

Abstract
Due to their electron-rich aromatic structure, nucleophilic (radio)fluorination of pyridines is challenging, especially at the meta position. In this paper, we describe the first example of direct fluorination of a pyridine N-oxide to produce a metafluorinated pyridine. Specifically, fluorination of 3-bromo-4-nitropyridine N-oxide produced in several minutes 3-fluoro-4-nitropyridine N-oxide in moderate yield at room temperature. This intermediate compound was later converted to 3-fluoro-4-aminopyridine easily by catalytic hydrogenation. Furthermore, this approach was succesfully applied for labeling with fluorine-18. The use of pyridine N-oxides for the preparation of fluoropyridines is unprecedented in the chemical literature and has the potential to offer a new way for the synthesis of these important structures in pharmaceuticals and radiopharmaceuticals.
Graphical Abstract
Fluorination and radiofluorination of pyridines is challenging, especially in meta position. Here we describe a new chemical reaction to generate meta fluorinated pyridines: halogen substitution on pyridine N-oxides followed by reduction.
Few methods exist to produce 3- or 5-fluoro substituted pyridines compatible with the constraints of 18F radiochemistry, which include the use of fluoride ions as fluorine source and short reaction times1. Methods that generally work well for making 2- and 4-fluoropyridines such as substitution of halides, nitro, trimethyl ammonium salts2 or sulfonium salts3, work in very low yield for the 3- and 5-positions. These reactions only work in cases of pyridines containing secondary strong electron withdrawing groups such as nitrile or carboxamide4–6. Pyridines that do not contain secondary electron withdrawing groups are only amenable to fluorination through recent methods such as the use of iodonium salts7, 8 and iodonium ylides9. However, these methods often require precursors that are difficult to prepare. Given the limited number of reactions yielding 3- and 5-fluoropyridines, there is a great interest to develop new methods that are fast, react in high yield and use readily available precursors. For the current project, we were faced with the challenge of developing a labeling method for [18F]3-fluoro-4-aminopyridine (10). 3-fluoro-4-aminopyridine is a fluorine-containing derivative of 4-aminopyridine, a clinically approved drug for multiple sclerosis that binds to potassium channels in the central nervous system, currently under investigation for imaging demyelination10–12. In this report, we describe how we synthesized this compound and discuss our findings on the different reactivity of 3-bromo-4-nitropyridine and 3-bromo-4-nitropyridine N-oxide towards fluorination.
For the synthesis of [18F]3-fluoro-4-aminopyridine we considered 3 possible routes from commercially available reagents (Scheme 1):
Scheme 1. Possible fluorination strategies for 3-fluoro-4-aminopyridine.

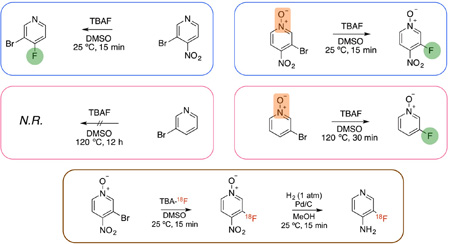
A. Nucleophilic aromatic substitution of 3-bromo-4-(boc-amino)pyridine (1) followed by acid deprotection. Fluorination did not proceed under the experimental conditions (A.1: 1 eq TBAF, 125 C,DMSO, 16 h). B. Nucleophilic aromatic substitution of 3-bromo-4-nitropyridine (3) or 3-iodo-4-nitropyridine (4) followed by reduction. Under the experimental conditions (B.1: 0.5 eq. TBAF, 25 C, DMSO, 15 min) the product formed was 3-bromo-4-fluoropyridine (6) or 3-iodo-4-fluoropyridine (7) instead of the desired 3-fluoro-4-nitropyridine (5). C. Nucleophilic aromatic substitution of 3-bromo-4-nitropyridine N-oxide (8) followed by reduction. Fluorination produced 3-fluoro-4-nitropyridine N-oxide (9) in 37% yield (C.1: 0.5 eq. TBAF, 25 C, DMSO, 5 min) and hydrogenation of 9 (3 mg of 10% Pd/C, 1 atm H2, MeOH, 25 C, 10 min) produced 3-fluoro-4-aminopyridine (10) quantitatively.
The first option consists of fluorination of Boc-protected 3-bromo-4-aminopyridine (1) followed by acid deprotection (Scheme 1A). Unfortunately, treatment of 1 with tetrabutylammonium fluoride (TBAF) did not produce the desired product even after several hours at high temperature.
We then considered fluorination of 3-bromo-4-nitropyridine (3) followed by reduction of the nitro group (Scheme 1B). Treatment of 3 with 0.5 equivalents of TBAF at room temperature produced the para-substituted product 3-bromo-4-fluoropyridine (6) in 71.1 ± 3.6% yield (relative to TBAF, n = 4) as determined by HPLC and NMR (Sup. Figs. 1A (HPLC) and 1B (NMR)). Under these conditions, less than 0.2% of 3-fluoro-4-nitropyridine (5) was produced demonstrating a clear preference for the substitution to occur at the para- or nitro-position. Similar results were obtained with 3-iodo-4-nitropyridine (4) (Sup. Fig. 2A and 2B). This type of reactivity is expected from previously published data on nitro-substituted pyridines2, 13 and consistent with the results from Abrahim et al5 who tested fluorination of a pyridine containing a nitro group in ortho and a bromo in meta and found exclusive substitution at the nitro/ortho position.
Finally, we decided to try fluorination of 3-bromo-4-nitropyridine N-oxide (8) hoping that the N-oxide would further increase the reactivity of the pyridine resulting in fluorination at the meta-position (Scheme 1C). In this case, treatment of 8 with 0.5 equivalents TBAF at room temperature produced the meta-fluorinated compound, 3-fluoro-4-nitropyridine N-oxide (9), as the main product in 20.7 ± 2.7% yield (relative to TBAF, n = 4) (Sup. Fig. 3A and 3B). Under these conditions, we detected less than 2% fluorination at the para position indicating that the presence of the N-oxide group favors meta fluorination.
To peek into the potential use of pyridine N-oxides as fluorination precursors, we compared the reactivity of 3-bromopyridine (11) and 3-bromopyridine N-oxide (13) with TBAF (Scheme 2). Not surprisingly, 3-bromopyridine did not undergo reaction even after treatment with 1.2 eq. of TBAF for 12h at 120 °C (Sup. Figs. 4A and 4B). In contrast, over 25% conversion of 3-bromopyridine N-oxide (13) to 3-fluoropyridine N-oxide (14) was detected after 30 min at 120 °C (Sup. Figs. 5A and 5B). This result indicates that even though the nitro group in para position contributes to the enhanced reactivity of the pyridine (likely by lowering its activation energy) it is not sufficient to explain the reactivity and regioselectivity of the N-oxide towards fluorination. This regioselectivity is particularly surprising given an early study on the nucleophilic displacement of monosubstituted pyridine N-oxides by sodium ethoxide in ethanol which found that the rate of displacement of nitro was 1,100–3,100 larger than bromo and that the order of reactivity was 2>4>>3 position14. This difference may be due to the different nature of the nucleophile and/or solvation effects and constitutes a research question of its own.
Scheme 2.

Fluorination of: A. 3-bromopyridine and B. 3-bromo-pyridine N-oxide.
After obtaining compound 9, this was then reduced using standard hydrogenation conditions to generate the desired final product, 3-fluoro-4-aminopyridine (10) quantitatively (Sup. Fig. 6). To our knowledge, pyridine N-oxides have not previously been used as precursors for radiofluorination and with the success of non-radioactive fluorination, we decided to test this approach for the production of [18F]3-fluoro-4-aminopyridine.
Radiochemical synthesis of [18F]3-fluoro-4-aminopyridine was conducted as shown in Scheme 3. The procedure was similar to the non-radioactive synthesis described above except that [18F]TBAF was prepared immediately before the reaction by trapping [18F]fluoride in a strong anion exchange cartridge and eluting it with TBA-HCO3. The reaction was carried out in DMSO at room temperature. After 15 min, HPLC analysis of the reaction crude showed an early peak that elutes with the solvent front corresponding to unreacted [18F]TBAF and a main peak corresponding to the desired product (Sup. Fig. 7). Under the test conditions, the isolated decay-corrected yield for the desired product was 10.4 ± 1.8% (n = 8). Upon further characterization, we noticed that co-injection of the reaction crude with a small amount of reference standard (9) consistently gave higher yields (25 ± 4%, decay corrected, n = 8) (Fig. 1A). This led us to hypothesize that the non-radioactive 3-fluoro-4-nitropyridine N-oxide (9) could be contributing to the radiolabeling yield by 19F/18F exchange. When we tested labeling of 3-fluoro-4-nitropyridine N-oxide with [18F]TBAF in the absence of the bromo precursor we obtained 33.1 ± 5.4% (n = 4) decay-corrected isolated yield (Fig. 1B). Even more remarkable is that this reaction reaches equilibrium within seconds. Fluorine exchange in organic compounds has been reported before15, however, to our knowledge this is the first example of fluoride exchange of a C-F bond in a heterocyclic compound.
Scheme 3.

Radiochemical synthesis of 3-fluoro-4-amino-pyridine.
Figure 1. Radioactive and UV HPLC traces.
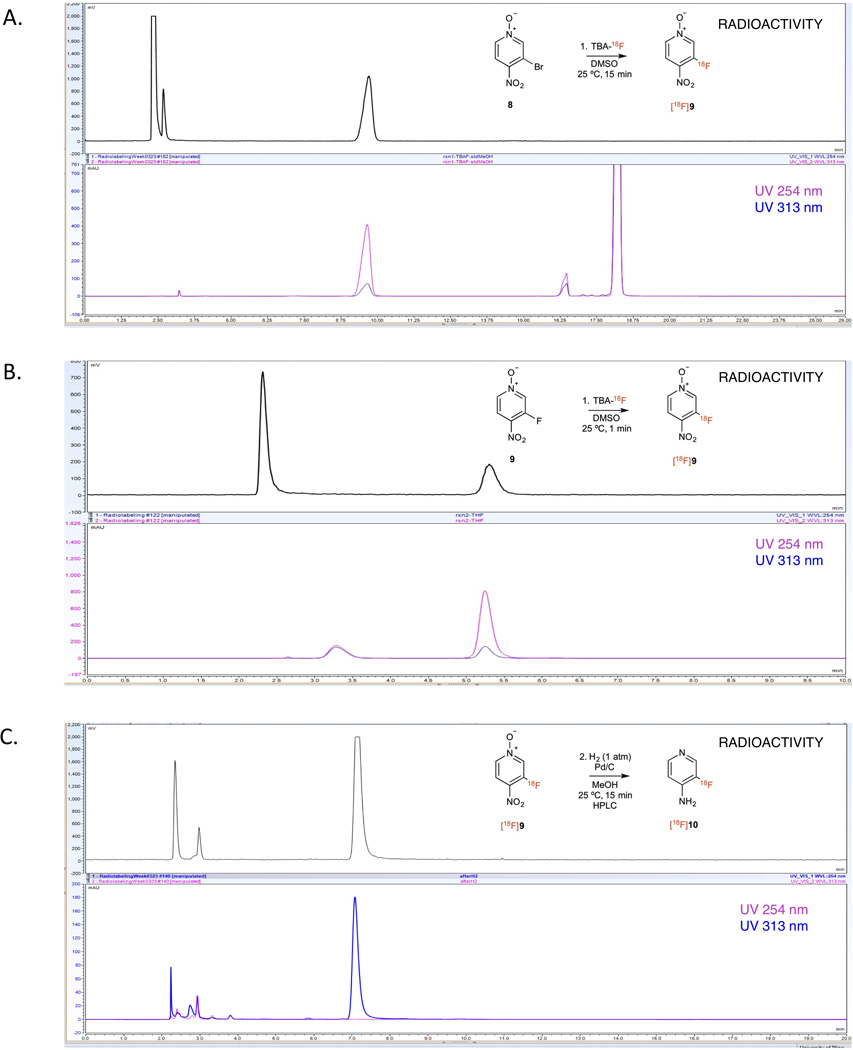
A. Coinjection of crude from reaction 1 and reference standard. Unreacted [18F]TBAF, Reaction product (9) and precursor (8) elute at 2.3, 9.65 and 18.25 min, respectively. B. Injection of reaction crude of 19F/18F reaction, unreacted [18F]TBAF and product elute at 2.3 and 5.3 min, respectively. C. Coinjection of crude from reaction 2 and reference standard. Reaction product (10) elutes at 7.05 min, starting material (9) (minor peak) elutes at 5.80 min.
After obtaining the labeled intermediate ([18F]9), this product was hydrogenated using the same conditions as the non-radioactive reactant. The final tracer was purified by HPLC (Fig. 1C) and the isolated yield was 55 ± 11% (corrected for decay, n = 8). Product identity was confirmed by coinjection of the final radiotracer with the cold standard into an analytical C-18 HPLC. A single peak was detected by radioTLC indicating high radiochemical purity. The total synthesis time including all purification steps was less than two hours and the final overall yield was around 2.5% (5.4% corrected for decay) with around 65% of the activity lost during the multiple purification and transfer steps. Automation of this procedure using a radiochemistry module and the use of solid phase extraction cartridges in place of HPLC should reduce losses, shorten the synthesis time and improve the overall yield.
To sumarize, in this project we set out to develop a method for the radiochemical synthesis of [18F]3-fluoro-4-aminopyridine (10). Initially, we considered direct fluorination of Boc-protected 3-bromo-4-aminopyridine (1), however this reaction did not produce the desired product even after stirring several hours at high temperature. We then reasoned that introduction of a nitro group on the pyridine (which could later be reduced to the amine) would make the substrate more reactive towards fluorination. Indeed, when we mixed 3-bromo- or 3-iodo-4-nitropyridine (3 or 4) with TBAF we saw it react cleanly, however the only reaction observed was the exchange of the nitro group at the 4-, or para, position (6, 7, respectively). In an attempt to further increase the reactivity of the substrate and hoping to get partial substitution at the meta position, we tried fluorination of 3-bromo-4-aminopyridine N-oxide (8) (which can also be reduced to generate the final product) and obtained the desired product in 20.7% average yield. Following these experiments, we tested radiofluorination of 3-bromo-4-nitropyridine N-oxide (8) using freshly prepared [18F]TBAF. This reaction produced the desired compound, [18F]3-fluoro-4-nitropyridine N-oxide (9), as the main product in 10.4% average radiochemical yield. In addition, this compound can also be produced by 19F/18F exchange, which occurs faster and in higher yield. As far as we know, this represents the first example of fluoride exchange of a C-F bond in a heterocyclic compound. Producing tracers by fluoride exchange limits the final specific activity as the radioactive product cannot be separated from the cold one, however, there are recent examples in which tracers produced by fluoride exchange achieved excellent specific radioactivities ranging from 1–3 Ci/µmol (37–111 GBq/µmol)16, 17. Finally, we tested fluorination of the corresponding pyridines not containing a nitro group and found that 3-bromopyridine N-oxide can also undergo halogen exchange suggesting a broad scope for this reaction.
Supplementary Material
Acknowledgments
This project was supported by grants NIH/NIBIB 1K99EB020075 to Pedro Brugarolas and NIH/NINDS 1R21NS084382 to Brian Popko. Prof. Brian Popko is gratefully acknowledged for his mentorship and financial support to the project. We thank Dr. D. Murali for providing training and advice in radiolabeling. Prof. Chin-Tu Chen and the Integrated Small Animal Imaging Research Resource at the University of Chicago are acknowledged for generously sharing laboratory space and equipment. Finally, Prof. Sergey Kozmin is acknowledged for providing valuable comments on the manuscript.
Footnotes
Electronic supplementary information (ESI) available: Full experimental methods, 1H, 13C and 19F NMR spectra, and HPLC chromatograms for all reactions. See DOI: 10.1039/c6cc02362b
Author contributions: P.B. conceived, designed, executed and analyzed experiments. R.F. assisted in the design and analysis of radiolabeling experiments. S.-H. Cheng assisted with radiolabeling. O.J. advised on the choice of chemistry and data analysis. P.B. wrote the paper and all authors revised it and approved it.
Notes and references
- 1.Preshlock S, Tredwell M, Gouverneur V. Chemical Reviews. 2016;116:719–766. doi: 10.1021/acs.chemrev.5b00493. [DOI] [PubMed] [Google Scholar]
- 2.Karramkam M, Hinnen F, Vaufrey F, Dollé F. Journal of Labelled Compounds and Radiopharmaceuticals. 2003;46:979–992. [Google Scholar]
- 3.Chun JH, Morse CL, Chin FT, Pike VW. Chem Commun (Camb) 2013;49:2151–2153. doi: 10.1039/c3cc37795d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beer HF, Haeberli M, Ametamey S, Schubiger PA. Journal of Labelled Compounds and Radiopharmaceuticals. 1995;36:933–945. [Google Scholar]
- 5.Abrahim A, Angelberger P, Kletter K, Müller M, Joukhadar C, Erker T, Langer O. Journal of Labelled Compounds and Radiopharmaceuticals. 2006;49:345–356. [Google Scholar]
- 6.Liu H, Liu S, Miao Z, Deng Z, Shen B, Hong X, Cheng Z. Journal of medicinal chemistry. 2013;56:895–901. doi: 10.1021/jm301740k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carroll MA, Nairne J, Woodcraft JL. Journal of Labelled Compounds and Radiopharmaceuticals. 2007;50:452–454. [Google Scholar]
- 8.Chun JH, Pike VW. Chem Commun (Camb) 2012;48:9921–9923. doi: 10.1039/c2cc35005j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rotstein BH, Stephenson NA, Vasdev N, Liang SH. Nature communications. 2014;5:4365. doi: 10.1038/ncomms5365. [DOI] [PubMed] [Google Scholar]
- 10.Brugarolas P, Sanchez-Rodriguez J, Caprariello AV, Lacroix J, Chen C-T, Appelbaum DE, Miller RH, Bezanilla F, Popko B. Abstracts of Papers of the American Chemical Society. 2014;248 [Google Scholar]
- 11.Brugarolas P, Sanchez-Rodriguez J, Lacroix J, Bezanilla F, Chen C-T, Appelbaum D, Popko B. Journal of Nuclear Medicine Meeting Abstracts. 2014;55:1124. [Google Scholar]
- 12.Brugarolas P, Sanchez-Rodriguez J, Lacroix J, Chen C-T, Bezanilla F, Popko B. Journal of Nuclear Medicine Meeting Abstracts. 2015;56:493. [Google Scholar]
- 13.Tredwell M, Gouverneur V. Angew Chem Int Ed Engl. 2012;51:11426–11437. doi: 10.1002/anie.201204687. [DOI] [PubMed] [Google Scholar]
- 14.Johnson RM. Journal of the Chemical Society B. Physical Organic. 1966:1058–1061. [Google Scholar]
- 15.Blom E, Karimi F, Långström B. Journal of Labelled Compounds and Radiopharmaceuticals. 2009;52:504–511. doi: 10.1002/jlcr.3033. [DOI] [PubMed] [Google Scholar]
- 16.Liu Z, Pourghiasian M, Benard F, Pan J, Lin KS, Perrin DM. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2014;55:1499–1505. doi: 10.2967/jnumed.114.137836. [DOI] [PubMed] [Google Scholar]
- 17.Liu Z, Amouroux G, Zhang Z, Pan J, Hundal-Jabal N, Colpo N, Lau J, Perrin DM, Benard F, Lin KS. Mol Pharm. 2015;12:974–982. doi: 10.1021/acs.molpharmaceut.5b00003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.