

Abstract
The blood–brain barrier (BBB) has become a major focus of attention in cerebral pathophysiology and disease progression in the central nervous system. Endothelial tight junctions, the basal lamina, and perivascular astrocytes are jointly referred to as BBB or neurovascular unit. Around the cerebral endothelial cells is the basal lamina composed primarily of laminin, fibronectin, and heparan sulfate. The basal lamina provides a structural barrier to extravasation of cellular blood elements and anchors endothelial cells to astrocytes. Barriers limiting transport into and out of the brain are found at the tight junction proteins and at the basal lamina. The relative contribution of these two sites has not been studied, but it is likely that both are disrupted to some extent in various injury scenarios. We have shown that activation of matrix metalloproteinases (MMPs) opens the BBB by degrading tight junction proteins (claudin-5 and occludin) and increases BBB permeability after stroke, and that an MMP inhibitor prevents degradation of tight junction proteins and attenuates BBB disruption.
Keywords: Claudin-5, Tight junction proteins, Blood-brain barrier, Endothelial cells, Matrix metalloproteinase and stroke
1. Introduction
The blood–brain barrier (BBB) is the regulated interface between the peripheral circulation and the central nervous system. Tight junctions between endothelial cells of the BBB restrict paracellular diffusion of water-soluble substances from blood to brain (1, 2). Tight junction proteins join endothelial cells together, forming an interface between blood and brain (3–5). Transmembrane tight junction proteins consist of three integral proteins: claudins, occludin, and junctional adhesion molecules (6, 7). Zona occludens and cingulin are considered to be cytoplasmic tight junctional accessory proteins, which connect tight junctions to the actin cytoskeleton (8, 9). The extracellular loops of occludin, claudins, and adhesion molecules originating from neighboring cells form the paracellular barrier of the tight junction, which selectively excludes most blood-borne substances from entering the brain.
Tight junctions are dynamic structures. Proteins of tight junction are subject to changes in expression, subcellular location, posttranslational modification, and protein–protein interactions under both physiological and pathophysiological conditions (2). Occludin, claudin-5, and zona occludens-1 (ZO-1), which are the main structural barrier proteins, are considered sensitive indicators of normal and disturbed functional states of the BBB (10–15). The claudins have been demonstrated to be one of the essential integral proteins in tight junction strands, and the relative composition of the claudin species in tight junctions directly determines the barrier function. Adrenomedullin, a vasodilator, improves BBB function in part through the induction of claudin-5 expression (16). Claudin-5 knockout mice have a selective permeability defect in the BBB that allows molecules of less than 800 kDa to cross the BBB (17, 18). In endothelial cell cultures and in in vivo BBB models, tight junction proteins are disrupted by ischemia/hypoxia (19–22). We have shown that MMPs open the BBB after stroke by degrading tight junction proteins. We have further shown that an MMP inhibitor prevents degradation of the tight junction proteins. Our findings support previous studies suggesting that tight junction proteins, including claudin-5, undergo structural changes after an ischemic injury (23) and directly implicate MMPs in stroke-induced degradation of tight junction proteins associated with BBB permeability.
Disruption of BBB tight junctions by diseases or drugs can lead to impaired BBB function and thus compromise the central nervous system. Therefore, understanding how BBB tight junctions are affected by various factors holds significant promise for the prevention and treatment of neurological diseases. Claudins are 20–24-kDa proteins; at least 24 have been identified in mammals (2). Claudins expressed in endothelial cells of neural tissues include claudin-1, −3, −5, and −12, and they are thought to be candidate molecules responsible for endothelial barrier function (24). Claudin-5 is primarily expressed in endothelial cells of BBB. There is clear evidence that the claudins constitute the backbone of tight junction strands (25). Commercial availability of antibodies to claudin-5 can be used with straightforward assay protocols to detect disruption of claudins by disease or drugs with either immunofluorescence or Western blotting. Confocal immunofluorescence detection allows confirmation by morphology of disruption of claudin-5, the location of claudin-5 in tight junctions of BBB, and the changes in expression and distribution of claudin-5 in different types of brain cells.
2. Materials
2.1. Double-Staining Immunofluorescence
10× Phosphate-buffered saline (PBS, see Note 1): 80 g of NaCl (137 mM), 2.0 g of KCl (27 mM), 14.4 g of Na2HPO4 (14 mM), and 2.4 g of KH2PO4 (43 mM). Bring total volume to 1 L with deionized water (see Note 2). Adjust pH to 7.3. Sterilize by autoclaving.
2% Paraformaldehyde containing 0.1 M sodium periodate and 0.075 M lysine in 100 mM phosphate buffer, pH 7.3 (2% PLP).
Pre-cooled acetone at 4°C.
Blocking buffer: 1× PBS containing 0.1% Tween 20, 1% BSA, and 5% normal serum. Blocking serum ideally should be derived from the same species in which the secondary antibody is raised.
Antibody dilution buffer: 1× PBS, 0.1% Tween 20, and 1% BSA.
- Primary antibodies:
- Mouse monoclonal anti-claudin-5 (22–24 kDa) (Invitrogen/Zymed, cat#: 35–2500; see Note 3).
- Rabbit anti-glial fibrillary acidic protein (GFAP) antibody (Sigma–Aldrich Chemicals, St. Louis, MO) for recognizing astrocytes.
- Secondary antibodies:
- Anti-mouse IgG labeled with Cy3 (red fluorescence).
- Anti-rabbit IgG labeled with FITC (green fluorescence).
Mounting medium: ProLong anti-fade (Molecular Probes, Eugene, OR).
Solvent tanks or Tek jars.
Pre-cleaned microscope slides (Superfrost/plus, Fisher Scientific, Pittsburgh, PA).
Slide racks to fit into the solvent tanks.
Humidifying chamber or airtight plastic container.
Microscope cover glass (22 × 60 mm), slightly narrower than microscope slides (Fisher Scientific, Pittsburgh, PA).
Pipettes and other general laboratory equipment.
Horizontal shaker.
Fluorescence and confocal microscopes.
2.2. Sodium Dodecyl Sulfate-Polyacrylamide Gel
1.5 M Tris–HCl, pH 8.8 (Bio-Rad, Hercules, CA).
10% Sodium dodecyl sulfate (SDS). Store at room temperature.
0.5 M Tris–HCl, pH 6.8 (Bio-Rad, Hercules, CA).
30% Acrylamide/bisacrylamide (29:1, 3.3% C, Bio-Rad). Acrylamide is a potent cumulative neurotoxin: wear gloves at all times.
N, N, N, N′-tetramethylethylenediamine (TEMED, Bio-Rad).
Ammonium persulfate: prepare 10% (100 mg/ml) solution in water immediately before use.
Water-saturated isobutanol. Combine equal parts of isobutanol and distilled water. Mix well and allow the phases to separate. Use the top layer. Store at room temperature.
Kaleidoscope pre-stained standards (Bio-Rad, Hercules, CA).
Electrophoresis and electrotransfer system for Western blot.
Orbital shaker and rocking platform. Refrigerated microcentrifuge.
2.3. Western Blotting for Claudin-5
Radio-immunoprecipitation assay (RIPA) buffer: 150 mM sodium chloride, 1.0% NP-40 or Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris–HCl, pH 8.0. RIPA buffer is useful for preparing whole-cell extracts containing total nuclear, cytoplasmic, and membrane proteins.
Halt protease inhibitor single-use cocktail and halt phosphatase inhibitor single-use cocktail (Thermo Scientific, Rockford, IL).
Micro BCA protein assay kit (Thermo Scentific).
Laemmli 2× loading (sample) buffer: 4% SDS, 10% 2-mercaptoethanol, 20% glycerol, 0.004% bromophenol blue, and 0.125 M Tris–HCl, pH 6.8. It can also be prepared at 4× and 6× strength to minimize dilution of the samples. The 2× buffer is to be mixed in a 1:1 ratio with the sample.
10× Running buffer (also called electrophoresis buffer) (1 L): 30.3 g Trizma base (0.25 M), 144 g glycine (1.92 M), and 10 g SDS (1%, add last). Dissolve the chemicals in deionized water and bring the total volume to 1 L. Do not adjust pH. Working solution: Dilute 100 ml 10× running buffer in 900 ml deionized water to make a 1× running buffer.
10× Transfer buffer (1 L): 30.3 g Trizma base (0.25 M) and 144 g glycine (1.92 M), pH should be 8.3, do not adjust. To make 1 L of 1× transfer buffer: 200 ml methanol, 100 ml 10× transfer buffer, and 700 ml deionized water (see Note 4).
Immobilon transfer membrane: Polyvinylidene fluoride (PVDF) membrane (pore size: 0.2 mm, Millipore). 3MM chromatography paper (Whatman).
PBS with Tween 20 (PBS-T): 1× PBS and 0.1% Tween 20. Dilute 100 ml 10× PBS with 900 ml deionized water.
Blocking buffer: 5% (w/v) nonfat dry milk in PBS-T. Filter through filter paper (Qualitative Circles, Whatman).
Antibody dilution buffer: 5% (w/v) nonfat dry milk in PBS-T.
Primary antibody: Mouse anti-claudin-5 (Invitrogen, cat#: 35–2500).
Secondary antibody: Anti-mouse IgG conjugated to horseradish peroxidase (Cell Signaling, cat. #: 7076).
SuperSignal West Pico Chemiluminescent Substrate system (Pierce) and Bio-Max ML film (Kodak, Rochester, NY).
Stripping buffer: 0.2 M glycine, pH 2.5, 0.05% Tween 20, and 0.1% SDS. Sterile filter the solution and keep at 4°C.
3. Methods
3.1. Preparation of Brain Sections for Double Staining of Immunofluorescence
Spontaneously hypertensive rats (SHR) that had undergone a middle cerebral artery occlusion (MCAO) with reperfusion for 3 and 24 h are deeply anesthetized and intracardially perfused with 400 ml of 2% PLP. Brains are removed rapidly, postfixed for 24 h in 2% PLP, and cryoprotected in 30% sucrose in PBS until brains sink to the bottom of the container. Freeze PLP-fixed brains in blocks in liquid nitrogen according to standard procedures. The samples may be stored at −80°C before sectioning.
Before cutting sections, allow the temperature of the block to equilibrate to the temperature of the cryostat (usually −20°C). Cut 10-μm-thick sections of the brain and place the sections on a Fisher Superfrost slide. Store the slides at −80°C in a sealed slide box until ready for staining. Thaw the slides at room temperature prior to fixing and staining.
Fix the slides in cold acetone for 10 min and keep refrigerated at 4°C. Wash in three changes of PBS-T. Label the slides with a solvent-resistant pen and demarcate the sections if required.
To blocking nonspecific binding of IgG, incubate the slides in blocking buffer in a humidified slide chamber for 1 h at room temperature.
Remove the blocking buffer without letting the sections dry and by washing. Apply a mixture of the claudin-5 (3 μg/ml) and GFAP (1:500) primary antibodies in antibody dilution buffer. Incubate the slides with primary antibodies in a humidified slide chamber for 48 h (or two nights) at 4°C (see Note 5).
Rinse the slides in PBS-T for 6 min three times.
From this step, the slides should be put under aluminum foil and the room lights dimmed. The secondary antibodies (goat anti-mouse IgG labeled with Cy3 for claudin-5; goat anti-rabbit IgG labeled with FITC for GFAP) are diluted at 1:500 in antibody dilution buffer.
Incubate the slides for 2 h (see Note 5) at room temperature with secondary antibodies in a humidified slide chamber.
Rinse the slides in PBS-T three times, 6 min each time, and mount in ProLong anti-fade (Molecular Probes, Eugene, OR). Seal the slides around the edges of the cover glasses with nail polish or Permount (Fisher Scientific) 1 h after mounting.
Photograph the slides with a fluorescence microscope (Olympus BX51, Olympus Optical Co. Ltd, Japan) and a confocal microscope (Zeiss LSM 510, Carl Zeiss MicroImaging, Thornwood, NY). Sections incubated in the absence of the primary antibody, with rabbit IgGs will not exhibit immunofluorescence. Examples of the signals for claudin-5 and astrocytes (GFAP) are shown in Fig. 1.
Fig. 1.
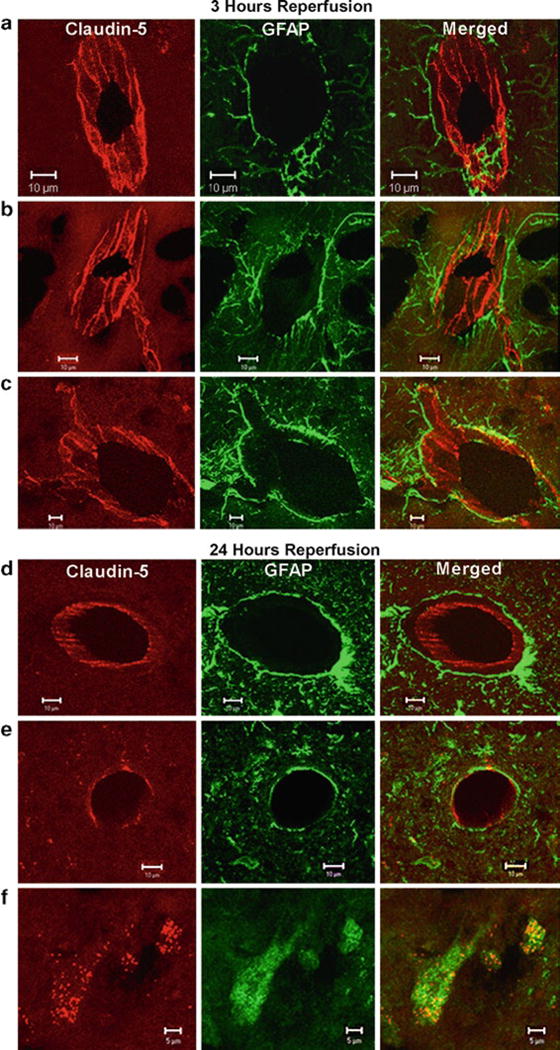
Confocal micrographs showing claudin-5 immunoreactivity in the sham-operated animals and ischemic and nonischemic hemispheres after 3 h of reperfusion (a–c). The sham-operated animals and the nonischemic side show that the claudin-5 (Cy-3, red) in blood vessels is separated from the astrocytes (GFAP-FITC, green) surrounding them (a and b). The merged images show that the claudin-5 and astrocytes are separate. In the ischemic hemisphere (c), there is fragmentation and degeneration of the claudin-5 immunoreactivity. Co-localization of claudin-5 and GFAP was seen in the ischemic hemisphere. Confocal micrographs showing claudin-5 immunoreactivity in the ischemic and nonischemic hemispheres after 24 h of reperfusion (d–f). Immunoreactivity of claudin-5 with GFAP in brain vessels in nonischemic hemisphere (d). Ischemia caused a loss of claudin-5 from the blood vessels at 24 h (e). There was co-localization of the claudin-5 in the GFAP-positive astrocytes (f). These photos were taken from penumbral areas in the caudate and lateral cortex. Scale bars indicate 10 μm in (a–e). Scale bars show 5 μm in (f) (reproduced from ref. 23).
3.2. Preparation of Samples for Assay of Claudin-5 by Western Blotting
SHR that had undergone an MCAO with reperfusion for 3 and 24 h are deeply anesthetized and intracardially perfused briefly with 0.9% saline to flush blood out of the brain. Remove brains rapidly and dissect the tissue of interest with clean tools, on ice preferably, as quickly as possible to prevent degradation by proteases (see Note 6).
Place the tissue in round-bottom microcentrifuge tubes and immerse in liquid nitrogen to “snap freeze.” Store the samples at −80°C for later use or keep on ice for immediate homogenization.
Add RIPA buffer containing protease and phosphatase inhibitors to the tissue (1:4 tissue to RIPA buffer) and homogenize with an electric homogenizer (see Note 7). Incubate the lysates on ice for 10 min.
Centrifuge for 20 min at 12,000 × g at 4°C in a refrigerated microcentrifuge. Gently remove the tubes from the centrifuge and place on ice. Collect the supernatant and place in a fresh tube on ice; discard the insoluble pellet.
Determination of protein concentration using Micro BCA protein assay kit. Bovine serum albumin (BSA) is a frequently used protein standard. Once you have determined the concentration of each sample, you can freeze them at −20°C or −80°C for later use, or prepare for loading onto a SDS-PAGE gel.
Preparation of samples for loading onto gels. Add Laemmli loading buffer 1:1 with samples (50 μg total protein will be loaded for the claudin-5 assay). Do not add reducing agents (mercaptoethanol or DTT) and do not boil (nonreducing & denaturing conditions, see Note 8). The samples are ready for separation by SDS-PAGE.
3.3. SDS-PAGE
When separated on a polyacrylamide gel, the procedure is abbreviated as SDS-PAGE. The technique is a standard means for separating proteins according to their apparent molecular weight.
Preparation of PAGE gel. These instructions assume the use of a Hoefer miniVE vertical electrophoresis system. It is critical that the glass plates for the gels are scrubbed clean with distilled water followed by 95% ethanol and then air-dried before use.
Prepare a 1.0-mm-thick, 15% separating gel by mixing 2.5 ml of 1.5 M Tris–HCl, pH 8.8, with 5 ml acrylamide/bis solution, 100 μl of 10% SDS, 2.348 ml of water, 50 μl 10% ammonium persulfate solution (APS), and 5 μl TEMED (see Note 9). Pour the gel to about 1 cm from the bottom of the comb and carefully layer 1 ml of water-saturated isobutanol on top of the gel. Allow 30 min for the gel to polymerize (see Note 10).
When the gel has polymerized, pour off the isobutanol and rinse with deionized water.
Prepare the stacking gel by mixing 1.25 ml of 0.5 M Tris–HCl, pH 6.8, with 0.833 ml acrylamide/bis solution, 50 μl of 10% SDS, 2.837 ml water, 25 μl APS, and 5 μl TEMED. Pour the stacking gel and insert the comb. The stacking gel should polymerize within 30 min.
Prepare the running buffer by diluting 50 ml of the 10× running buffer with 450 ml of water in a graduated cylinder and mix.
When the stacking gel has polymerized, place in a gel rig and immerse in running buffer. Carefully remove the comb and flush the wells thoroughly with running buffer (see Note 11).
Load the samples into the wells. Include one well for prestained molecular weight markers. A range of molecular weight markers that will enable the determination of the protein size is also used to monitor the progress of an electrophoretic run. We use 8 μl Bio-Rad Kaleidoscope pre-stained standards (cat. #161-0324).
Place the safety lid on the tank and plug the leads into an approved power supply. Run the gel with constant voltage at 100 V for 75–90 min. Allow the dye front (blue, 7.6 kDa) to run to the bottom edge of the gel. The molecular weight of claudin-5 is about 22–24 kDa (see Note 12).
3.4. Western Blotting for Claudin-5
Preparation of membrane: Cut a piece of PVDF membrane (pore size, 0.2 μm, slightly larger than gel). Wet for 5 min in methanol on a rocker at room temperature. Remove methanol and equilibrate the membrane in 1× blotting buffer for at least 10 min.
When the SDS-PAGE gel has finished running, cut a corner of the gel to identify the proper orientation of the gel. Equilibrate the gel in transfer buffer for 30 min prior to blotting to facilitate the removal of electrophoresis buffer salts and detergents from the gel.
-
Assemble “sandwich” for the Hoefer blot module. Saturate the sponges and filter papers (slightly larger than gel) in 1× blotting buffer. Assemble the transfer apparatus in the following order: Black cathode (−) side of the Blot Module – Sponge – 3MM filter paper – gel – PVDF membrane – 3MM filter paper – sponge – Red Anode (+) side of the Blot Module. Ensure that no bubbles are trapped in the resulting sandwich.
Make sure that the PVDF membrane is between the gel and the anode. It is vitally important to ensure that this orientation or the proteins will be lost from the gel into the buffer rather than transferred to the PVDF.
Place the cassette into the transfer tank. Set the buffer tank on top of a magnetic stirrer with a stir bar. Add the pre-cooled transfer buffer into the module such that it completely covers the sandwich. Fill the tank with distilled water, preferably chilled to 4°C, to dissipate heat from the module during the transfer.
Place the safety lid on the tank and plug the leads into an approved power supply. Transfer for 1 h at 25 V.
Once the transfer is complete, disassemble the apparatus and cut the PVDF membrane to the same size as the gel by tracing around the gel with a razor blade. The excess gel and PVDF can then be discarded. The colored molecular weight markers should be clearly visible on the membrane. Note: Perform all operations with the membrane quickly and do not allow it to become dry.
Immediately place the membrane in blocking buffer (5% (w/v) nonfat dry milk in PBS-T) at room temperature for 1 h on an orbital shaker.
Discard the blocking buffer.
Make a 1:500 dilution of the primary claudin-5 antibody in PBS-T containing 5% nonfat milk and incubate the washed membrane overnight at 4°C on a rocker in a Kapak bag.
Wash the membrane three times for 5 min each in a large volume of PBS-T on an orbital shaker.
Incubate the membrane with an anti-mouse secondary antibody at 1:5,000 dilution for 75 min at room temperature on a rocker.
Discard the secondary antibody and wash the membrane three times for 10 min each with a large volume of PBS-T on an orbital shaker at room temperature.
During the final wash, prepare 5 ml working solution of SuperSignal West Pico Chemiluminescent Substrate system. Once the final wash is removed from the blot, transfer the membrane into the working solution and incubate for 5 min.
Drain the detect reagent, blot the membrane with Kimwipes, and then place it between the leaves of a plastic paper protector that has been cut to the size of an X-ray film cassette.
In a dark room, place the membrane with the protein side up in an X-ray film cassette. Place the film on top of the membrane and expose the film for a suitable exposure time, typically, initially 10–20 s, and then re-expose for the optimal time as needed. An example of the results produced is shown in Fig. 2.
Fig. 2.
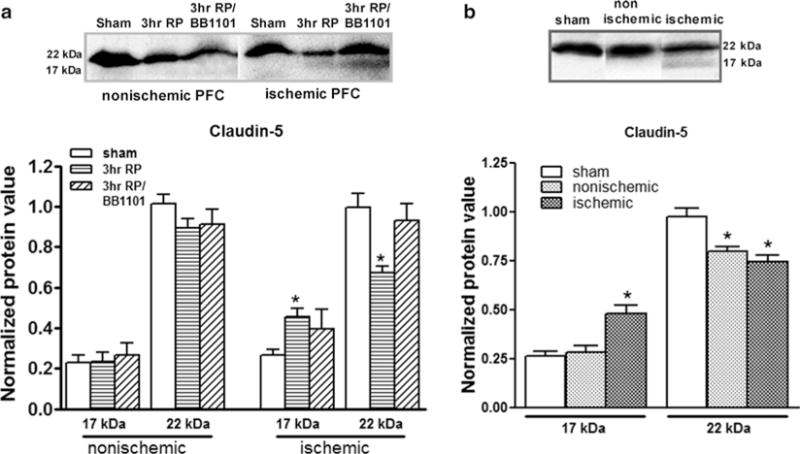
(a) Western blots for claudin-5. The Western blots were performed with whole-cell extracts from rat brains with a 90-min MCAO followed by 3 hours reperfusion (3hr RP). The Western blot showed significant reductions (p < 0.01, n = 6) of claudin-5 (22 kDa) at 3 h in the ischemic piriform cortex (PFC). In addition, a new 17-kDa band for claudin-5 was found in the ischemic hemisphere at 3 h post-reperfusion. The 17-kDa protein showed a significant increase (p < 0.01). Treatment with BB-1101, a broad-spectrum MMP inhibitor (3hr RP/BB1101), significantly inhibited degradation of claudin-5 (22 kDa) (p < 0.02, n = 6). (b) Western blots showing degradation of claudin-5 after a 90-min MCAO followed by 24-h reperfusion. A decrease in the higher molecular weight form (22 kDa) of claudin-5 was observed in both the ischemic and nonischemic hemispheres compared to sham-operated animals (p < 0.05, p < 0.01, respectively, n = 6). A significant increase in the 17-kDa claudin-5 was seen in the ischemic hemisphere (p < 0.002) (reproduced from ref. 23).
3.5. Stripping and Re-probing Blots for Other Tight Junction Proteins or Loading Control (See Notes 13 and 14)
Rinse the membrane with 1× PBS-T.
Cut the membrane to a slightly bigger size than the blot and place in a Kapak bag. Add 10 ml of stripping buffer and seal the bag.
Immerse in an 80°C water bath for 20 min. Rinse the membrane in 1× PBS-T. Save the membrane in PBS-T and store at 4°C until re-probing. Before re-probing, block the membrane for 1 h in blocking buffer.
Footnotes
There are numerous recipes for PBS. We recommend the recipe listed here because we found it to produce minor background for immunohistochemistry.
All solutions should be prepared in deionized water that has a resistivity of 18.2 MΩ-cm and a total organic content of less than five parts per billion.
We have found this claudin-5 antibody to be excellent for both Western blotting and immunofluorescence. Similar antibodies are available from other commercial sources.
For transfer of proteins larger than 80 kDa, it is recommended that SDS is included at a final concentration of 0.1%. If your protein of interest is small, consider removing SDS from the transfer buffer and keep the methanol concentration at 20%.
Claudin-5 and other claudins are 20–24-kDa integral membrane proteins with four transmembrane domains. We have found that 48-h incubation of primary antibody and 2-h incubation of secondary antibody generated very clear immunostaining of claudin-5.
As soon as lysis occurs, proteolysis, dephosphorylation, and denaturation begin. These events can be slowed down tremendously by keeping the samples on ice at all times and adding protease and phosphatase inhibitors to the lysis buffer immediately before use.
Volumes of lysis buffer must be determined in relation to the amount of tissue present (protein extract should not be too diluted to avoid loss of protein and large volumes of samples to be loaded onto gels). Ideally, protease and phosphatase inhibitors should be added to RIPA buffer immediately before adding to tissue samples. The buffer (with inhibitors) should be ice cold prior to homogenization.
We have found this antibody against claudin-5 to produce a specific band of 22 kDa when samples are prepared under nonreducing and non-denaturing conditions.
TEMED is best stored at room temperature in a desiccator. Buy small bottles as it may decline in quality (gels will take longer to polymerize) after opening.
Acrylamide is a potent cumulative neurotoxin: wear gloves at all times.
Gels can be purchased precast, or produced in the laboratory (recipes can be found in laboratory handbooks). Either way, choose carefully the percentage of your gel as this will determine the rate of migration and degree of separation between proteins. The smaller the size of the protein of interest, the higher the percentage of acrylamide that is required to achieve adequate separation and focusing of protein bands (see http://www.abcam.com/technical for details).
Proceed to the transfer step immediately since proteins that have been electrophoretically separated may slowly diffuse within gels after electrophoresis.
Loading controls are required when a comparison is to be made between the levels of a protein in different samples. The loading control bands can be used to quantify the protein amounts in each lane and check for even transfer from the gel to the membrane across the whole gel. For whole-cell/cytoplasmic samples, beta-actin, GAPDH, and tubulin are markers commonly used as loading controls. In our case, we used Brilliant Blue R (Sigma–Aldrich) staining on the same PVDF membranes to normalize protein loading and transfer, since many control proteins commonly used in conventional relative protein level analysis, such as GAPDH and beta-actin, have been reported to be affected by ischemia (26). The results are reported as normalized band intensity for quantifying relative protein levels (Fig. 2) (23). As the Brilliant Blue R stain is not reversible, stain membranes with Brilliant Blue R only when there are no further plans to do Western blots.
This protocol can be adapted for other claudins and other tight junction proteins, such as occludin (65 kDa) and ZO-1 (220 kDa), when a gradient polyacrylamide gel (4–20%) is used.
References
- 1.Vracko R. Basal lamina scaffold-anatomy and significance for maintenance of orderly tissue structure. Am J Pathol. 1974;77(2):314–46. [PMC free article] [PubMed] [Google Scholar]
- 2.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57(2):173–85. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 3.Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004;16(1):1–13. doi: 10.1016/j.nbd.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 4.Brightman MW, Reese TS. Junctions between intimately apposed cell membranes in the vertebrate brain. J Cell Biol. 1969;40(3):648–77. doi: 10.1083/jcb.40.3.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liebner S, et al. Correlation of tight junction morphology with the expression of tight junction proteins in blood-brain barrier endothelial cells. Eur J Cell Biol. 2000;79(10):707–17. doi: 10.1078/0171-9335-00101. [DOI] [PubMed] [Google Scholar]
- 6.Papadopoulos MC, et al. Occludin expression in microvessels of neoplastic and non-neoplastic human brain. Neuropathol Appl Neurobiol. 2001;27(5):384–95. doi: 10.1046/j.0305-1846.2001.00341.x. [DOI] [PubMed] [Google Scholar]
- 7.Furuse M, et al. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993;123(6 Pt 2):1777–88. doi: 10.1083/jcb.123.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haskins J, et al. ZO-3, a novel member of the MAGUK protein family found at the tight junction, interacts with ZO-1 and occludin. J Cell Biol. 1998;141(1):199–208. doi: 10.1083/jcb.141.1.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Itoh M, et al. Involvement of ZO-1 in cadherin-based cell adhesion through its direct binding to alpha catenin and actin filaments. J Cell Biol. 1997;138(1):181–92. doi: 10.1083/jcb.138.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirase T, et al. Regulation of tight junction permeability and occludin phosphorylation by Rhoa-p160ROCK-dependent and - independent mechanisms. J Biol Chem. 2001;276(13):10423–31. doi: 10.1074/jbc.M007136200. [DOI] [PubMed] [Google Scholar]
- 11.Hirase T, et al. Occludin as a possible determinant of tight junction permeability in endothelial cells. J Cell Sci. 1997;110(Pt 14):1603–13. doi: 10.1242/jcs.110.14.1603. [DOI] [PubMed] [Google Scholar]
- 12.Kanda T, Numata Y, Mizusawa H. Chronic inflammatory demyelinating polyneuropathy: decreased claudin-5 and relocated ZO-1. J Neurol Neurosurg Psychiatry. 2004;75(5):765–9. doi: 10.1136/jnnp.2003.025692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wen H, et al. Selective decrease in paracellular conductance of tight junctions: role of the first extracellular domain of claudin-5. Mol Cell Biol. 2004;24(19):8408–17. doi: 10.1128/MCB.24.19.8408-8417.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dobrogowska DH, Vorbrodt AW. Immunogold localization of tight junctional proteins in normal and osmotically-affected rat blood-brain barrier. J Mol Histol. 2004;35(5):529–39. doi: 10.1007/10.1007/s10735-004-1318-3. [DOI] [PubMed] [Google Scholar]
- 15.Matter K, Balda MS. Holey barrier: claudins and the regulation of brain endothelial permeability. J Cell Biol. 2003;161(3):459–60. doi: 10.1083/jcb.200304039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Honda M, et al. Adrenomedullin improves the blood-brain barrier function through the expression of claudin-5. Cell Mol Neurobiol. 2006;26(2):109–18. doi: 10.1007/s10571-006-9028-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Furuse M, et al. Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol. 1998;141(7):1539–50. doi: 10.1083/jcb.141.7.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nitta T, et al. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol. 2003;161(3):653–60. doi: 10.1083/jcb.200302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huber JD, Egleton RD, Davis TP. Molecular physiology and pathophysiology of tight junctions in the blood-brain barrier. Trends Neurosci. 2001;24(12):719–25. doi: 10.1016/s0166-2236(00)02004-x. [DOI] [PubMed] [Google Scholar]
- 20.Mark KS, Davis TP. Cerebral microvascular changes in permeability and tight junctions induced by hypoxia-reoxygenation. Am J Physiol Heart Circ Physiol. 2002;282(4):H1485–94. doi: 10.1152/ajpheart.00645.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Witt KA, et al. Effects of hypoxia-reoxygenation on rat blood-brain barrier permeability and tight junctional protein expression. Am J Physiol Heart Circ Physiol. 2003;285(6):H2820–31. doi: 10.1152/ajpheart.00589.2003. [DOI] [PubMed] [Google Scholar]
- 22.Yamagata K, et al. Hypoxia-induced changes in tight junction permeability of brain capillary endothelial cells are associated with IL-1beta and nitric oxide. Neurobiol Dis. 2004;17(3):491–9. doi: 10.1016/j.nbd.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 23.Yang Y, et al. Matrix metalloproteinase- mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007;27(4):697–709. doi: 10.1038/sj.jcbfm.9600375. [DOI] [PubMed] [Google Scholar]
- 24.Wolburg H, et al. Localization of claudin-3 in tight junctions of the blood-brain barrier is selectively lost during experimental autoimmune encephalomyelitis and human glioblastoma multiforme. Acta Neuropathol (Berl) 2003;105(6):586–92. doi: 10.1007/s00401-003-0688-z. [DOI] [PubMed] [Google Scholar]
- 25.Forster C. Tight junctions and the modulation of barrier function in disease. Histochem Cell Biol. 2008;130(1):55–70. doi: 10.1007/s00418-008-0424-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nishida Y, et al. Screening for control genes in mouse hippocampus after transient forebrain ischemia using high-density oligonucleotide array. J Pharmacol Sci. 2006;101(1):52–7. doi: 10.1254/jphs.fp0050881. [DOI] [PubMed] [Google Scholar]