

Abstract
Mammalian pre-implantation development is a complex process involving dramatic changes in the transcriptional architecture1–4. We report here a comprehensive analysis of transcriptome dynamics from oocyte to morula in both human and mouse embryos, using single-cell RNA sequencing. Based on single-nucleotide variants in human blastomere messenger RNAs and paternal-specific single-nucleotide polymorphisms, we identify novel stage-specific monoallelic expression patterns for a significant portion of polymorphic gene transcripts (25 to 53%). By weighted gene co-expression network analysis5,6, we find that each developmental stage can be delineated concisely by a small number of functional modules of co-expressed genes. This result indicates a sequential order of transcriptional changes in pathways of cell cycle, gene regulation, translation and metabolism, acting in a step-wise fashion from cleavage to morula. Cross-species comparisons with mouse pre-implantation embryos reveal that the majority of human stage-specific modules (7out of 9) are notably preserved, but developmental specificity and timing differ between human and mouse. Furthermore, we identify conserved key members (or hub genes) of the human and mouse networks. These genes represent novel candidates that are likely to be key in driving mammalian pre-implantation development. Together, the results provide a valuable resource to dissect gene regulatory mechanisms underlying progressive development of early mammalian embryos.
Mammalian pre-implantation development involves sequential decay of maternally stored RNAs in parallel with massive induction of new transcripts from the embryonic genome in a process called zygotic (or embryonic) genome activation (ZGA or EGA)7–9. However, previous gene expression profiling of human pre-implantation has been limited by blastomere sample size and quantitation platforms10–13. Recent advances in single-cell RNA sequencing (RNA-seq) technology14–17 have provided the unprecedented opportunity to study gene regulation in early human embryos under high resolution. We mapped approximately 700 million sequencing reads derived from 33 single cells of multiple stages, ranging from mature oocytes to 8-cell blastomeres. To control the quality of single-cell RNA-seq from technical errors, we focused on libraries (26 out of a total 33, or 79%) that share at least 80% similarity with other libraries derived from identical pre-implantation stages (Supplementary Table 1). In addition, we performed RNA-seq for three samples of individual morula embryos. The typical number of detectable genes (reads per kilobase per million (RPKM) > 0.1) ranged approximately from 8,500 to 12,000 genes in individual cells, demonstrating that our technique has greater sensitivity and coverage compared with previous microarray methods10–13. Unsupervised hierarchal clustering analysis and principal component analysis revealed that cells of different pre-implantation stages form distinct clusters (Fig. 1a, b). Furthermore, single-cell resolution revealed that blastomeres from the same 8-cell embryo are more similar to each other than blastomeres from a separate 8-cell embryo (Fig. 1a, b).
Figure 1. High-resolution single-cell transcriptome analysis of human pre-implantation embryos.
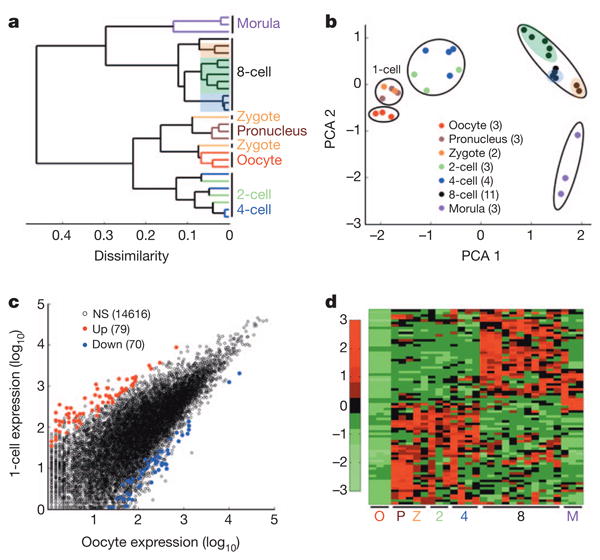
a, Unsupervised hierarchical clustering. b, Principal component analysis of single blastomere expression patterns for seven stages of oocytes and pre-implantation embryos. Sister cells from the same 8-cell embryo are highlighted together. The number of samples for each stage are indicated in the PCA legend in parenthesis as well as in Supplementary Table 1. c, Scatterplot showing the number of activated (red) and reduced (blue) genes in 1-cell embryos (n = 5) compared to oocytes (n = 3). d, Heatmap showing relative expression patterns of 1-cell activated genes (n = 79) across all pre-implantation stages. NS, not significant.
Intriguingly, cluster analyses showed that both pronuclear and zygote (1-cell stage) embryos clustered closely together but away from oocytes and cleavage embryos, indicating that the 1-cell stage exhibits a distinct transcriptome pattern. Comparisons between mature oocytes and 1-cell embryos identified 149 differentially expressed genes (false discovery rate (FDR) < 5%, > 2-fold change), 79 and 70 of which were up- and downregulated in 1-cell embryos, respectively (Fig. 1c). Interestingly, approximately half of the upregulated genes remain highly expressed in 2- and 4-cell stages but are downregulated at the 8-cell stage, whereas the other half is further upregulated at the 8-cell stage (Fig. 1d). By performing similar single-cell RNA-seq in mouse pre-implantation embryos (Supplementary Table 1), we observed 520 transcripts that were upregulated in pronuclei compared to mature oocytes (Supplementary Fig. 1), confirming that both human and mouse species exhibit a conserved minor wave of ZGA before the major wave of ZGA (or EGA). These results indicated that our approach has overall greater quantitation for identifying gene expression changes between different stages of pre-implantation development.
Single-cell RNA-seq enabled base-resolution scrutiny without confounding effects from cell population heterogeneity. In this unique study, all embryos were derived from intra-cytoplasmic sperm injection of different egg donors with the same sperm donor. Based on the paternal genotype as assayed by exome sequencing of the sperm donor's blood sample, we were able to follow the paternal genome in embryos through single-nucleotide polymorphism (SNP) analysis18–20. We identified paternally or maternally expressed genes either by inferring phased paternal haplotype information21,22 or by leveraging sites which are paternally homozygous in exome sequencing, but heterozygous in embryo transcripts (Fig. 2a and Supplementary Table 2). In total, we determined the parent-of-origin expression for approximately 850 to 1,400 gene transcripts (or 15–20% of all detected gene transcripts per stage). For further validation of allele-specific expression in early embryos, we analysed the status of imprinting genes that exhibit known parent-of-origin expression patterns (see Supplementary Text and Supplementary Figs 2 and 3). Unexpectedly, we found that 53% of 8-cell embryo transcripts and 23% of morula transcripts exhibit monoallelic maternal expression patterns (Fig. 2b), even though major EGA in human early embryos already occurred during the transition of 4- to 8-cell stage (Supplementary Fig. 4). For example, within a single haplotype region, we observed simultaneous maternal transcript activation and degradation in the ASB6 locus and C9orf78 locus, respectively (Fig. 2c). In a different scenario, the paternal genotype is homozygous in three consecutive SNPs (rs3829009, rs6990278 and rs8537) at the cell-cycle regulator CDCA2 gene locus. In this example, the phased paternal SNP pattern is not detected in 2- and 4-cell embryos but seen in 8-cell embryos, indicating that the detected transcripts in 2- and 4-cell embryos must be of maternal origin and also that CDCA2 undergoes paternal activation at the 8-cell stage (Fig. 2d). However, paternal activation at this locus appears transient as the paternal allele could not be detected in the morula stage. In fact, we observed a moderate correlation between expression of the paternal allele and overall transcript expression (r = 0.53, P = 0.06), suggesting that the paternal allele is regulated dynamically.
Figure 2. Tracing parent-of-origin allelic RNA transcripts through SNV analysis in pre-implantation embryos.

a, Schematic for deducing maternal and paternal transcript origin from the presence or absence of homozygous paternal alleles. Embryo number 3 has unclear contribution from the maternal allele. b, Pie charts showing the number and per cent of all gene transcripts exhibiting single-nucleotide variants (SNVs) and their assignments into one of three categories as illustrated in a. c, Example of maternal activation and degradation from a deduced partial haplotype. d, Example of paternal activation by comparing embryos that showed biallelic expression in one stage but monoallelic (maternal) expression in a previous stage. rs3829009 is highlighted because the minor alleleisanArg884Ser missense variant. SNPs are adenine (green), guanine (red) and thymine (brown).
Notably, the rs3829009 SNP in CDCA2 is a missense variant that results in a benign Arg to Ser amino acid change23 (Fig. 2d). As RNA-seq produces base-resolution information for codons, we further examined the global prevalence of potentially deleterious transcript expression. Using multiple single-cells from the same 8-cell embryo, we consistently identified 1,225 homozygous SNP variants, 3.5% of which are predicted as damaging non-synonymous variants (Supplementary Fig. 5). This result suggests that single-cell RNA-seq would be a powerful approach for genome-scale screening of potentially deleterious variants in embryos.
Thus far the analyses focused on individual gene-transcript changes during each transitional stage, but did not reveal the crucial shift of gene networks in pre-implantation development. To understand the co-expression relationships between genes at a systems level, we performed weighted gene co-expression network analysis (WGCNA)5,6. This unsupervised and unbiased analysis identified distinct co-expression modules corresponding to clusters of correlated transcripts (Fig. 3a). Notably, 9 out of 25 co-expression modules showed stage-specific expression values, that is, these modules are comprised of genes that tend to be overexpressed in a single developmental stage (r > 0.7, P < 10−3, Supplementary Fig. 6).
Figure 3. Network analysis of human pre-implantation development.
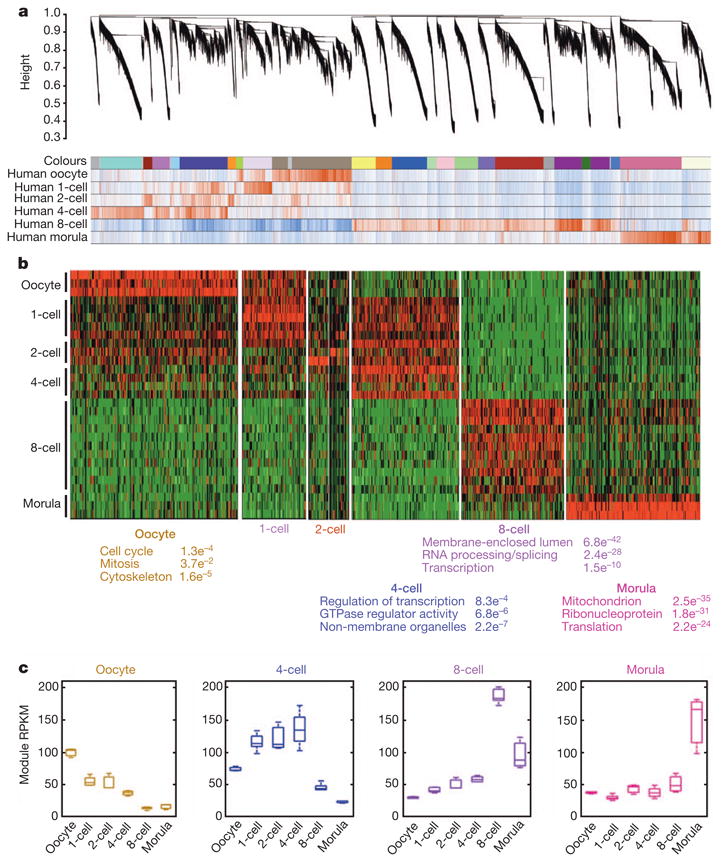
a, Hierarchical cluster tree showing co-expression modules identified using WGCNA. Modules correspond to branches and are labelled by colours as indicated by the first colour band underneath the tree. Remaining colour bands reveal highly correlated (red) or anti-correlated (blue) transcripts for the particular stage. b, Heatmap showing relative expression of 7,313 genes in 7 representative stage-specific modules across all samples. As each developmental window only has one or two highly correlated modules, the modules were assigned biological names. Top three representative gene ontology terms and their associated functional enrichment P values are shown below. c, Boxplots showing the distribution of module expression (mean RPKM of all genes within a given module) for different cell types.
These stage-specific modules probably represent core gene networks operating in each transitional stage. In total, 8,301 genes were part of human oocyte to morula stage-specific modules, revealing a step-wise requirement of new transcripts that are involved in gene transcription (4-cell), post-transcriptional RNA processing (8-cell), and then protein translation and cell energetics (morula) (Fig. 3b). As expected, the oocyte module undergoes gradual degradation over the course of development, whereas modules in the 4- to 8-cell transition show sharp degradation (threefold) and activation (fourfold) (Fig. 3c). Collectively, our systems analysis revealed that the transcriptional organization for human pre-implantation development can be summarized using a small number of stage-specific modules with well-defined function.
Importantly, these modules are highly robust and reproducible as can be seen from our module preservation analysis24 (Fig. 4a). This module preservation analysis uses a permutation test to define a test statistic, Zsummary, that summarizes the evidence that the network topology of the module is preserved in an independent data set (see Methods). Seven out of nine modules were detected with highly significant preservation scores (Zsummary > 10) in two publically available human preimplantation data sets (but with substantially less RNA transcripts). Prior to the direct study of transcription changes during human preimplantation process, mouse models provided crucial insights into the pre-implantation gene regulatory network2–4. However, it seems that only one study (involving far fewer transcripts) performed a cross-species comparison in the context of a pluripotency network11. Therefore, we performed single-cell RNA-seq in mouse blastomeres from oocyte to morula embryos for a direct cross-species comparative analysis (Supplementary Table 1). Cross-species module preservation analysis showed that 7 out of 9 human stage-specific modules were at least moderately preserved (Zsummary > 5) (Fig. 4a and Supplementary Table 1). We further validated the cross-species preservation using other mouse pre-implantation data sets11,25, which confirmed most human stage-specific modules are preserved in mouse (Fig. 4a). As expected, intra-species module preservation was more significant (Zsummary > 10) than inter-species preservation (Zsummary > 5, Fig. 4a). Thus, these preserved gene networks represent a strongly conserved transcriptional architecture of key developmental programs.
Figure 4. Stage-specific gene activation is preserved in human and mouse pre-implantation development.

a, Heatmap of human module preservation scores in available independent data sets labelled on the x axis. b, Heatmap showing the significance of gene overlaps between independently constructed human and mouse modules. The x axis shows only human stage-specific modules (n = 9) and the y axis shows all mouse modules (n = 15). Each cell contains the number of intersecting genes and P value of the intersection. Colour legend represents −log10 P value based on the hypergeometric test. c, Schematic drawing of the sequential transcriptome switches of four major pathways in human and mouse pre-implantation embryos. Gene lists for gene ontology terms are derived from human and mouse stage-specific modules with significant overlap (P < 10−4) as shown in b. d, Module visualization of network connections and associated function. Highly connected intramodular hub genes are indicated by a red dot and independently validated hub genes are highlighted in blue (for human only) and orange (for both human and mouse). Data in part a are from refs 10, 11 and 25.
By applying WGCNA independently to our mouse data set, we found that mouse development also involves stage-specific co-expression modules (Supplementary Fig. 7). Gene ontology analyses showed that mouse stage-specific modules share many functional similarities to their human counterparts, including the conserved sequential activation of functional enrichment changes (Supplementary Fig. 8). As expected, overlaps between human and mouse stage-specific modules were highly significant (P < 10−4, Fig. 4b, see also Supplementary Fig. 9), and the functional enrichment of overlapped stage-specific genes mostly reflected the overall network function (Fig. 4c). However, when we examined overlap of pre-major ZGA in human and mouse, we found that most genes were enriched for protein transport and GTPase signalling, whereas their respective modules as a whole were more enriched for cell-cycle genes (Fig. 4c and Supplementary Fig. 8).
Notably, stage-specific modules in human and mouse overlapped across multiple stages. For example, the mouse oocyte and 1-cell module genes overlapped significantly with genes that were specific to human oocyte, 1-cell and 4-cell stages (P < 10−6, Fig. 4b). This result suggested that mouse pre-major ZGA genes are spread over the longer gestational pre-major EGA window in humans. Likewise, post-major ZGA modules in mouse were found to have significant overlap and spread throughout all post-major EGA human stages (Fig. 4c). Although there is divergence in the timing of the major ZGA between human and mouse, these two species re-converge in both timing and function at the 8-cell to morula transition, during which many mitochondrion-related transcripts are upregulated (Fig. 4c and Supplementary Fig. 8). Collectively, our results show that human and mouse share many core transcriptional programs in early development, but diverge in their stage-specificity and timing, probably reflecting species-specific differences in human and mouse gestational periods.
Using the WGCNA measure of intramodular gene connectivity (kME), we identified 491 intramodular hub genes across all stage-specific modules (kME > 0.9, P < 10−22). Intramodular hub genes are centrally located in their respective modules and may thus be critical components within the network. Remarkably, 337 (69%, P < 10−16) hub genes in stage-specific modules can be validated (that is, independently identified as a hub gene in a separate data set), demonstrating that pre-implantation hub genes are highly reproducible (Supplementary Table 3). For example, KPNA7 (kME = 0.89, P < 10−22) is consistently identified as a hub gene in multiple human and mouse pre-major EGA networks (kME > 0.91). Notably, approximately half of Kpna7-deficient mouse embryos fail to develop to blastocysts, whereas the other half show delayed developmental progress26. Furthermore, SIN3A, a transcriptional co-repressor, is a 4-cell intramodular hub gene that was validated in every independent data set (Fig. 4d and Supplementary Table 3). Indeed, analysis of upstream regulators of human 8-cell module genes revealed overrepre-sentation in MYC, MAX and MXI1 targets, which are absent in the 4-cell stage modules. This finding is consistent with the previously characterized activation of Myc-enriched genes upon Sin3a depletion27. Together, these results demonstrate that some intramodular hubs are probably key players in pre-implantation development.
In summary, we have demonstrated that single-cell RNA sequencing has markedly improved transcriptome quantification of rare human pre-implantation embryo samples at both individual transcript and system levels. The findings extend our knowledge of the transcriptional architecture, sequential order of gene activation, and genetic programming for early human embryogenesis. Our cross-species systems analysis demonstrates that the human pre-implantation transcriptional organization is highly preserved, highlighting an evolutionarily conserved molecular process including key genes that drive mammalian pre-implantation development. It remains to be resolved whether the differences in a portion of co-expressed modules can account for species-specific function, such as seen previously in systems comparisons of human and mouse brain28. Furthermore, we expect that single-cell RNA-seq can also quantitatively delineate the structures, isoforms, and allele-specific expression patterns of both coding genes and non-coding regulatory RNAs8,13,25. Moreover, compared to exome or genomic sequencing, RNA-seq analysis has the advantage of quantitatively revealing expression defects that are due to genetic and/or epigenetic alternations in gene regulatory domains that are beyond the detection of DNA sequencing, such as in the case of imprinting disorders. Thus, single-cell RNA-seq of a single blastomere could be a promising approach for pre-implantation genetic diagnosis in the near future.
Methods
Ethics statement
Oocytes were obtained from female patients (between ages 26 and 35) at the Center for Clinical Reproductive Medicine at the Jiangsu People's Hospital, Nanjing, China, with written informed consent and institutional approval. Sperm was obtained from an anonymous healthy donor at an in vitro fertilization sperm bank in Changsha, China, with informed consent. This study was approved by the Institutional Review Board (IRB) on Human Subject Research and Ethics Committee in the First Affiliated Hospital to Nanjing Medical University, China. None of the donors received any financial re-imbursement. The identity of the subjects was anonymous to all of the scientists in this project from the beginning. Investigators at UCLA were only involved in data analysis and manuscript writing. According to the UCLA IRB review of their involvement in the study, UCLA investigators were granted exemption approval (IRB no. 12-001361).
Human embryo collection and culture
Vitrified mature oocytes were warmed with a thawing kit before RNA isolation (Jieying Laboratory). All thawing steps were performed at room temperature (23–25 °C). Oocyte survival was evaluated based on integrity of the oocyte membrane and the zona pellucida. Fertilization is achieved through intracytoplasmic sperm injection (ICSI) to synchronize the development time for each batch of viable oocytes for this study. Embryos were cultured in Cleavage Medium (SAGE) in a low-oxygen humidified atmosphere containing 5% CO2, 5% O2 and 90% N2 at 37 °C. Stages of human embryo development were assessed through microscopy and collected separately.
Mice embryos collection
For timed pregnancy, PMSG (7-10UI) was intraper-itoneally injected into C57BL/6 female mice aged 4 to 6 weeks. Next, hCG (7-10UI) was intraperitoneally injected after 46 to 48h. Pregnant mice were euthanized at various time points to obtain embryos as follows: metaphase II oocyte (12 h), zygote (24 to 26 h), 2-cell (30 to 32 h), 4-cell embryo (day 2), 8-cell embryo (day 3), and morula (day 4). Animal experiments were reviewed and approved by the Animal Experiment Administration Committee of Tongji University School of Medicine, China.
RNA isolation and library construction
Both human and mouse blastomeres were prepared using identical protocols. Single blastomeres were isolated by removing the zona pellucida using acidic tyrode solution (Sigma, catalogue no. T1788), then separated by gentle mouth pipetting in a calcium-free medium. Single cells were washed twice with 1× PBS containing 0.1% BSA before placing in lysis buffer. RNA was isolated from single cells or single morula embryos and amplified as described previously14. Library construction was performed following Illumina manufacturer suggestions. Libraries were sequenced on the Illumina Hiseq2000 platform and sequencing reads that contained polyA, low quality, and adapters were pre-filtered before mapping. Filtered reads were mapped to the hg19 genome and mm9 genome using default parameters from BWA aligner29, and reads that failed to map to the genome were re-mapped to their respective mRNA sequences to capture reads that span exons.
Transcriptional profiling
In both human and mouse cases, data normalization was performed by transforming uniquely mapped transcript reads to RPKM30. Genes with low expression in all stages (average RPKM < 0.5) were filtered out, followed by quantile normalization. For differential expression, we compared every time point to its previous time point using default parameters in DESeq using normalized read counts. Genes were called differentially expressed if they exhibited a Benjamini and Hochberg–adjusted P value (FDR) < 5% and a mean fold change of > 2.
Paternal exon sequencing
The paternal genomic DNA was extracted from 10 ml peripheral blood of the sperm donor (serial number D5422) using QIAamp DNA Blood Mini Kit. Then 1ug of the genomic DNA was fragmented through sonication. Exome enrichment was performed using the Agilent SureSelect Human All Exon Kit (50 Mb). Sequencing was performed on the Illumina Hiseq2000 platform according to the manufacturer's instructions and reads were mapped to the hg19 using default parameters for the BWA aligner29.
SNP analysis
SNP calling was performed using the GATK software using default parameters16. The SNP database we used for reference was from the 1000 Genomes Project which contains SNP variants from the Chinese population (CHB). Only SNP sites with coverage of 10 reads or more were considered. SNP annotation was performed used Annovar31 and SNP variant effect on protein functional change was predicted using SIFT23. Only predictions with high confidence (SIFT score <0.05) were considered.
Paternal and maternal gene calling
We performed paternal and maternal gene calling in two ways. In the first, we deduced the parental haplotype and used linkage information to determine the parent-of-origin. We used heterozygous SNPs from paternal genotype (first allele ≥ 5 reads, second allele ≥ 3reads, both quality values >20) and identified at least two embryos which contain paternal SNPs (both alleles ≥ 5 reads, both quality values >20). We asked whether the linkage between two adjacent loci are supported by correct links in at least two embryos, and whether the correct links are greater than the wrong links at each loci (for example, the number of correct links >3 × wrong links). These results are in Supplementary Table 2.
In the second approach, we used homozygous SNPs from the paternal genotype so we could trace exactly which allele the embryo must carry. If embryos are heterozygous at this site, we infer that the alternative allele is maternally derived. In addition, if there is absence of the paternal allele and expression of an alternative allele, these transcripts are also maternally derived. We have provided a schematic for these scenarios in Fig. 2a.
Weighted gene co-expression network analysis
Both human and mouse data sets were independently constructed using the following method. A signed weighted correlation network was constructed by first creating a matrix of pairwise correlations between all pairs of genes across the measured samples6. Next, the adjacency matrix was constructed by raising the co-expression measure, 0.5 + 0.5 × correlation matrix, to the power β = 12. The power of 12, which is the default value, is interpreted as a soft-threshold of the correlation matrix. Based on the resulting adjacency matrix, we calculated the topological overlap, which is a robust and biologically meaningful measure of network interconnectedness32 (that is, the strength of two genes' co-expression relationship with respect to all other genes in the network). Genes with highly similar co-expression relationships were grouped together by performing average linkage hierarchical clustering on the topological overlap. We used the Dynamic Hybrid Tree Cut algorithm33 to cut the hierarchal clustering tree, and defined modules as branches from the tree cutting. We summarized the expression profile of each module by representing it as the first principal component (referred to as module eigengene). Modules whose eigengenes were highly correlated (correlation above 0.7) were merged.
Module preservation statistics
To evaluate the human modules in mouse developmental data24, we mapped human genes to mouse genes (orthologous genes) as annotated from the Mouse Genome Informatics (MGI) database34. An advantage of WGCNA is that it provides powerful module preservation statistics which assess whether the density (how tight interconnections among genes in a module are) and connectivity patterns of individual modules (for example, intramodular hub gene status) are preserved between two data sets. To assess the preservation our human modules (reference network) in the test network (either human or mouse), we used the R function ‘modulePreservation’ in the WGCNA R package, as this quantitative measure of module preservation enables rigorous argument that a module is not preserved24. By averaging the several preservation statistics generated through many permutations of the original data, a Zsummary value is calculated, which summarizes the evidence that a module is preserved and indicative of module robustness and reproducibility. In general, modules with Zsummary scores > 10 are interpreted as strongly preservation (that is, densely connected, distinct, and reproducible modules), Zsummary scores between 2 and 10 are weak to moderately preserved, and Zsummary scores <2 are not preserved.
Identification and visualization of hub genes
Module eigengenes lead to a natural measure of module membership (also known as module eigengene based connectivity kME). Specifically, an approximate measure of module membership for gene i with respect to module q is defined as follows MMq(i) = cor(x(i), Eq), where x(i) is the expression profile of gene i and Eq is the eigengene of module q. As we use signed networks here, we expect that module genes have significant positive module membership values. The advantage of using a correlation to quantify module membership is that this measure is naturally scaled to lie in the interval [−1, 1] and a corresponding statistical significance measure (P value) can be easily computed. Genes with highest module membership values are referred to as intramodular hub genes (for example, kME > 0.9, P < 10−22). Intramodular hub genes are centrally located inside the module and represent the expression profiles of the entire module35. We used VisANT36 to visualize the top 150 gene connections (based on topological overlap) among the top 100 hub genes (that is, genes with the highest kME).
Hub gene validation
We used WGCNA to independently construct a network in published data sets, and generated an independent list of hub genes (kME > 0.9) for each data set. For human–human comparisons and mouse–mouse comparisons, we determined the overlap of hub genes from the same developmental stage. However, for human–mouse module overlaps, we used all modules with significant gene overlap (P < 10−4, see Fig. 4b) to compute intramodular hub gene overlap. For example, we overlap hub genes found in the human 4-cell module to both mouse oocyte and 1-cell networks since both these networks have significant overlaps with the 4-cell module.
Enrichment of upstream regulators
The Ingenuity Upstream Regulator Analytic was used to determine enrichment of upstream regulators for all human stage-specific modules. Regulators that were not transcriptional regulators were not considered.
Gene ontology analysis
Functional annotation was performed with the Database for Annotation, Visualization and Integrated Discovery (DAVID) Bioinformatics Resource37. Gene ontology terms shown in this study summarized all similar sub-terms into an overarching term, and Benjamani-Hochberg adjusted P values are shown for the representative term.
Supplementary Material
Acknowledgments
We thank many of our colleagues for invaluable discussions and comments on our study, in particular, P. Pajukanta at UCLA for very helpful suggestions on SNP analyses and D. Geschwind for critically reading the manuscript. This work was supported by 973 Grant Programs 2012CB966300, 2011CB966204 and 2011CB965102 from the Ministry of Science and Technology in China; the International Science and Technology Cooperation Program of China (no. 2011DFB30010); and the National Natural Science Foundation of China (81271258).
Footnotes
Author Information: Reprints and permissions information is available at www.nature.com/reprints.
Supplementary Information is available in the online version of the paper.
Author Contributions: Z.X., K.H., J.L. and G.F. designed the study. Z.X., L.C., C.J., Y.F., Z.L., Q.Z., L.C. and Y.E.S. carried out experiments or contributed critical reagents and protocols. K.H., C.C. and S.H. analysed the data and performed statistical analyses. K.H. and G.F. wrote the manuscript in discussion with all the authors. All the authors read and approved the manuscript.
The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper.
All sequencing data generated for this work have been deposited in the NCBI Gene Expression Omnibus (GEO) under accession number GSE44183.
References
- 1.Niakan KK, Han J, Pedersen RA, Simon C, Pera RA. Human pre-implantation embryo development. Development. 2012;139:829–841. doi: 10.1242/dev.060426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hamatani T, Carter MG, Sharov AA, Ko MS. Dynamics of global gene expression changes during mouse preimplantation development. Dev Cell. 2004;6:117–131. doi: 10.1016/s1534-5807(03)00373-3. [DOI] [PubMed] [Google Scholar]
- 3.Wang QT, et al. A genome-wide study of gene activity reveals developmental signaling pathways in the preimplantation mouse embryo. Dev Cell. 2004;6:133–144. doi: 10.1016/s1534-5807(03)00404-0. [DOI] [PubMed] [Google Scholar]
- 4.Zeng F, Baldwin DA, Schultz RM. Transcript profiling during preimplantation mouse development. Dev Biol. 2004;272:483–496. doi: 10.1016/j.ydbio.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 5.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005 doi: 10.2202/1544-6115.1128. http://dx.doi.org/10.2202/1544-6115.1128. [DOI] [PubMed]
- 7.Walser CB, Lipshitz HD. Transcript clearance during the maternal-to-zygotic transition. Curr Opin Genet Dev. 2011;21:431–443. doi: 10.1016/j.gde.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 8.Schier AF. The maternal-zygotic transition: death and birth of RNAs. Science. 2007;316:406–407. doi: 10.1126/science.1140693. [DOI] [PubMed] [Google Scholar]
- 9.Schultz RM. The molecular foundations of the maternal to zygotic transition in the preimplantation embryo. Hum Reprod Update. 2002;8:323–331. doi: 10.1093/humupd/8.4.323. [DOI] [PubMed] [Google Scholar]
- 10.Vassena R, et al. Waves of early transcriptional activation and pluripotency program initiation during human preimplantation development. Development. 2011;138:3699–3709. doi: 10.1242/dev.064741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie D, et al. Rewirable gene regulatory networks in the preimplantation embryonic development of three mammalian species. Genome Res. 2010;20:804–815. doi: 10.1101/gr.100594.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang P, et al. Transcriptome profiling of human pre-implantation development. PLoS ONE. 2009;4:e7844. doi: 10.1371/journal.pone.0007844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dobson AT, et al. The unique transcriptome through day 3 of human preimplantation development. Hum Mol Genet. 2004;13:1461–1470. doi: 10.1093/hmg/ddh157. [DOI] [PubMed] [Google Scholar]
- 14.Tang F, et al. RNA-Seq analysis to capture the transcriptome landscape of a single cell. Nature Protocols. 2010;5:516–535. doi: 10.1038/nprot.2009.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Islam S, et al. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res. 2011;21:1160–1167. doi: 10.1101/gr.110882.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramskold D, et al. Full-length mRNA-seq from single-cell levels of RNA and individual circulating tumor cells. Nature Biotech. 2012;30:777–782. doi: 10.1038/nbt.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hashimshony T, Wagner F, Sher N, Yanai I. CEL-Seq: single-cell RNA-seq by multiplexed linear amplification. Cell Rep. 2012;2:666–673. doi: 10.1016/j.celrep.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 18.The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nature Genet. 2010;42:969–972. doi: 10.1038/ng.680. [DOI] [PubMed] [Google Scholar]
- 21.Gabriel SB, et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 22.Altshuler DM, et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature Protocols. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 24.Langfelder P, Luo R, Oldham MC, Horvath S. Is my network module preserved and reproducible? PLOS Comput Biol. 2011;7:e1001057. doi: 10.1371/journal.pcbi.1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang F, et al. Deterministic and stochastic allele specific gene expression in single mouse blastomeres. PLoS ONE. 2011;6:e21208. doi: 10.1371/journal.pone.0021208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu J, et al. Novel importin-a family member Kpna7 is required for normal fertility and fecundity in the mouse. J Biol Chem. 2010;285:33113–33122. doi: 10.1074/jbc.M110.117044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dannenberg JH, et al. mSin3A corepressor regulates diverse transcriptional networks governing normal and neoplastic growth and survival. Genes Dev. 2005;19:1581–1595. doi: 10.1101/gad.1286905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller JA, Horvath S, Geschwind DH. Divergence of human and mouse brain transcriptome highlights Alzheimer disease pathways. Proc Natl Acad Sci USA. 2010;107:12698–12703. doi: 10.1073/pnas.0914257107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 31.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yip AM, Horvath S. Gene network interconnectedness and the generalized topological overlap measure. BMC Bioinformatics. 2007;8:22. doi: 10.1186/1471-2105-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Langfelder P, Zhang B, Horvath S. Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics. 2008;24:719–720. doi: 10.1093/bioinformatics/btm563. [DOI] [PubMed] [Google Scholar]
- 34.Eppig JT, Blake JA, Bult CJ, Kadin JA, Richardson JE. The Mouse Genome Database (MGD): comprehensive resource for genetics and genomics of the laboratory mouse. Nucleic Acids Res. 2012;40:D881–D886. doi: 10.1093/nar/gkr974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horvath S, Dong J. Geometric interpretation of gene coexpression network analysis. PLOS Comput Biol. 2008;4:e1000117. doi: 10.1371/journal.pcbi.1000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu Z, Mellor J, Wu J, DeLisi C. VisANT: an online visualization and analysis tool for biological interaction data. BMC Bioinformatics. 2004;5:17. doi: 10.1186/1471-2105-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources Nature Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.