptor is predominant isoform in the CDs and in the renal medulla and most effects on ENaC involved activation of the ETRB receptor.
Expression of the ETRB receptor in CDs was shown by multiple techniques in different species. While mice with CD-specific (or entire nephron) knockouts of ETRA receptor did not have altered blood pressure or sodium excretion (Ge et al., 2005; Stuart et al., 2012), CD-specific knockout of the ETRB receptor causes hypertension and sodium retention. However, CD-specific knockout of the ETRB receptor increases blood pressure to a lesser extent than CD ET-1 knockout (Ge et al., 2006). Combined knockout of both ETA and ETB receptors in CDs had increased blood pressure, which increased further with high salt intake. The degree of blood pressure elevation during normal Na+ intake was similar to CD-specific ET-1 knockout mice and higher than in CD ETRB knockout animals.
4.2. ET-1 regulation of ENaC in CDs
Kurokawa et al. reported more than twenty years ago that in rabbit CCDs perfused in vitro, low concentrations of ET-1 gradually increased total cellular membrane resistance because of inhibition of the luminal Na+ channel (Kurokawa et al., 1993). Another evidence of the key role of ENaC in ET-1 mediated processes was provided by Gariepy et al. who demonstrated that ETRB knockout rats develop hypertension on a high salt diet and that normal blood pressure in these salt-sensitive, hypertensive rats restores after amiloride treatment (Gariepy, Ohuchi, Williams, Richardson, & Yanagisawa, 2000). Moreover, electrophysiological study showed that picomolar concentrations of ET-1 attenuate ENaC open probability (Po) in an amphibian distal nephron cell line and this effect is prevented by ETRB antagonist but not ETRA antagonist (Gallego & Ling, 1996). Similar results were demonstrated in mammalian fibroblast cells by stably expressing genes for the three ENaC subunits (Gilmore, Stutts, & Milgram, 2001). Patch clamp studies of ENaC activity in isolated split-opened tubules provided compelling support for ET-1 acutely regulating ENaC activity and helped to identify signaling pathways and time-line of these effects. Bugaj et al. demonstrated in native rat CD principal cells that ET-1 dynamically decreases ENaC Po via ETRB receptors by about three-fold within 5 min. Authors demonstrated that subsequent intracellular pathways involved Src tyrosine kinase activity and MAPK1/2 signaling (Bugaj et al., 2008). Following electrophysiological studies in CDs isolated from CD-specific ET-1, ETRA, ETRB, and ETRA/ETRB knockout mice further confirmed that ENaC regulation by ET-1 via ETB receptors contributes to the antihypertensive and natriuretic effects of the local endothelin system in the mammalian CDs (Bugaj, Mironova, Kohan, & Stockand, 2012). Shown on Figure 2 is a basic scheme of ENaC regulation by ET-1.
Figure 2.
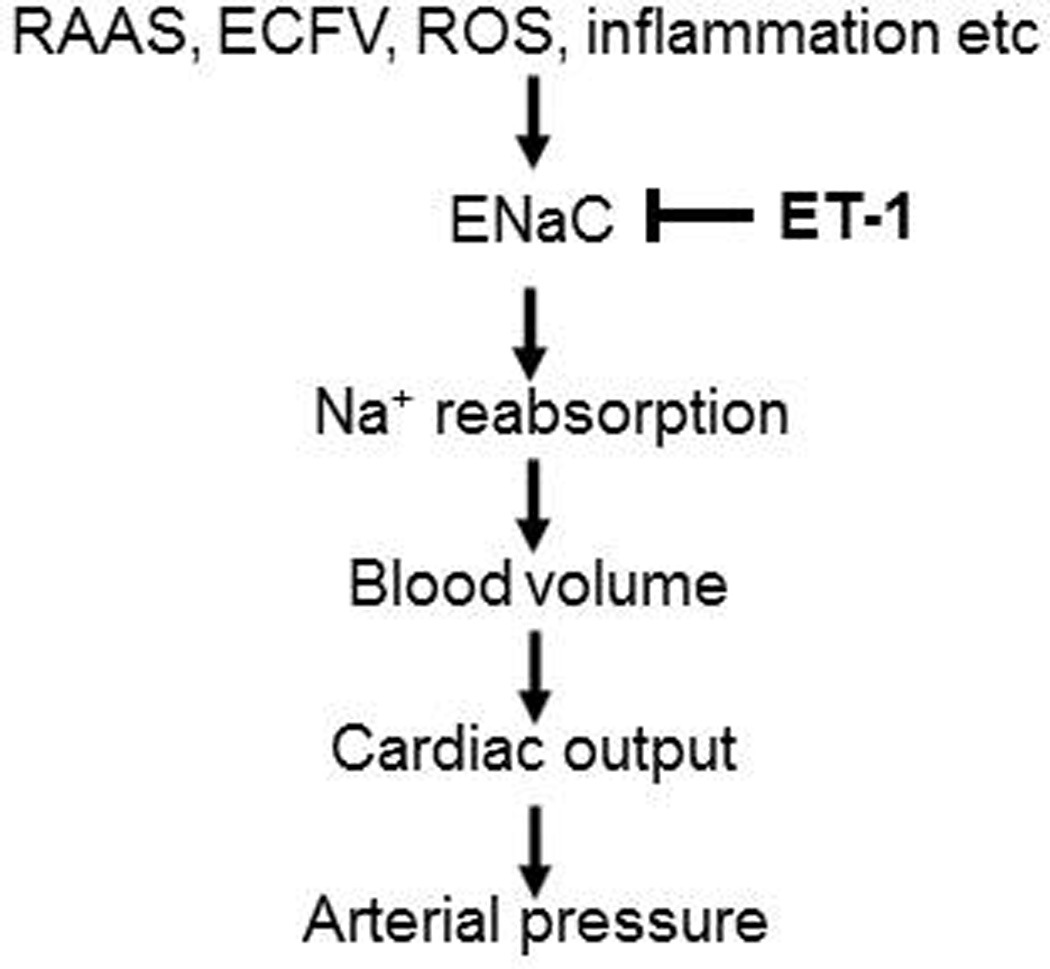
Simplified hypothesis to explain mechanisms of ENaC regulation by ET-1 in the setting of development of hypertension. Changes in extracellular fluid volume (ECFV), inflammation, oxidative stress, the Renin-Angiotensin-Aldosterone System (RAAS) and other systems and mechanisms positively modulate ENaC activity. Endothelin system represent feedback mechanism, which plays protective role in the maintenance of ENaC-mediated sodium absorption in the kidney and blood pressure, respectively.
5. Lung, smooth muscle and distal colon: ET-1 and ENaC
Pulmonary arterial hypertension (PAH) is one of human pathologies for which ET-1 is clearly a key mediator (Shao, Park, & Wort, 2011). The pathobiology of PAH is associated with increased pulmonary vascular resistance resulting from hyperplasia of vascular cells within vascular wall. ET-1 contributes to vascular remodeling by having direct effects on all three layers of vessel wall: endothelial cells, smooth muscle cells and fibroblasts. Excessive local pulmonary ET-1 production in patients with pulmonary hypertension has been established (Cacoub et al., 1997) and increased circulative levels of ET-1 are in correlation with increased mortality in PAH patients (Cacoub, Dorent, Nataf, & Carayon, 1993). Remarkably, PAH is one of a very short list of human diseases for which treatment with ET receptors antagonists have been approved. Endothelin-receptor antagonism with Bosentan, which is a dual ETRA and ETRB antagonist, was beneficial for the therapy for PAH (Rubin et al., 2002; Sitbon et al., 2003). Currently, two orally active antagonists: dual antagonist Bosentan and ETRA selective antagonist Ambrisentan are approved as a treatment for PAH (Rubin, 2012).
The lung is the site in human body where fluid content is tightly controlled within certain limits and, accordingly, treatment of pulmonary edema by ENaC activators/stimulators has been in a focus of scientific interest of multiple laboratories (Fronius, 2013). TGF-β, a key mediator of acute kidney injury, was shown to drive the internalization of the ENaC from the alveolar epithelial cell surface, leading to the persistence of alveolar flooding (Peters et al., 2014). In addition, these channels are potential contributors to cystic fibrosis lung disease (Hobbs, Da, & Tarran, 2013; Donaldson & Galietta, 2013). Thus, multiple lung pathologies are associated with ET-1 signaling and regulation of ENaC, but it appears that there is little overlap between the diseases linked with either endothelin system or ENaC complex. ENaC abnormal function is described in the context of disintegrating fluid homeostasis control, whereas endothelin system is primarily involved in lung pathologies accompanying vascular remodeling. Hence, the interaction between increased pulmonary production of ET-1 and activity of ENaC has not been sufficiently investigated yet. It is tempting to suggest that since ENaC mediated pulmonary pathologies are generally explicated by increased ENaC activity, the excessive ET-1 production is supposed to attenuate ENaC action and correspondently should contribute to prevention of pulmonary channelopathies. Nonetheless, multiple mechanisms have been described to be relevant for the regulation of ENaC in pulmonary epithelial cells (Eaton, Helms, Koval, Bao, & Jain, 2009). ENaC in the lung is under control of β-Adrenergic agents, puinergic agonists, steroids, inflammatory chemokines, reactive oxygen and nitrogen species and low oxygen tension (Eaton et al., 2009). Epithelial pulmonary cells of alveolar surface are exposed to air-filled compartment and ENaCs were reported to be directly activated by shear stress (Fronius, Bogdan, Althaus, Morty, & Clauss, 2010). The receptor for advanced glycation end-products (RAGE) regulated lung fluid balance via PKC-gp91phox signaling to ENaC (Downs, Kreiner, Johnson, Brown, & Helms, 2014). Kinases SGK, PKC and PKA up-regulate ENaC activity in a variety of epithelial cell systems (Baines, 2013; Eaton, Yue, Eaton, & Bao, 2014). Some of these signaling molecules and pathways are well known components of signal transduction by ET-1.
ET-1 system contributes to the pathogenesis of vascular diseases and is known to activate at least three major signaling pathways in vascular smooth muscle cells: phosphoinoisitide cascade, MAPK signaling cascades and Phosphoinositide 3-kinase (PI-3 kinase) pathway (Bouallegue et al., 2007). It is of note that β-ENaC subunit is expressed in smooth muscle cells where they are supposed to act as mechanotransducers to regulate myogenic response and hypertension (Drummond, 2012).
Abnormal activation of endothelin system is widely accepted as a common mechanism which is engaged in the progression of solid tumors (Rosano, Spinella, & Bagnato, 2013). The cancers where ET-1 signaling was shown to be an active player include colon cancer. Accordingly, the majority of information with regard to ET-1 action in the colon is related to its cancer-promoting effect. The signaling via ETRA is involved in colon cancer progression and metastasis (Nie et al., 2014; Sorby, Kleiveland, Andersen, Bukholm, & Jacobsen, 2011). On the contrary, not only the connection between ENaC activity and colon cancer progression has not been revealed, but even the concept of ENaC system playing significant role in cancer progression is unproven.
The involvement of distal colon ENaC in regulation of sodium reabsorption and regulation of blood pressure has been proposed (Rossier, 2014). The ENaC-β and ENaC-γ expression was shown to be diminished by high salt intake in colonic epithelial cells (Lienhard, Lauterburg, Escher, Frey, & Frey, 2012). Whether ET-1 mediated inhibition of ENaC underlies the ability of colonic epithelial cells to contribute to the protection of the mammalian body against salt overload remains to be determined. It must be taken into consideration that whereas ET-1 and ETRA, but not ETRB, were expressed at a high level in primary and cultured colon carcinoma cells, in normal colon tissues ET-1 level was very low or undetectable (Liakou et al., 2012). Whether high salt diets triggers increased production of ET-1 in colon tissues, as it does in kidney, has not been established and, accordingly, the connection between ET-1 signaling and inhibition of ENaC function in colon cells, although probable, cannot be postulated.
Preeclampsia (PE), a systemic disorder which is one of the leading cause of maternal mortality worldwide, is characterized by hypertension, proteinuria and edema (Al-Jameil, Aziz, Fareed, & Tabassum, 2014). It is instigated by abnormal insufficient placenta which releases in the blood stream tumor necrosis factor alpha (TNFα) and soluble fms-like tyrosine kinase (sFlt-1) causing the subsequent overexpression of vascular and renal ET-1 (Speed & Pollock, 2013). Multiple experimental models of PE are associated with elevated tissue levels of ET-1 and it appears that ET-1 serves as a final common pathway linking factor produced during placental ischemia to cause elevation of blood pressure (Palei, Spradley, Warrington, George, & Granger, 2013).
There is sufficient evidence indicating the involvement of ENaC with the progression of PE. One of the causes of PE is an inadequate cytotrophoblast migration and since β-ENaC mediates cytotrophoblast migration, an altered expression of ENaC contributes to placenta ischemia and hypertension (Warrington et al., 2014). A genetic variant of the β-subunit of ENaC was shown to be associated with the pathogenesis of PE and hypertension (Dhanjal, Owen, Anthony, Davidson, & Rayner, 2006; Jones, Owen, & Rayner, 2012).
Therefore, ET-1 is considered to be a key pathological factor in PE (Jain, 2012) and uncovering ET-1-mediated signaling pathways which contribute to hypertension in PE is of particular importance. The ET-1-mediated inhibition of ENaC could be one of the underappreciated mechanisms by which ET-1 is implicated in pathobiology of PE. ETRA antagonists showed some promise for the treatment of pregnancy-induced hypertension, but the potential problem of teratogenicity cannot be ignored (Speed & Pollock, 2013). Similar to colon cancer, the connection between overexpression of ET-1 and ENaC activity in PE has not been sufficiently investigated so far. It seems that the initial studies testing effects of ET-1 antagonists upon ENaC activity in cell cultures derived from placenta and animal models of preeclampsia are due.
6. Molecular mechanisms of inhibition of ENaC by ET-1
There are several proposed molecular mechanisms of ET-1 effects on ENaC in CDs. Both Src kinases and MAPK1/2 signaling were shown to mediate ET-1-dependent decreases in ENaC Po in NIH 3T3 cells stably expressing genes for all three (α, β, and γ) ENaC subunits (Gilmore et al., 2001) and the split-open collecting duct (Bugaj et al., 2008). The inhibitory effect of ET-1 on ENaC could be completely blocked when cells were pretreated with the selective Src family kinase inhibitor, PP2. Further studies revealed that basal Src family kinase activity strongly regulates ENaC inhibition. Inhibition of MAPK also abolishes ET-1 effects on ENaC (Bugaj et al., 2008). No roles for phospholipase C (PLC) or protein kinase C (PKC) in the rapid response of ENaC to ET-1 were identified.
Potential role for NO in the control of ENaC by ET-1 was proposed (Kohan, 2013) since it was reported that ET-1 increases NOS expression and NO production in the CDs (Stricklett, Hughes, & Kohan, 2006; Schneider, Ge, Pollock, Pollock, & Kohan, 2008; Sullivan, Goodchild, Cai, Pollock, & Pollock, 2007; Hyndman & Pollock, 2013). Inhibition of ENaC by NO in cultured cells was previously reported (Yu, Bao, Self, Eaton, & Helms, 2007; Helms et al., 2005). Furthermore, it was shown that NO reduces Cl− absorption in the mouse CCDs through an ENaC-dependent mechanism (Pech et al., 2013). However, role of NO in ET-1 mediated effects on ENaC was not directly studied yet and remains to be investigated.
Among the signaling molecules which contribute significantly to ET-1-mediated inhibition of ENaC are protein kinase A (PKA) and guanine nucleotide exchange factor (GEF) βPix. This mechanism of inhibition of ENaC by ET-1 is likely to be coupled to retention of ability of NEDD4-2 to modulate ENaC despite serum and glucocorticoid regulated kinases- (SGK) mediated phosphorylation. SGK and PKA are important signaling molecules that has been shown to up-regulate ENaC activity (Baines, 2013). Other kinases shown to up-regulate ENaC include CK2, GRK2, IKKβ and PKD1, whilst PKC, ERK1/2 and AMPK are inhibitory (Baines, 2013). Aldosterone-induced SGK1 is widely considered as one of principal regulators of ENaC (Pao, 2012; Soundararajan, Lu, & Pearce, 2012). SGK1 not only associates with the apical membrane to regulate open probability of ENaC, but also is recruited by scaffold proteins to multiunit signaling complexes to modulate ENaC expression (Pao, 2012). For SGK1 to be functionally active it must be phosphorylated within its carboxy-terminal domain by serine-threonine kinase mammalian target of rapamycin (mTOR) (Lu et al., 2010). ENaC is known to be regulated by ubiquitylation following the binding of ubiquitin ligase Nedd4-2 protein-protein interaction domain WW to a PY motif in the ENaC subunits (Snyder, 2009; Rotin & Staub, 2012; Ronzaud & Staub, 2014). It appears that the phosphorylation of Nedd4-2 and two of ENaC subunits (β and γ) are main convergence targets for the action of kinases regulating ENaC. Phosphorylation of Nedd4-2 by SGK1 reduces Nedd4-2 ability to bind to ENaC, due to the interaction of phosphorylated Nedd4-2 with 14-3-3 proteins, and hence increases ENaC activity (Wiemuth et al., 2010). 14-3-3 proteins associate with SGK1-phosphorylated Nedd4-2 to maintain its phosphorylated/inactive state and thereby obstruct its physical association with ENaC (Bhalla et al., 2005; Ichimura et al., 2005; Liang, Peters, Butterworth, & Frizzell, 2006; Liang, Butterworth, Peters, Walker, & Frizzell, 2008; Nagaki et al., 2006). PKA also phosphorylates Nedd4-2 and, accordingly, regulates ENaC by decreasing Nedd4-2 binding to ENaC and congruently interfering with Nedd4-2 E3 ubiquitin-protein ligase activity that targets ENaC for degradation (Snyder, Olson, Kabra, Zhou, & Steines, 2004). The proposed mechanism of negative regulation of ENaC by ET-1 suggests that ET-1-mediated formation of the complex comprising of βPix, which acts as a scaffold in this case, and adaptor protein 14-3-3, is sufficient to prevent ubiquitin ligase Nedd4-2 from association with 14-3-3. Accordingly, phosphorylated Nedd4-2 retains ability to modify ENaC which results in the decrease of ENaC function. It has been shown that SGK1 and Nedd4-2 physically associate with each other and with ENaC (Soundararajan et al., 2012). We have shown that ET-1 mediated recruitment of 14-3-3β by β1Pix impairs the interaction of 14-3-3β with the ubiquitin ligase Nedd4-2, thereby promoting ubiquitination and degradation of ENaC (Pavlov et al., 2010). It is of note that βPix-mediated ET-1-triggered inhibition of ENaC activity is dispensable of βPix action as GEF (Pavlov et al., 2010), but depends on its ability to act as a scaffold protein (Staruschenko & Sorokin, 2012). The cellular model shown in Figure 3 reveals a proposed signal transduction pathway coupling the ET receptors to ENaC.
Figure 3.
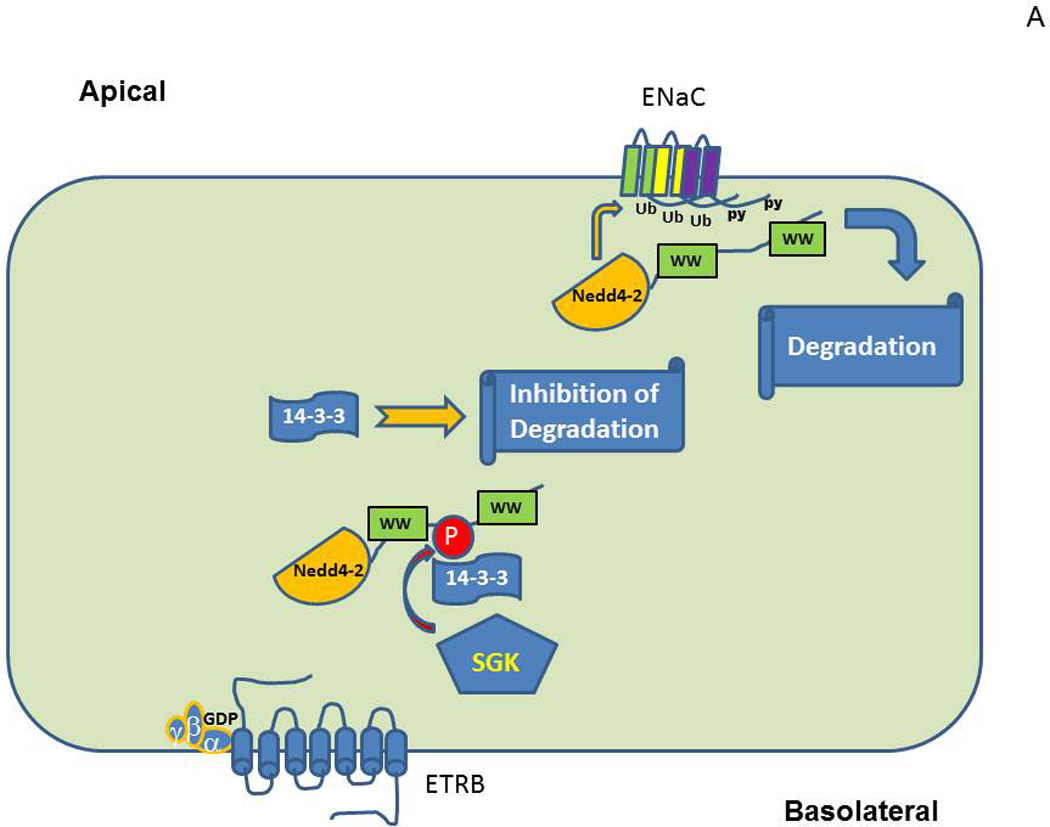
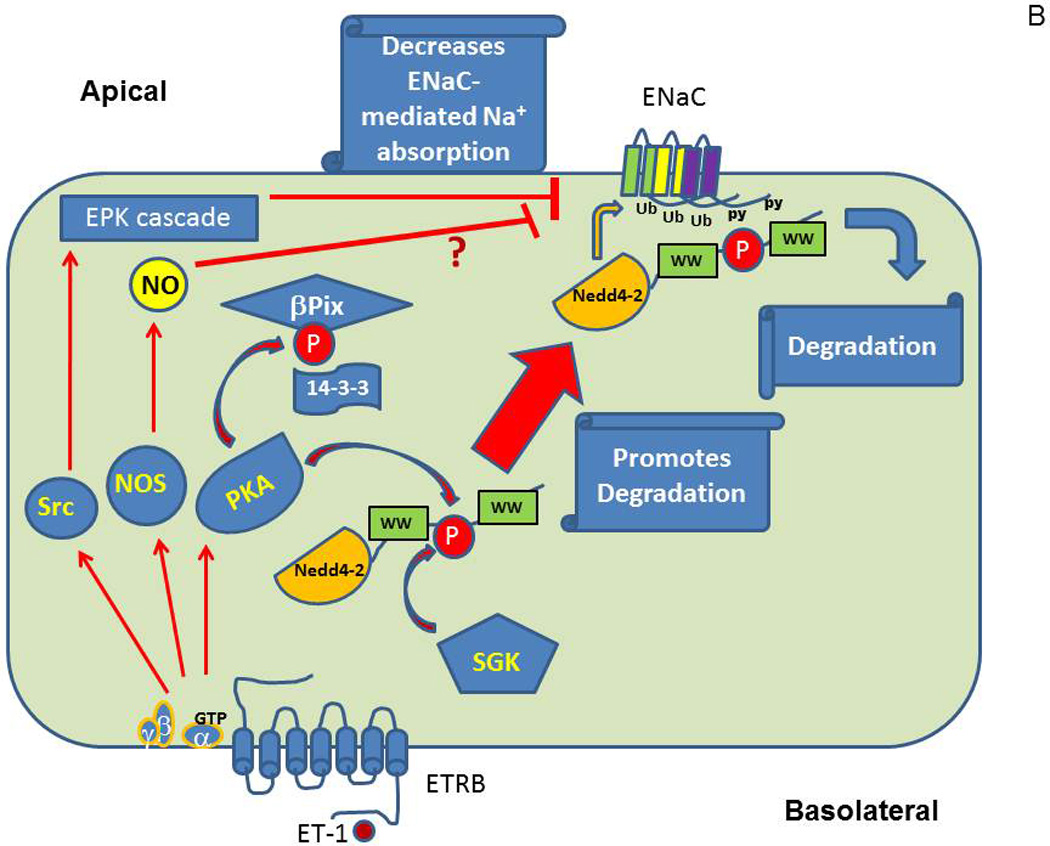
Proposed molecular mechanisms of ET-1-mediated inhibition of ENaC in CDs. (A) Quiescent cells with unoccupied ETRB. (B) Cells after activation of ET-1 signaling cascade.
In addition to described above mechanisms, it was reported that ET-1 gene is negatively regulated by the circadian clock protein Period 1 (Per1) (Richards et al., 2014). Interestingly, transcription of α-ENaC subunit is also regulated by Per1 (Gumz et al., 2009; Gumz et al., 2010). Besides, Per1 knockout mice have a hypotensive phenotype with a significant increase in cortical and medullary ET-1 production (Stow et al., 2012). These data suggest that the lower blood pressure in Per1 knockout mice may be linked to increased ET-1-dependent natriuresis/diuresis and ENaC is involved in this molecular mechanism. Epigenetic factors altering endothelin signaling (Welch, Jacobs, Wingo, & Cain, 2013; Stow, Jacobs, Wingo, & Cain, 2011) might be also involved in the control of ENaC activity since this channel is under control of several epigenetic mechanisms (Kone, 2013). However, these pathways and their connection remain to be studied in more details to make any definite conclusions.
7. Conclusions and Future Directions
The precise regulation of ENaC is important for normal Na+ and fluid homeostasis in kidneys, lung and colon. These organs are also well established bridgeheads for ET-1 actions and signaling. Even though inhibition of ENaC by ET-1 has been studied and reliably demonstrated in the context of renal cells, there is a high probability that modulation of ENaC by ET-1 also takes place in pulmonary channelopathies, preeclampsia and distal colon. Whereas antagonists of ETRA represent probable candidates for being able to serve as modulators of ENaC function, no systematic analysis of their efficiency to contribute to resolution of renal, lung and colon channelopaties caused by ENaC dysfunction has been carried out so far.
Molecular mechanisms of inhibition of ENaC by ET-1 are likely to be coupled to its effects through Src kinases and MAPK1/2 and retention of ability of NEDD4-2 to modulate ENaC despite SGK1-mediated phosphorylation. The current understanding of ET-1 effect upon ENaC function is based mostly upon cultured cell based techniques. The major challenge is to confirm the validity of these mechanisms in the context of whole organism. Future breakthroughs in the field are likely to come from analysis of renal function in models of channelopaties with animals in which precise modification of genes encoding proteins, involved in ET-1-mediated ENaC inhibition has been achieved. The recent advances in targeted genome editing techniques suggest that this task could be achieved within next few years (Cheng & Alper, 2014).
Acknowledgments
This work is supported by grants from National Institute of Health to Andrey Sorokin (DK098159) and Alexander Staruschenko (HL108880).
REFERENCES
- Agapitov AV, Haynes WG. Role of endothelin in cardiovascular disease. Journal of the Renin-Angiotensin-Aldosterone System. 2002;3:1–15. doi: 10.3317/jraas.2002.001. [DOI] [PubMed] [Google Scholar]
- Ahn D, Ge Y, Stricklett PK, Gill P, Taylor D, Hughes AK, et al. Collecting duct-specific knockout of endothelin-1 causes hypertension and sodium retention. Journal of Clinical Investigation. 2004;114:504–511. doi: 10.1172/JCI21064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Jameil N, Aziz KF, Fareed KM, Tabassum H. A brief overview of preeclampsia. Journal of Clinical Medicine Research. 2014;6:1–7. doi: 10.4021/jocmr1682w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez dlR, Navarro-Gonzalez JF, Giraldez T. ENaC modulators and renal disease. Current Molecular Pharmacology. 2013;6:35–43. doi: 10.2174/1874467211306010005. [DOI] [PubMed] [Google Scholar]
- Arai H, Hori S, Aramori I, Ohkubo H, Nakanishi S. Cloning and expression of a cDNA encoding an endothelin receptor. Nature. 1990;348:730–732. doi: 10.1038/348730a0. [DOI] [PubMed] [Google Scholar]
- Araki S, Haneda M, Togawa M, Kikkawa R. Endothelin-1 activates c-Jun NH2-terminal kinase in mesangial cells. Kidney International. 1997;51:631–639. doi: 10.1038/ki.1997.92. [DOI] [PubMed] [Google Scholar]
- Badr KF, Murray JJ, Breyer MD, Takahashi K, Inagami T, Harris RC. Mesangial cell, glomerular and renal vascular responses to endothelin in the rat kidney. Elucidation of signal transduction pathways. Journal of Clinical Investigation. 1989;83:336–342. doi: 10.1172/JCI113880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines D. Kinases as targets for ENaC regulation. Current Molecular Pharmacology. 2013;6:50–64. doi: 10.2174/18744672112059990028. [DOI] [PubMed] [Google Scholar]
- Bankir L, Bouby N, Ritz E. Vasopressin: a novel target for the prevention and retardation of kidney disease? Nature Reviews Nephrology. 2013;9:223–239. doi: 10.1038/nrneph.2013.22. [DOI] [PubMed] [Google Scholar]
- Barton M. Reversal of proteinuric renal disease and the emerging role of endothelin. Nature Clinical Practice Nephrology. 2008;4:490–501. doi: 10.1038/ncpneph0891. [DOI] [PubMed] [Google Scholar]
- Benigni A, Zoja C, Corna D, Orisio S, Longaretti L, Bertani T, et al. A specific endothelin subtype A receptor antagonist protects against injury in renal disease progression. Kidney International. 1993;44:440–444. doi: 10.1038/ki.1993.263. [DOI] [PubMed] [Google Scholar]
- Berger S, Bleich M, Schmid W, Cole TJ, Peters J, Watanabe H, et al. Mineralocorticoid receptor knockout mice: pathophysiology of Na+ metabolism. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:9424–9429. doi: 10.1073/pnas.95.16.9424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla V, Daidie D, Li H, Pao AC, LaGrange LP, Wang J, et al. Serum- and glucocorticoid-regulated kinase 1 regulates ubiquitin ligase neural precursor cell-expressed, developmentally down-regulated protein 4-2 by inducing interaction with 14-3-3. Molecular Endocrinology. 2005;19:3073–3084. doi: 10.1210/me.2005-0193. [DOI] [PubMed] [Google Scholar]
- Bhalla V, Hallows KR. Mechanisms of ENaC regulation and clinical implications. Journal of the American Society of Nephrology. 2008;19:1845–1854. doi: 10.1681/ASN.2008020225. [DOI] [PubMed] [Google Scholar]
- Birch RE, Schwiebert EM, Peppiatt-Wildman CM, Wildman SS. Emerging key roles for P2X receptors in the kidney. Frontiers in Physiology. 2013;4:262. doi: 10.3389/fphys.2013.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazer-Yost BL, Liu X, Helman SI. Hormonal regulation of ENaCs: insulin and aldosterone. American Journal of Physiology. 1998;274:C1373–C1379. doi: 10.1152/ajpcell.1998.274.5.C1373. [DOI] [PubMed] [Google Scholar]
- Boffa JJ, Tharaux PL, Dussaule JC, Chatziantoniou C. Regression of renal vascular fibrosis by endothelin receptor antagonism. Hypertension. 2001;37:490–496. doi: 10.1161/01.hyp.37.2.490. [DOI] [PubMed] [Google Scholar]
- Bouallegue A, Daou GB, Srivastava AK. Endothelin-1-induced signaling pathways in vascular smooth muscle cells. Current Vascular Pharmacology. 2007;5:45–52. doi: 10.2174/157016107779317161. [DOI] [PubMed] [Google Scholar]
- Bourque SL, Davidge ST, Adams MA. The interaction between endothelin-1 and nitric oxide in the vasculature: new perspectives. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology. 2011;300:R1288–R1295. doi: 10.1152/ajpregu.00397.2010. [DOI] [PubMed] [Google Scholar]
- Bugaj V, Mironova E, Kohan DE, Stockand JD. Collecting duct-specific endothelin B receptor knockout increases ENaC activity. American Journal of Physiology - Cell Physiology. 2012;302:C188–C194. doi: 10.1152/ajpcell.00301.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugaj V, Pochynyuk O, Mironova E, Vandewalle A, Medina JL, Stockand JD. Regulation of the epithelial Na+ channel by endothelin-1 in rat collecting duct. American Journal of Physiology - Renal Physiology. 2008;295:F1063–F1070. doi: 10.1152/ajprenal.90321.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacoub P, Dorent R, Nataf P, Carayon A. Endothelin-1 in pulmonary hypertension. The New England Journal of Medicine. 1993;329:1967–1968. doi: 10.1056/NEJM199312233292618. [DOI] [PubMed] [Google Scholar]
- Cacoub P, Dorent R, Nataf P, Carayon A, Riquet M, Noe E, et al. Endothelin-1 in the lungs of patients with pulmonary hypertension. Cardiovascular Research. 1997;33:196–200. doi: 10.1016/s0008-6363(96)00189-7. [DOI] [PubMed] [Google Scholar]
- Chahdi A, Sorokin A. Endothelin-1 induces p66Shc activation through EGF receptor transactivation: Role of β1Pix/Gαi3 interaction. Cellular Signalling. 2010a;22:325–329. doi: 10.1016/j.cellsig.2009.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahdi A, Sorokin A. The role of β1Pix/caveolin-1 interaction in endothelin signaling through Gα subunits. Biochemical and Biophysical Research Communications. 2010b;391:1330–1335. doi: 10.1016/j.bbrc.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SS, Grunder S, Hanukoglu A, Rosler A, Mathew PM, Hanukoglu I, et al. Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type 1. Nature Genetics. 1996;12:248–253. doi: 10.1038/ng0396-248. [DOI] [PubMed] [Google Scholar]
- Cheng JK, Alper HS. The genome editing toolbox: a spectrum of approaches for targeted modification. Current Opinion in Biotechnology. 2014;30C:87–94. doi: 10.1016/j.copbio.2014.06.005. [DOI] [PubMed] [Google Scholar]
- Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature. 1996;379:557–560. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- Dhanjal MK, Owen EP, Anthony JA, Davidson JS, Rayner BL. Association of pre-eclampsia with the R563Q mutation of the beta-subunit of the epithelial sodium channel. BJOG: An International Journal of Obstetrics and Gynaecology. 2006;113:595–598. doi: 10.1111/j.1471-0528.2006.00899.x. [DOI] [PubMed] [Google Scholar]
- Dhein S, Hochreuther S, Aus Dem SC, Bollig K, Hufnagel C, Raschack M. Long-term effects of the endothelin(A) receptor antagonist LU 135252 and the angiotensin-converting enzyme inhibitor trandolapril on diabetic angiopathy and nephropathy in a chronic type I diabetes mellitus rat model. Journal of Pharmacology and Experimental Therapeutics. 2000;293:351–359. [PubMed] [Google Scholar]
- Donaldson SH, Galietta L. New pulmonary therapies directed at targets other than CFTR. Cold Spring Harbor Perspectives in Medicine. 2013;3 doi: 10.1101/cshperspect.a009787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorado F, Velasco S, Esparis-Ogando A, Pericacho M, Pandiella A, Silva J, et al. The mitogen-activated protein kinase Erk5 mediates human mesangial cell activation. Nephrology Dialysis Transplantation. 2008;23:3403–3411. doi: 10.1093/ndt/gfn333. [DOI] [PubMed] [Google Scholar]
- Downs CA, Kreiner LH, Johnson NM, Brown LA, Helms MN. RAGE regulates lung fluid balance via PKC-gp91phox signaling to epithelial sodium channels (ENaC) American Journal of Respiratory Cell and Molecular Biology. 2014 doi: 10.1165/rcmb.2014-0002OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond HA. βENaC is a molecular component of a VSMC mechanotransducer that contributes to renal blood flow regulation, protection from renal injury, and hypertension. Frontiers in Physiology. 2012;3:341. doi: 10.3389/fphys.2012.00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Sours-Brothers S, Coleman R, Ding M, Graham S, Kong DH, et al. Canonical transient receptor potential 1 channel is involved in contractile function of glomerular mesangial cells. Journal of the American Society of Nephrology. 2007;18:1437–1445. doi: 10.1681/ASN.2006091067. [DOI] [PubMed] [Google Scholar]
- Eaton AF, Yue Q, Eaton DC, Bao HF. ENaC activity and expression is decreased in the lungs of protein kinase C-α knockout mice. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2014;307:L374–L385. doi: 10.1152/ajplung.00040.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton DC, Helms MN, Koval M, Bao HF, Jain L. The contribution of epithelial sodium channels to alveolar function in health and disease. Annual Review of Physiology. 2009;71:403–423. doi: 10.1146/annurev.physiol.010908.163250. [DOI] [PubMed] [Google Scholar]
- Ecelbarger CA, Kim GH, Terris J, Masilamani S, Mitchell C, Reyes I, et al. Vasopressin-mediated regulation of epithelial sodium channel abundance in rat kidney. American Journal of Physiology - Renal Physiology. 2000;279:F46–F53. doi: 10.1152/ajprenal.2000.279.1.F46. [DOI] [PubMed] [Google Scholar]
- Edwards RM, Stack EJ, Pullen M, Nambi P. Endothelin inhibits vasopressin action in rat inner medullary collecting duct via the ETB receptor. Journal of Pharmacology and Experimental Therapeutics. 1993;267:1028–1033. [PubMed] [Google Scholar]
- Feraille E, Doucet A. Sodium-potassium-adenosinetriphosphatase-dependent sodium transport in the kidney: hormonal control. Physiological Reviews. 2001;81:345–418. doi: 10.1152/physrev.2001.81.1.345. [DOI] [PubMed] [Google Scholar]
- Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. European Heart Journal. 2012;33:829–837. doi: 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foschi M, Chari S, Dunn MJ, Sorokin A. Biphasic activation of p21ras by endothelin-1 sequentially activates the ERK cascade and phosphatidylinositol 3-kinase. The EMBO Journal. 1997;16:6439–6451. doi: 10.1093/emboj/16.21.6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foschi M, Sorokin A, Pratt P, McGinty A, La Villa G, Franchi F, et al. PreproEndothelin-1 expression in human mesangial cells: evidence for a p38 mitogen-activated protein kinase/protein kinases-C-dependent mechanism. Journal of the American Society of Nephrology. 2001;12:1137–1150. doi: 10.1681/ASN.V1261137. [DOI] [PubMed] [Google Scholar]
- Fronius M. Treatment of pulmonary edema by ENaC activators/stimulators. Current Molecular Pharmacology. 2013;6:13–27. doi: 10.2174/1874467211306010003. [DOI] [PubMed] [Google Scholar]
- Fronius M, Bogdan R, Althaus M, Morty RE, Clauss WG. Epithelial Na+ channels derived from human lung are activated by shear force. Respiratory Physiology & Neurobiology. 2010;170:113–119. doi: 10.1016/j.resp.2009.11.004. [DOI] [PubMed] [Google Scholar]
- Fukuda K, Yanagida T, Okuda S, Tamaki K, Ando T, Fujishima M. Role of endothelin as a mitogen in experimental glomerulonephritis in rats. Kidney International. 1996;49:1320–1329. doi: 10.1038/ki.1996.188. [DOI] [PubMed] [Google Scholar]
- Gallego MS, Ling BN. Regulation of amiloride-sensitive Na+ channels by endothelin-1 in distal nephron cells. American Journal of Physiology. 1996;271:F451–F460. doi: 10.1152/ajprenal.1996.271.2.F451. [DOI] [PubMed] [Google Scholar]
- Gariepy CE, Ohuchi T, Williams SC, Richardson JA, Yanagisawa M. Salt-sensitive hypertension in endothelin-B receptor-deficient rats. Journal of Clinical Investigation. 2000;105:925–933. doi: 10.1172/JCI8609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Ahn D, Stricklett PK, Hughes AK, Yanagisawa M, Verbalis JG, et al. Collecting duct-specific knockout of endothelin-1 alters vasopressin regulation of urine osmolality. American Journal of Physiology - Renal Physiology. 2005;288:F912–F920. doi: 10.1152/ajprenal.00432.2004. [DOI] [PubMed] [Google Scholar]
- Ge Y, Bagnall A, Stricklett PK, Strait K, Webb DJ, Kotelevtsev Y, et al. Collecting duct-specific knockout of the endothelin B receptor causes hypertension and sodium retention. American Journal of Physiology - Renal Physiology. 2006;291:F1274–F1280. doi: 10.1152/ajprenal.00190.2006. [DOI] [PubMed] [Google Scholar]
- Ge Y, Stricklett PK, Hughes AK, Yanagisawa M, Kohan DE. Collecting duct-specific knockout of the endothelin A receptor alters renal vasopressin responsiveness, but not sodium excretion or blood pressure. American Journal of Physiology - Renal Physiology. 2005;289:F692–F698. doi: 10.1152/ajprenal.00100.2005. [DOI] [PubMed] [Google Scholar]
- Gilmore ES, Stutts MJ, Milgram SL. SRC family kinases mediate epithelial Na+ channel inhibition by endothelin. The Journal of Biological Chemistry. 2001;276:42610–42617. doi: 10.1074/jbc.M106919200. [DOI] [PubMed] [Google Scholar]
- Gomez-Garre D, Largo R, Liu XH, Gutierrez S, Lopez-Armada MJ, Palacios I, et al. An orally active ETA/ETB receptor antagonist ameliorates proteinuria and glomerular lesions in rats with proliferative nephritis. Kidney International. 1996;50:962–972. doi: 10.1038/ki.1996.397. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Cobos JC, Trebak M. TRPC channels in smooth muscle cells. Frontiers in Bioscience. 2010;15:1023–1039. doi: 10.2741/3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunder S, Firsov D, Chang SS, Jaeger NF, Gautschi I, Schild L, et al. A mutation causing pseudohypoaldosteronism type 1 identifies a conserved glycine that is involved in the gating of the epithelial sodium channel. The EMBO Journal. 1997;16:899–907. doi: 10.1093/emboj/16.5.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Z, Inscho EW. Endothelin and the renal vasculature. Contributions to Nephrology. 2011;172:35–49. doi: 10.1159/000328720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumz ML, Cheng KY, Lynch IJ, Stow LR, Greenlee MM, Cain BD, et al. Regulation of αENaC expression by the circadian clock protein Period 1 in mpkCCDc14 cells. Biochimica et Biophysica Acta - Gene Regulatory Mechanisms. 2010;1799:622–629. doi: 10.1016/j.bbagrm.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumz ML, Stow LR, Lynch IJ, Greenlee MM, Rudin A, Cain BD, et al. The circadian clock protein Period 1 regulates expression of the renal epithelial sodium channel in mice. Journal of Clinical Investigation. 2009;119:2423–2434. doi: 10.1172/JCI36908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo LJ, Alli AA, Eaton DC, Bao HF. ENaC is regulated by natriuretic peptide receptor-dependent cGMP signaling. American Journal of Physiology - Renal Physiology. 2013;304:F930–F937. doi: 10.1152/ajprenal.00638.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson JH, Nelson-Williams C, Suzuki H, Schild L, Shimkets R, Lu Y, et al. Hypertension caused by a truncated epithelial sodium channel gamma subunit: genetic heterogeneity of Liddle syndrome. Nature Genetics. 1995a;11:76–82. doi: 10.1038/ng0995-76. [DOI] [PubMed] [Google Scholar]
- Hansson JH, Schild L, Lu Y, Wilson TA, Gautschi I, Shimkets R, et al. A de novo missense mutation of the beta subunit of the epithelial sodium channel causes hypertension and Liddle syndrome, identifying a proline-rich segment critical for regulation of channel activity. Proceedings of the National Academy of Sciences of the United States of America. 1995b;92:11495–11499. doi: 10.1073/pnas.92.25.11495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada T, Horinouchi T, Higa T, Hoshi A, Higashi T, Terada K, et al. Endothelin-1 activates extracellular signal-regulated kinases 1/2 via transactivation of platelet-derived growth factor receptor in rat L6 myoblasts. Life Sciences. 2014;104:24–31. doi: 10.1016/j.lfs.2014.04.002. [DOI] [PubMed] [Google Scholar]
- He C, Miao X, Li J, Qi H. Angiotensin II induces endothelin-1 expression in human hepatic stellate cells. Digestive Diseases and Sciences. 2013;58:2542–2549. doi: 10.1007/s10620-013-2685-y. [DOI] [PubMed] [Google Scholar]
- Helms MN, Yu L, Malik B, Kleinhenz DJ, Hart CM, Eaton DC. Role of SGK1 in nitric oxide inhibition of ENaC in Na+-transporting epithelia. American Journal of Physiology - Cell Physiology. 2005;289:C717–C726. doi: 10.1152/ajpcell.00006.2005. [DOI] [PubMed] [Google Scholar]
- Hobbs CA, Da TC, Tarran R. Does epithelial sodium channel hyperactivity contribute to cystic fibrosis lung disease? The Journal of Physiology. 2013;591:4377–4387. doi: 10.1113/jphysiol.2012.240861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horinouchi T, Terada K, Higashi T, Miwa S. Endothelin receptor signaling: new insight into its regulatory mechanisms. Journal of Pharmacological Sciences. 2013;123:85–101. doi: 10.1254/jphs.13r02cr. [DOI] [PubMed] [Google Scholar]
- Hua H, Munk S, Whiteside CI. Endothelin-1 activates mesangial cell ERK1/2 via EGF-receptor transactivation and caveolin-1 interaction. American Journal of Physiology - Renal Physiology. 2003;284:F303–F312. doi: 10.1152/ajprenal.00127.2002. [DOI] [PubMed] [Google Scholar]
- Hyndman KA, Pollock JS. Nitric oxide and the A and B of endothelin of sodium homeostasis. Current Opinion in Nephrology and Hypertension. 2013;22:26–31. doi: 10.1097/MNH.0b013e32835b4edc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura T, Yamamura H, Sasamoto K, Tominaga Y, Taoka M, Kakiuchi K, et al. 14-3-3 proteins modulate the expression of epithelial Na+ channels by phosphorylation-dependent interaction with Nedd4-2 ubiquitin ligase. The Journal of Biological Chemistry. 2005;280:13187–13194. doi: 10.1074/jbc.M412884200. [DOI] [PubMed] [Google Scholar]
- Ilatovskaya DV, Pavlov TS, Levchenko V, Staruschenko A. ROS production as a common mechanism of ENaC regulation by EGF, insulin, and IGF-1. American Journal of Physiology - Cell Physiology. 2013;304:C102–C111. doi: 10.1152/ajpcell.00231.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai T, Hirata Y, Emori T, Yanagisawa M, Masaki T, Marumo F. Induction of endothelin-1 gene by angiotensin and vasopressin in endothelial cells. Hypertension. 1992;19:753–757. doi: 10.1161/01.hyp.19.6.753. [DOI] [PubMed] [Google Scholar]
- Imamura T, Huang J, Dalle S, Ugi S, Usui I, Luttrell LM, et al. β-arrestin-mediated recruitment of the Src family kinase Yes mediates endothelin-1-stimulated glucose transport. The Journal of Biological Chemistry. 2001;276:43663–43667. doi: 10.1074/jbc.M105364200. [DOI] [PubMed] [Google Scholar]
- Iwasaki H, Eguchi S, Ueno H, Marumo F, Hirata Y. Endothelin-mediated vascular growth requires p42/p44 mitogen-activated protein kinase and p70 S6 kinase cascades via transactivation of epidermal growth factor receptor. Endocrinology. 1999;140:4659–4668. doi: 10.1210/endo.140.10.7023. [DOI] [PubMed] [Google Scholar]
- Jain A. Endothelin-1: a key pathological factor in pre-eclampsia? Reproductive BioMedicine Online. 2012;25:443–449. doi: 10.1016/j.rbmo.2012.07.014. [DOI] [PubMed] [Google Scholar]
- Jasti J, Furukawa H, Gonzales EB, Gouaux E. Structure of acid-sensing ion channel 1 at 1.9 A resolution and low pH. Nature. 2007;449:316–323. doi: 10.1038/nature06163. [DOI] [PubMed] [Google Scholar]
- Jones ES, Owen EP, Rayner BL. The association of the R563Q genotype of the ENaC with phenotypic variation in Southern Africa. American Journal of Hypertension. 2012;25:1286–1291. doi: 10.1038/ajh.2012.125. [DOI] [PubMed] [Google Scholar]
- Kawanabe Y, Hashimoto N, Masaki T. Involvements of voltage-independent Ca2+ channels and phosphoinositide 3-kinase in endothelin-1-induced PYK2 tyrosine phosphorylation. Molecular Pharmacology. 2003;63:808–813. doi: 10.1124/mol.63.4.808. [DOI] [PubMed] [Google Scholar]
- Kodama H, Fukuda K, Takahashi T, Sano M, Kato T, Tahara S, et al. Role of EGF Receptor and Pyk2 in endothelin-1-induced ERK activation in rat cardiomyocytes. Journal of Molecular and Cellular Cardiology. 2002;34:139–150. doi: 10.1006/jmcc.2001.1496. [DOI] [PubMed] [Google Scholar]
- Kohan DE. Role of collecting duct endothelin in control of renal function and blood pressure. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology. 2013;305:R659–R668. doi: 10.1152/ajpregu.00345.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohan DE, Hughes AK, Perkins SL. Characterization of endothelin receptors in the inner medullary collecting duct of the rat. The Journal of Biological Chemistry. 1992;267:12336–12340. [PubMed] [Google Scholar]
- Kohan DE, Inscho EW, Wesson D, Pollock DM. Physiology of endothelin and the kidney. Comprehensive Physiology. 2011;1:883–919. doi: 10.1002/cphy.c100039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohan DE, Padilla E, Hughes AK. Endothelin B receptor mediates ET-1 effects on cAMP and PGE2 accumulation in rat IMCD. American Journal of Physiology. 1993;265:F670–F676. doi: 10.1152/ajprenal.1993.265.5.F670. [DOI] [PubMed] [Google Scholar]
- Kohan DE, Pollock DM. Endothelin antagonists for diabetic and non-diabetic chronic kidney disease. British Journal of Clinical Pharmacology. 2013;76:573–579. doi: 10.1111/bcp.12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohan DE, Rossi NF, Inscho EW, Pollock DM. Regulation of blood pressure and salt homeostasis by endothelin. Physiological Reviews. 2011;91:1–77. doi: 10.1152/physrev.00060.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konczalla J, Wanderer S, Mrosek J, Schuss P, Platz J, Guresir E, et al. Crosstalk between the angiotensin and endothelin-system in the cerebrovasculature. Current Neurovascular Research. 2013;10:335–345. doi: 10.2174/15672026113109990030. [DOI] [PubMed] [Google Scholar]
- Kone BC. Epigenetics and the control of the collecting duct epithelial sodium channel. Seminars in Nephrology. 2013;33:383–391. doi: 10.1016/j.semnephrol.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp UC. Endothelin in the control of renal sympathetic nerve activity. Contributions to Nephrology. 2011;172:107–119. doi: 10.1159/000328688. [DOI] [PubMed] [Google Scholar]
- Kramer BK, Ritthaler T, Ackermann M, Holmer S, Schricker K, Riegger GA, et al. Endothelium-mediated regulation of renin secretion. Kidney International. 1994;46:1577–1579. doi: 10.1038/ki.1994.451. [DOI] [PubMed] [Google Scholar]
- Kudo T, Baird A. Inhibition of aldosterone production in the adrenal glomerulosa by atrial natriuretic factor. Nature. 1984;312:756–757. doi: 10.1038/312756a0. [DOI] [PubMed] [Google Scholar]
- Kurokawa K, Yoshitomi K, Ikeda M, Uchida S, Naruse M, Imai M. Regulation of cortical collecting duct function: effect of endothelin. American Heart Journal. 1993;125:582–588. doi: 10.1016/0002-8703(93)90207-p. [DOI] [PubMed] [Google Scholar]
- Levin ER. Endothelins as cardiovascular peptides. American Journal of Nephrology. 1996;16:246–251. doi: 10.1159/000169004. [DOI] [PubMed] [Google Scholar]
- Li L, Garikepati RM, Tsukerman S, Kohan D, Wade JB, Tiwari S, et al. Reduced ENaC activity and blood pressure in mice with genetic knockout of the insulin receptor in the renal collecting duct. American Journal of Physiology - Renal Physiology. 2013;304:F279–F288. doi: 10.1152/ajprenal.00161.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liakou P, Tepetes K, Germenis A, Leventaki V, Atsaves V, Patsouris E, et al. Expression patterns of endothelin-1 and its receptors in colorectal cancer. Journal of Surgical Oncology. 2012;105:643–649. doi: 10.1002/jso.23017. [DOI] [PubMed] [Google Scholar]
- Liang X, Butterworth MB, Peters KW, Walker WH, Frizzell RA. An obligatory heterodimer of 14-3-3β and 14-3-3ε is required for aldosterone regulation of the epithelial sodium channel. The Journal of Biological Chemistry. 2008;283:27418–27425. doi: 10.1074/jbc.M803687200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Peters KW, Butterworth MB, Frizzell RA. 14-3-3 isoforms are induced by aldosterone and participate in its regulation of epithelial sodium channels. The Journal of Biological Chemistry. 2006;281:16323–16332. doi: 10.1074/jbc.M601360200. [DOI] [PubMed] [Google Scholar]
- Lienhard D, Lauterburg M, Escher G, Frey FJ, Frey BM. High salt intake down-regulates colonic mineralocorticoid receptors, epithelial sodium channels and 11β-hydroxysteroid dehydrogenase type 2. PLoS ONE. 2012;7:e37898. doi: 10.1371/journal.pone.0037898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104:545–556. doi: 10.1016/s0092-8674(01)00241-0. [DOI] [PubMed] [Google Scholar]
- Lin YJ, Kwok CF, Juan CC, Hsu YP, Shih KC, Chen CC, et al. Angiotensin II enhances endothelin-1-induced vasoconstriction through upregulating endothelin type A receptor. Biochemical and Biophysical Research Communications. 2014;451:263–269. doi: 10.1016/j.bbrc.2014.07.119. [DOI] [PubMed] [Google Scholar]
- Little PJ. GPCR responses in vascular smooth muscle can occur predominantly through dual transactivation of kinase receptors and not classical Gαq protein signalling pathways. Life Sciences. 2013;92:951–956. doi: 10.1016/j.lfs.2013.03.017. [DOI] [PubMed] [Google Scholar]
- Lopez-Ongil S, Diez-Marques ML, Griera M, Rodriguez-Puyol M, Rodriguez-Puyol D. Crosstalk between mesangial and endothelial cells: angiotensin II down-regulates endothelin-converting enzyme 1. Cellular Physiology and Biochemistry. 2005;15:135–144. doi: 10.1159/000083646. [DOI] [PubMed] [Google Scholar]
- Lu M, Wang J, Jones KT, Ives HE, Feldman ME, Yao LJ, et al. mTOR complex-2 activates ENaC by phosphorylating SGK1. Journal of the American Society of Nephrology. 2010;21:811–818. doi: 10.1681/ASN.2009111168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch IJ, Welch AK, Kohan DE, Cain BD, Wingo CS. Endothelin-1 inhibits sodium reabsorption by ETA and ETB receptors in the mouse cortical collecting duct. American Journal of Physiology - Renal Physiology. 2013;305:F568–F573. doi: 10.1152/ajprenal.00613.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamenko M, Zaika O, Doris PA, Pochynyuk O. Salt-dependent inhibition of epithelial Na+ channel-mediated sodium reabsorption in the aldosterone-sensitive distal nephron by bradykinin. Hypertension. 2012;60:1234–1241. doi: 10.1161/HYPERTENSIONAHA.112.200469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamenko M, Zaika O, Pochynyuk O. Direct regulation of ENaC by bradykinin in the distal nephron. Implications for renal sodium handling. Current Opinion in Nephrology and Hypertension. 2014;23:122–129. doi: 10.1097/01.mnh.0000441053.81339.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamenko M, Zaika O, Prieto MC, Jensen VB, Doris PA, Navar LG, et al. Chronic angiotensin II infusion drives extensive aldosterone-independent epithelial Na+ channel activation. Hypertension. 2013;62:1111–1122. doi: 10.1161/HYPERTENSIONAHA.113.01797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott GF, Hurst RD, Whiteside CI. Isolated rat glomerular cells demonstrate L-type Ca2+-channel activity. Cell Calcium. 1993;14:387–396. doi: 10.1016/0143-4160(93)90043-6. [DOI] [PubMed] [Google Scholar]
- Meyers KE, Sethna C. Endothelin antagonists in hypertension and kidney disease. Pediatric Nephrology. 2013;28:711–720. doi: 10.1007/s00467-012-2316-4. [DOI] [PubMed] [Google Scholar]
- Mironova E, Boiko N, Bugaj V, Kucher V, Stockand JD. Regulation of Na excretion and arterial blood pressure by purinergic signaling intrinsic to the distal nephron: consequences and mechanisms. Acta Physiologica. 2014 doi: 10.1111/apha.12372. [DOI] [PubMed] [Google Scholar]
- Mironova E, Bugaj V, Roos KP, Kohan DE, Stockand JD. Aldosterone-independent regulation of the epithelial Na+ channel (ENaC) by vasopressin in adrenalectomized mice. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:10095–10100. doi: 10.1073/pnas.1201978109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moe O, Tejedor A, Campbell WB, Alpern RJ, Henrich WL. Effects of endothelin on in vitro renin secretion. American Journal of Physiology. 1991;260:E521–E525. doi: 10.1152/ajpendo.1991.260.4.E521. [DOI] [PubMed] [Google Scholar]
- Motte S, McEntee K, Naeije R. Endothelin receptor antagonists. Pharmacology & Therapeutics. 2005;93:434–442. doi: 10.1016/j.pharmthera.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Nadler SP, Zimpelmann JA, Hebert RL. Endothelin inhibits vasopressin-stimulated water permeability in rat terminal inner medullary collecting duct. Journal of Clinical Investigation. 1992;90:1458–1466. doi: 10.1172/JCI116013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaki K, Yamamura H, Shimada S, Saito T, Hisanaga S, Taoka M, et al. 14-3-3 mediates phosphorylation-dependent inhibition of the interaction between the ubiquitin E3 ligase Nedd4-2 and epithelial Na+ channels. Biochemistry. 2006;45:6733–6740. doi: 10.1021/bi052640q. [DOI] [PubMed] [Google Scholar]
- Neuhofer W, Pittrow D. Role of endothelin and endothelin receptor antagonists in renal disease. European Journal of Clinical Investigation. 2006;36:78–88. doi: 10.1111/j.1365-2362.2006.01689.x. [DOI] [PubMed] [Google Scholar]
- Nie S, Zhou J, Bai F, Jiang B, Chen J, Zhou J. Role of endothelin A receptor in colon cancer metastasis: in vitro and in vivo evidence. Molecular Carcinogenesis. 2014;53:E85–E91. doi: 10.1002/mc.22036. [DOI] [PubMed] [Google Scholar]
- Oishi R, Nonoguchi H, Tomita K, Marumo F. Endothelin-1 inhibits AVP-stimulated osmotic water permeability in rat inner medullary collecting duct. American Journal of Physiology. 1991;261:F951–F956. doi: 10.1152/ajprenal.1991.261.6.F951. [DOI] [PubMed] [Google Scholar]
- Orth SR, Amann K, Gehlen F, Unger L, Wagner J, Raschack M, et al. Adult human mesangial cells (HMCs) express endothelin-B-receptors which mediate endothelin-1-induced cell growth. Journal of Cardiovascular Pharmacology. 2000;36:S232–S237. doi: 10.1097/00005344-200036051-00069. [DOI] [PubMed] [Google Scholar]
- Otsuka A, Mikami H, Katahira K, Tsunetoshi T, Minamitani K, Ogihara T. Changes in plasma renin activity and aldosterone concentration in response to endothelin injection in dogs. Acta Endocrinologica. 1989;121:361–364. doi: 10.1530/acta.0.1210361. [DOI] [PubMed] [Google Scholar]
- Palei AC, Spradley FT, Warrington JP, George EM, Granger JP. Pathophysiology of hypertension in pre-eclampsia: a lesson in integrative physiology. Acta Physiologica. 2013;208:224–233. doi: 10.1111/apha.12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao AC. SGK regulation of renal sodium transport. Current Opinion in Nephrology and Hypertension. 2012;21:534–540. doi: 10.1097/MNH.0b013e32835571be. [DOI] [PubMed] [Google Scholar]
- Park JG, Bose A, Leszyk J, Czech MP. PYK2 as a mediator of endothelin-1/G α11 signaling to GLUT4 glucose transporters. The Journal of Biological Chemistry. 2001;276:47751–47754. doi: 10.1074/jbc.C100524200. [DOI] [PubMed] [Google Scholar]
- Pavlov TS, Chahdi A, Ilatovskaya DV, Levchenko V, Vandewalle A, Pochynyuk O, et al. Endothelin-1 inhibits the epithelial Na+ channel through βPix/14-3-3/Nedd4-2. Journal of the American Society of Nephrology. 2010;21:833–843. doi: 10.1681/ASN.2009080885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov TS, Ilatovskaya DV, Levchenko V, Li L, Ecelbarger CM, Staruschenko A. Regulation of ENaC in mice lacking renal insulin receptors in the collecting duct. The FASEB Journal. 2013a;27:2723–2732. doi: 10.1096/fj.12-223792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov TS, Levchenko V, O'Connor PM, Ilatovskaya DV, Palygin O, Mori T, et al. Deficiency of renal cortical EGF increases ENaC activity and contributes to salt-sensitive hypertension. Journal of the American Society of Nephrology. 2013b;24:1053–1062. doi: 10.1681/ASN.2012080839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce D, Soundararajan R, Trimpert C, Kashlan OB, Deen PM, Kohan DE. Collecting duct principal cell transport processes and their regulation. Clinical Journal of the American Society of Nephrology. 2014 doi: 10.2215/CJN.05760513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pech V, Thumova M, Dikalov SI, Hummler E, Rossier BC, Harrison DG, et al. Nitric oxide reduces Cl absorption in the mouse cortical collecting duct through an ENaC-dependent mechanism. American Journal of Physiology - Renal Physiology. 2013;304:F1390–F1397. doi: 10.1152/ajprenal.00292.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters DM, Vadasz I, Wujak L, Wygrecka M, Olschewski A, Becker C, et al. TGF-β directs trafficking of the epithelial sodium channel ENaC which has implications for ion and fluid transport in acute lung injury. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E374–E383. doi: 10.1073/pnas.1306798111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pochynyuk O, Bugaj V, Rieg T, Insel PA, Mironova E, Vallon V, et al. Paracrine regulation of the epithelial Na+ channel in the mammalian collecting duct by purinergic P2Y2 receptor tone. The Journal of Biological Chemistry. 2008;283:36599–36607. doi: 10.1074/jbc.M807129200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pochynyuk O, Rieg T, Bugaj V, Schroth J, Fridman A, Boss GR, et al. Dietary Na+ inhibits the open probability of the epithelial sodium channel in the kidney by enhancing apical P2Y2-receptor tone. The FASEB Journal. 2010;24:2056–2065. doi: 10.1096/fj.09-151506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock DM. Endothelin receptor subtypes and tissue distribution. In: Highsmith RF, editor. Endothelin: molecular biology, physiology, and pathology. Totowa, New Jercey: Humana Press; 1998. pp. 1–29. [Google Scholar]
- Pollock DM, Keith TL, Highsmith RF. Endothelin receptors and calcium signaling. The FASEB Journal. 1995;9:1196–1204. doi: 10.1096/fasebj.9.12.7672512. [DOI] [PubMed] [Google Scholar]
- Quinn S, Harvey BJ, Thomas W. Rapid aldosterone actions on epithelial sodium channel trafficking and cell proliferation. Steroids. 2014;81:43–48. doi: 10.1016/j.steroids.2013.11.005. [DOI] [PubMed] [Google Scholar]
- Raja SG, Raja SM. Treating pulmonary arterial hypertension: current treatments and future prospects. Therapeutic Advances in Chronic Disease. 2011;2:359–370. doi: 10.1177/2040622311420773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport RM. Acute nitric oxide synthase inhibition and endothelin-1-dependent arterial pressure elevation. Frontiers in Pharmacology. 2014;5:57. doi: 10.3389/fphar.2014.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards J, Welch AK, Barilovits SJ, All S, Cheng KY, Wingo CS, et al. Tissue-specific and time-dependent regulation of the endothelin axis by the circadian clock protein Per1. Life Sciences. 2014 doi: 10.1016/j.lfs.2014.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieg T, Bundey RA, Chen Y, Deschenes G, Junger W, Insel PA, et al. Mice lacking P2Y2 receptors have salt-resistant hypertension and facilitated renal Na+ and water reabsorption. The FASEB Journal. 2007;21:3717–3726. doi: 10.1096/fj.07-8807com. [DOI] [PubMed] [Google Scholar]
- Rieg T, Gerasimova M, Boyer JL, Insel PA, Vallon V. P2Y2 receptor activation decreases blood pressure and increases renal Na+ excretion. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology. 2011;301:R510–R518. doi: 10.1152/ajpregu.00148.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronzaud C, Staub O. Ubiquitylation and control of renal Na+ balance and blood pressure. Physiology. 2014;29:16–26. doi: 10.1152/physiol.00021.2013. [DOI] [PubMed] [Google Scholar]
- Rosano L, Spinella F, Bagnato A. Endothelin 1 in cancer: biological implications and therapeutic opportunities. Nature Reviews Cancer. 2013;13:637–651. doi: 10.1038/nrc3546. [DOI] [PubMed] [Google Scholar]
- Rossier BC. Epithelial sodium channel (ENaC) and the control of blood pressure. Current Opinion in Pharmacology. 2014;15:33–46. doi: 10.1016/j.coph.2013.11.010. [DOI] [PubMed] [Google Scholar]
- Rotin D, Staub O. Nedd4-2 and the regulation of epithelial sodium transport. Frontiers in Physiology. 2012;3:212. doi: 10.3389/fphys.2012.00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin LJ. Endothelin receptor antagonists for the treatment of pulmonary artery hypertension. Life Sciences. 2012;91:517–521. doi: 10.1016/j.lfs.2012.07.033. [DOI] [PubMed] [Google Scholar]
- Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogh A, et al. Bosentan therapy for pulmonary arterial hypertension. The New England Journal of Medicine. 2002;346:896–903. doi: 10.1056/NEJMoa012212. [DOI] [PubMed] [Google Scholar]
- Rufanova VA, Alexanian A, Wakatsuki T, Lerner A, Sorokin A. Pyk2 mediates endothelin-1 signaling via p130Cas/BCAR3 cascade and regulates human glomerular mesangial cell adhesion and spreading. Journal of Cellular Physiology. 2009;219:45–56. doi: 10.1002/jcp.21649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T, Yanagisawa M, Takuwa Y, Miyazaki H, Kimura S, Goto K, et al. Cloning of a cDNA encoding a non-isopeptide-selective subtype of the endothelin receptor. Nature. 1990;348:732–735. doi: 10.1038/348732a0. [DOI] [PubMed] [Google Scholar]
- Sanghi A, Zaringhalam M, Corcoran CC, Saeed F, Hoffert JD, Sandoval PC, et al. A knowledge base of vasopressin actions in kidney. American Journal of Physiology – Renal Physiology. 2014 doi: 10.1152/ajprenal.00012.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider MP, Ge Y, Pollock DM, Pollock JS, Kohan DE. Collecting duct-derived endothelin regulates arterial pressure and Na excretion via nitric oxide. Hypertension. 2008;51:1605–1610. doi: 10.1161/HYPERTENSIONAHA.107.108126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao D, Park JE, Wort SJ. The role of endothelin-1 in the pathogenesis of pulmonary arterial hypertension. Pharmacological Research. 2011;63:504–511. doi: 10.1016/j.phrs.2011.03.003. [DOI] [PubMed] [Google Scholar]
- Shimkets RA, Warnock DG, Bositis CM, Nelson-Williams C, Hansson JH, Schambelan M, et al. Liddle's syndrome: heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell. 1994;79:407–414. doi: 10.1016/0092-8674(94)90250-x. [DOI] [PubMed] [Google Scholar]
- Simon DB, Karet FE, Rodriguez-Soriano J, Hamdan JH, DiPietro A, Trachtman H, et al. Genetic heterogeneity of Bartter's syndrome revealed by mutations in the K+ channel, ROMK. Nature Genetics. 1996;14:152–156. doi: 10.1038/ng1096-152. [DOI] [PubMed] [Google Scholar]
- Simonson MS, Dunn MJ. Ca2+ signaling by distinct endothelin peptides in glomerular mesangial cells. Experimental Cell Research. 1991;192:148–156. doi: 10.1016/0014-4827(91)90169-u. [DOI] [PubMed] [Google Scholar]
- Simonson MS, Dunn MJ. Endothelin peptides and the kidney. Annual Review of Physiology. 1993;55:249–265. doi: 10.1146/annurev.ph.55.030193.001341. [DOI] [PubMed] [Google Scholar]
- Simonson MS, Herman WH. Protein kinase C and protein tyrosine kinase activity contribute to mitogenic signaling by endothelin-1. Cross-talk between G protein-coupled receptors and pp60c–src. The Journal of Biological Chemistry. 1993;268:9347–9357. [PubMed] [Google Scholar]
- Simonson MS, Wann S, Mene P, Dubyak GR, Kester M, Nakazato Y, et al. Endothelin stimulates phospholipase C, Na+/H+ exchange, c-fos expression, and mitogenesis in rat mesangial cells. Journal of Clinical Investigation. 1989;83:708–712. doi: 10.1172/JCI113935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitbon O, Badesch DB, Channick RN, Frost A, Robbins IM, Simonneau G, et al. Effects of the dual endothelin receptor antagonist bosentan in patients with pulmonary arterial hypertension: a 1-year follow-up study. Chest. 2003;124:247–254. doi: 10.1378/chest.124.1.247. [DOI] [PubMed] [Google Scholar]
- Snyder PM. Down-regulating destruction: phosphorylation regulates the E3 ubiquitin ligase Nedd4-2. Science Signaling. 2009;2:e41. doi: 10.1126/scisignal.279pe41. [DOI] [PubMed] [Google Scholar]
- Snyder PM, Olson DR, Kabra R, Zhou R, Steines JC. cAMP and serum and glucocorticoid-inducible kinase (SGK) regulate the epithelial Na+ channel through convergent phosphorylation of Nedd4-2. The Journal of Biological Chemistry. 2004;279:45753–45758. doi: 10.1074/jbc.M407858200. [DOI] [PubMed] [Google Scholar]
- Sonkusare S, Palade PT, Marsh JD, Telemaque S, Pesic A, Rusch NJ. Vascular calcium channels and high blood pressure: pathophysiology and therapeutic implications. Vascular Pharmacology. 2006;44:131–142. doi: 10.1016/j.vph.2005.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorby LA, Kleiveland CR, Andersen SN, Bukholm IR, Jacobsen MB. The endothelin axis in the metastatic process of colon carcinoma. Anticancer Research. 2011;31:861–869. [PubMed] [Google Scholar]
- Sorokin A. Endothelin signaling and actions in the renal mesangium. Contributions to Nephrology. 2011;172:50–62. doi: 10.1159/000328680. [DOI] [PubMed] [Google Scholar]
- Sorokin A, Foschi M, Dunn MJ. Endothelin signalling and regulation of protein kinases in glomerular mesangial cells. Clinical Science. 2002;103:132S–136S. doi: 10.1042/CS103S132S. [DOI] [PubMed] [Google Scholar]
- Sorokin A, Kohan DE. Physiology and pathology of endothelin-1 in renal mesangium. American Journal of Physiology - Renal Physiology. 2003;285:F579–F589. doi: 10.1152/ajprenal.00019.2003. [DOI] [PubMed] [Google Scholar]
- Sorokin A, Kozlowski P, Graves L, Philip A. Protein-tyrosine kinase Pyk2 mediates endothelin-induced p38 MAPK activation in glomerular mesangial cells. The Journal of Biological Chemistry. 2001;276:21521–21528. doi: 10.1074/jbc.M008869200. [DOI] [PubMed] [Google Scholar]
- Soundararajan R, Lu M, Pearce D. Organization of the ENaC-regulatory machinery. Critical Reviews in Biochemistry and Molecular Biology. 2012;47:349–359. doi: 10.3109/10409238.2012.678285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soundararajan R, Pearce D, Hughey RP, Kleyman TR. Role of epithelial sodium channels and their regulators in hypertension. The Journal of Biological Chemistry. 2010;285:30363–30369. doi: 10.1074/jbc.R110.155341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speed JS, Pollock DM. Endothelin, kidney disease, and hypertension. Hypertension. 2013;61:1142–1145. doi: 10.1161/HYPERTENSIONAHA.113.00595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Li D, Edwards N, Hynes AM, Wood K, Al-Hamed M, et al. Identification of compound heterozygous KCNJ1 mutations (encoding ROMK) in a kindred with Bartter's syndrome and a functional analysis of their pathogenicity. Physiological Reports. 2013;1:e00160. doi: 10.1002/phy2.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staruschenko A. Regulation of transport in the connecting tubule and cortical collecting duct. Comprehensive Physiology. 2012;2:1541–1584. doi: 10.1002/cphy.c110052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staruschenko A, Adams E, Booth RE, Stockand JD. Epithelial Na+ channel subunit stoichiometry. Biophysical Journal. 2005;88:3966–3975. doi: 10.1529/biophysj.104.056804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staruschenko A, Palygin O, Ilatovskaya DV, Pavlov TS. Epidermal growth factors in the kidney and relationship to hypertension. American Journal of Physiology - Renal Physiology. 2013;305:F12–F20. doi: 10.1152/ajprenal.00112.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staruschenko A, Sorokin A. Role of βPix in the Kidney. Frontiers in Physiology. 2012;3:154. doi: 10.3389/fphys.2012.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockand JD. New ideas about aldosterone signaling in epithelia. American Journal of Physiology - Renal Physiology. 2002;282:F559–F576. doi: 10.1152/ajprenal.00320.2001. [DOI] [PubMed] [Google Scholar]
- Stockand JD. The role of the epithelial Na+ channel (ENaC) in high AVP but low aldosterone states. Frontiers in Physiology. 2012;3:304. doi: 10.3389/fphys.2012.00304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockand JD, Mironova E, Bugaj V, Rieg T, Insel PA, Vallon V, et al. Purinergic inhibition of ENaC produces aldosterone escape. Journal of the American Society of Nephrology. 2010;21:1903–1911. doi: 10.1681/ASN.2010040377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stow LR, Jacobs ME, Wingo CS, Cain BD. Endothelin-1 gene regulation. The FASEB Journal. 2011;25:16–28. doi: 10.1096/fj.10-161612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stow LR, Richards J, Cheng KY, Lynch IJ, Jeffers LA, Greenlee MM, et al. The circadian protein period 1 contributes to blood pressure control and coordinately regulates renal sodium transport genes. Hypertension. 2012;59:1151–1156. doi: 10.1161/HYPERTENSIONAHA.112.190892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stricklett PK, Hughes AK, Kohan DE. Endothelin-1 stimulates NO production and inhibits cAMP accumulation in rat inner medullary collecting duct through independent pathways. American Journal of Physiology - Renal Physiology. 2006;290:F1315–F1319. doi: 10.1152/ajprenal.00450.2005. [DOI] [PubMed] [Google Scholar]
- Stuart D, Chapman M, Rees S, Woodward S, Kohan DE. Myocardial, smooth muscle, nephron, and collecting duct gene targeting reveals the organ sites of endothelin A receptor antagonist fluid retention. J Journal of Pharmacology and Experimental Therapeutics. 2013;346:182–189. doi: 10.1124/jpet.113.205286. [DOI] [PubMed] [Google Scholar]
- Stuart D, Rees S, Woodward SK, Koesters R, Strait KA, Kohan DE. Disruption of the endothelin A receptor in the nephron causes mild fluid volume expansion. BMC Nephrology. 2012;13:166. doi: 10.1186/1471-2369-13-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan JC, Goodchild TT, Cai Z, Pollock DM, Pollock JS. Endothelin(A) (ET(A)) and ET(B) receptor-mediated regulation of nitric oxide synthase 1 (NOS1) and NOS3 isoforms in the renal inner medulla. Acta Physiologica. 2007;191:329–336. doi: 10.1111/j.1748-1716.2007.01754.x. [DOI] [PubMed] [Google Scholar]
- Takemoto F, Uchida S, Ogata E, Kurokawa K. Endothelin-1 and endothelin-3 binding to rat nephrons. American Journal of Physiology. 1993;264:F827–F832. doi: 10.1152/ajprenal.1993.264.5.F827. [DOI] [PubMed] [Google Scholar]
- Teitelbaum I, Berl T. Increased cytosolic Ca2+ inhibits AVP-stimulated adenylyl cyclase activity in rat IMCT cells by activation of PKC. American Journal of Physiology. 1994;266:F486–F490. doi: 10.1152/ajprenal.1994.266.3.F486. [DOI] [PubMed] [Google Scholar]
- Tomita K, Nonoguchi H, Terada Y, Marumo F. Effects of ET-1 on water and chloride transport in cortical collecting ducts of the rat. American Journal of Physiology. 1993;264:F690–F696. doi: 10.1152/ajprenal.1993.264.4.F690. [DOI] [PubMed] [Google Scholar]
- Tykocki NR, Watts SW. The interdependence of endothelin-1 and calcium: a review. Clinical Science. 2010;119:361–372. doi: 10.1042/CS20100145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacca F, Bagnato A, Catt KJ, Tecce R. Transactivation of the epidermal growth factor receptor in endothelin-1-induced mitogenic signaling in human ovarian carcinoma cells. Cancer Research. 2000;60:5310–5317. [PubMed] [Google Scholar]
- Vignon-Zellweger N, Heiden S, Emoto N. Renal function and blood pressure: molecular insights into the biology of endothelin-1. Contributions to Nephrology. 2011;172:18–34. doi: 10.1159/000328164. [DOI] [PubMed] [Google Scholar]
- Wang W, Li C, Nejsum LN, Li H, Kim SW, Kwon TH, et al. Biphasic effects of ANP infusion in conscious, euvolumic rats: roles of AQP2 and ENaC trafficking. American Journal of Physiology - Renal Physiology. 2006;290:F530–F541. doi: 10.1152/ajprenal.00070.2005. [DOI] [PubMed] [Google Scholar]
- Wang Y, Simonson MS, Pouyssegur J, Dunn MJ. Endothelin rapidly stimulates mitogen-activated protein kinase activity in rat mesangial cells. Biochemical Journal. 1992;287:589–594. doi: 10.1042/bj2870589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnock DG, Kusche-Vihrog K, Tarjus A, Sheng S, Oberleithner H, Kleyman TR, et al. Blood pressure and amiloride-sensitive sodium channels in vascular and renal cells. Nature Reviews Nephrology. 2014;10:146–157. doi: 10.1038/nrneph.2013.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrington JP, Coleman K, Skaggs C, Hosick PA, George EM, Stec DE, et al. Heme oxygenase-1 promotes migration and β-epithelial Na+ channel expression in cytotrophoblasts and ischemic placentas. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology. 2014;306:R641–R646. doi: 10.1152/ajpregu.00566.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch AK, Jacobs ME, Wingo CS, Cain BD. Early progress in epigenetic regulation of endothelin pathway genes. British Journal of Pharmacology. 2013;168:327–334. doi: 10.1111/j.1476-5381.2012.01826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welling PA. Regulation of renal potassium secretion: molecular mechanisms. Seminars in Nephrology. 2013;33:215–228. doi: 10.1016/j.semnephrol.2013.04.002. [DOI] [PubMed] [Google Scholar]
- Welling PA, Ho K. A comprehensive guide to the ROMK potassium channel: form and function in health and disease. American Journal of Physiology - Renal Physiology. 2009;297:F849–F863. doi: 10.1152/ajprenal.00181.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen D, Cornelius RJ, Sansom SC. Interacting influence of diuretics and diet on BK channel-regulated K homeostasis. Current Opinion in Pharmacology. 2014;15:28–32. doi: 10.1016/j.coph.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel M, Knels L, Kummer W, Koch T. Distribution of endothelin receptor subtypes ETA and ETB in the rat kidney. Journal of Histochemistry & Cytochemistry. 2006;54:1193–1203. doi: 10.1369/jhc.5A6888.2006. [DOI] [PubMed] [Google Scholar]
- Wiemuth D, Lott JS, Ly K, Ke Y, Teesdale-Spittle P, Snyder PM, et al. Interaction of serum- and glucocorticoid regulated kinase 1 (SGK1) with the WW-domains of Nedd4-2 is required for epithelial sodium channel regulation. PLoS ONE. 2010;5:e12163. doi: 10.1371/journal.pone.0012163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Suzuki H, Kubo Y, Matsumoto A, Uemura H. Endothelin A receptor-like immunoreactivity on the basal infoldings of rat renal tubules and collecting ducts. Archives of Histology and Cytology. 2008;71:77–87. doi: 10.1679/aohc.71.77. [DOI] [PubMed] [Google Scholar]
- Yu L, Bao HF, Self JL, Eaton DC, Helms MN. Aldosterone-induced increases in superoxide production counters nitric oxide inhibition of epithelial Na channel activity in A6 distal nephron cells. American Journal of Physiology - Renal Physiology. 2007;293:F1666–F1677. doi: 10.1152/ajprenal.00444.2006. [DOI] [PubMed] [Google Scholar]
- Yukimura T, Notoya M, Mizojiri K, Mizuhira V, Matsuura T, Ebara T, et al. High resolution localization of endothelin receptors in rat renal medulla. Kidney International. 1996;50:135–147. doi: 10.1038/ki.1996.296. [DOI] [PubMed] [Google Scholar]
- Zaika O, Mamenko M, O'Neil RG, Pochynyuk O. Bradykinin acutely inhibits activity of the epithelial Na+ channel in mammalian aldosterone-sensitive distal nephron. American Journal of Physiology - Renal Physiology. 2011;300:F1105–F1115. doi: 10.1152/ajprenal.00606.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaika O, Mamenko M, Staruschenko A, Pochynyuk O. Direct activation of ENaC by angiotensin II: recent advances and new insights. Current Hypertension Reports. 2013;15:17–24. doi: 10.1007/s11906-012-0316-1. [DOI] [PMC free article] [PubMed] [Google Scholar]