

Abstract
Giant unilamellar vesicles (GUVs), composed of a phospholipid bilayer, are often used as a model system for cell membranes. However the study of proteo-membrane interactions in this system is limited as the incorporation of integral and lipid-anchored proteins into GUVs remains challenging. Here, we present a simple generic method to incorporate proteins into GUVs. The basic principle is to break proteo-liposomes by an osmotic shock. They subsequently reseal into larger vesicles which, if necessary, can endure the same to obtain even bigger proteo-GUVs. This process does not require specific lipids or reagents, works under physiological conditions with high concentrations of protein, the proteins remains functional after incorporation and the resulting proteo-GUVs can be micromanipulated. Moreover, our protocol is valid for a wide range of protein substrates. We have successfully reconstituted three structurally different proteins, two trans-membrane proteins, TolC and the neuronal t-SNARE, and one lipid-anchored peripheral protein, GABARAP-Like 1 (GL1). In each case, we verified that the protein remains active after incorporation and in their correctly folded state. We also measured their mobility by performing diffusion measurements via fluorescence recovery after photobleaching (FRAP) experiments on micromanipulated single GUVs. The diffusion coefficients are in agreement with previous data.
Graphical abstract
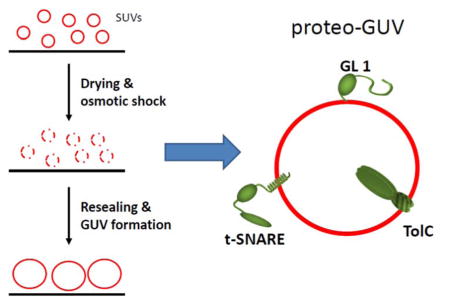
INTRODUCTION
The backbone of cellular membranes is made of a lipid bilayer that hosts and regulates protein machineries transiently or permanently. Protein activities are affected by the membrane properties and the embedded proteins in turn can influence the characteristics of the membrane. These cross-interactions play a huge part in many different biological processes. To study these cross-interactions in vitro, various membrane models were developed to mimic in vivo systems. Common examples are supported bilayers, unilamellar vesicles (50 nm–100 μm) and nanodiscs (reviewed in 1). Giant unilamellar vesicles (GUVs) (3 to 100 μm), in particular, have a prominent role due to their large size, resembling the dimensions of a cell, making them an appealing tool for micromanipulation and microscopy studies.
Several protocols have been developed to form GUVs. The most common method is to grow them from a dry lipid film by electroformation in sucrose or buffer solutions, on platinum wires and indium tin oxide coated glass (ITO glass) 2, 3, 4, 5, but other protocols have been published which grow GUVs for example by swelling in solution6, 7 or from gel films8, 9, by infrared heating10, solvent exchange11, evaporation12 or by the water in oil emulsion technique 13, 14.
In the past decade, it appeared that making proteo-GUVS, i.e. GUVs containing proteins, was a very challenging task. Several protocols have been proposed 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, mostly based on the ones established for protein-free GUVs. All these approaches have been successfully used with specific proteins.
Here we describe a new protocol to form proteo-GUVs that is applicable to three different types of proteins: a lipid anchored protein named gamma-aminobutyric acid receptor-associated protein (GL1), a single transmembrane protein called t-SNARE (target- membrane-located soluble N-ethylmaleimide-sensitive factor attachment protein receptor composed of Syntaxin 1 and SNAP25) and a channel protein named TolC required to form a homotrimer to be able to insert in membranes (Fig. 1).
Figure 1. Schematics of the GL1, t-SNARE and TolC.
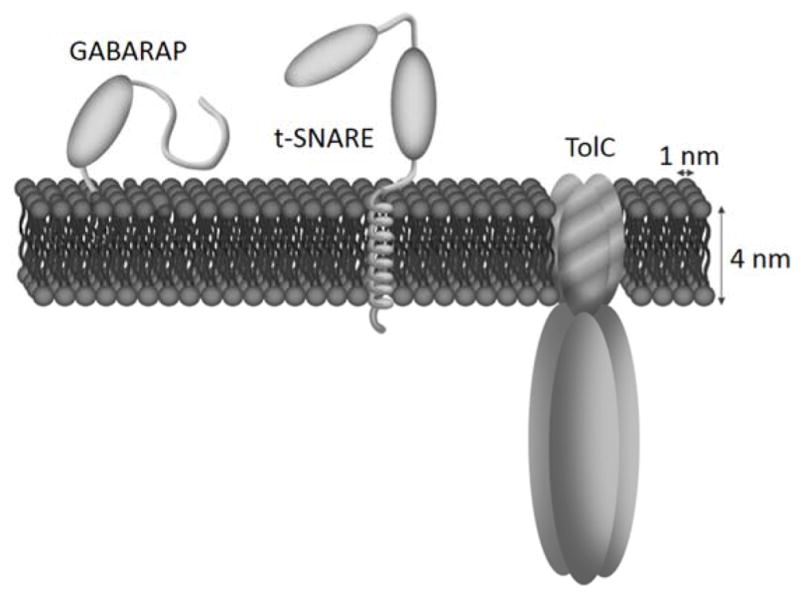
GL1 is a protein anchored to DOPE lipid, the t-SNARE has a single trans-membrane α-helix and TolC is a trimeric barrel establishing a 14 nm long pore including a transmembrane region made of a 4 nm-wide beta-barrel complex. The plain ellipsoids represent a structured part of the protein outer membrane regions (including both Syntaxin1A and SNAP25 helices in the case of t-SNARE).
The general idea of this new method, that we call “osmotic shock method” is based on hydration of dried small proteo-liposomes with pure water resulting supposedly in an osmotic shock that triggers the formation of proteo-GUVs. The unilamellarity of these proteo-GUVs has been checked by fluorescence. The main advantages of the osmotic shock method are that the density of the proteins is well controlled (up to a few tenths of percent), the functionality of the protein is conserved, the production costs are low and the final proteo-GUVs can be prepared in any saline buffer at physiological osmolarity. The resulting GUVs can easily be separated and micromanipulated. The diffusion coefficients of the reconstituted proteins obtained by fluorescence recovery after photobleaching experiments gave consistent results with previous data.
Experimental section
Materials
Phospholipids: dioleoyl-phosphatidylcholine (DOPC), dioleoyl-Phosphatidylserine (DOPS), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (POPS, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), cholesterol, dioleoyl-phosphoethanolamine-nitro-benzoxadiazolyl (NBD-PE), dioleoyl-glycero-phosphoethanolamine-lissamine rhodamine B sulfonyl (LR-PE) were purchased from Avanti polar lipids. ATTO647-dioleoyl-phosphoethanolamine (DOPE-ATTO647), ATTO488-dioleoyl-phosphoethanolamine (DOPE-ATTO488) and the dye ATTO488 maleimide (ATTO488) were purchased from ATTO-Tec. Alexa Fluor488 C5-maleimide (Alexa488) and Alexa Fluor® 647 C2 Maleimide were purchased from Life Technologies. Trizma hydrochloride (TrisHCl), sodium chloride, glycerol, Octyl β-D-glucopyranoside and TCEP were purchased from Sigma-Aldrich. 1,4-Dithiothreitol (DTT) was purchased from ROCHE. Dialysis cassettes were purchased from Thermo Fisher. Petri-dishes with a glass bottom (Mattek) were used for the experiments. PreScission protease, a HiLoad Superdex 75 column and PD MidiTrap G-25 columns were bought at GE Healthcare.
Proteins expression and purification
GL1-Like-1 (hereafter referred to simply as GL1) is a lipid-anchored protein. In order to facilitate our GUV experiments, we have mutated GL1 to include an amino terminal cysteine (for dye-labeling described below) and we have truncated GL1 at the COOH terminus such that the reactive glycine (G116) is fully exposed and ready for interaction with Atg7 and Atg3. Atg3 and Atg7 are two autophagic enzymes involved in the ubiquitin-like reaction that covalently bind GL1 to the membrane. GL1, Atg3, and Atg7 were then expressed and purified as previously described27.
The transmembrane t-SNARE complex, which includes full length, wildtype mouse His6-SNAP25 and rat Syntaxin1A, was produced by expression of the polycistronic plasmid pTW34 in the BL-21 gold (DE3) Escherichia coli bacterial strain, and purified as described before.28, 29
The transmembrane t-SNARE S193C with a single Cysteine residue was mutated using plasmid pTW34 as template. All the native Cysteine residues on Syntaxin1A and SNAP25 were mutated to Serine, and the Serine of residue 193 on Syntaxin1A was mutated to Cysteine, using the QuickChange site directed mutagenesis kit from Stratagene. The resultant plasmid was expressed in the BL-21 gold (DE3) Escherichia coli bacterial strain, and purified as described before.29, 30
The plasmid for the cytosolic domain of VAMP2 (CDV S28C) was produced by cloning residues 1 to 96 of VAMP2 into a pCDFDuet-1 vector containing GST-PreScission-CDV 29, 31, followed by a single site-specific point mutation at residue 28 (S28C) using the QuickChange site directed mutagenesis kit from Stratagene. Similar to previous report,30, 32 this plasmid was expressed in BL21 gold (DE3) Escherichia coli bacterial strain and purified with a buffer containing 25 mM Tris (pH 7.4), 400 mM KCl, 10% Glycerol (w/v), 1 mM TCEP. The GST tag was cleaved by incubating the protein (attached to glutathione beads) with PreScission protease overnight at 4°C. The protein was eluted, and then purified by size exclusion chromatography on a HiLoad Superdex 75 (16/60, GE Healthcare) column.
The TolC protein was expressed in E. coli strain C43 (DE3) and purified in β-OG following a protocol previously described and valid for most outer membrane factors of tripartite bacterial efflux pumps 33.
Proteins labeling
GL1 was labeled with Alexa Fluor488 C5-maleimide through the amino terminal cysteine. 100 μM of GL1 protein were mixed with 600 μM TCEP. After 5 min incubation at room temperature, 800 μM of the fluorescent dye dissolved in DMSO was added. The mixture was protected from the light and slowly checked at room temperature for 2 hours or overnight at 4C. The labeled GL1 was then dialyzed (100 mM NaCl, 50 mM Trizma hydrochloride, 1 mM DTT) overnight at 4°C to remove the excess of free dye in order to reduce background fluorescence.
The transmembrane t-SNARE S193C and soluble CDV S28C were labeled with Alexa Fluor® 488 C5 Maleimide or Alexa Fluor® 647 C2 Maleimide (Life Technologies), similarly to previous report 29. The protein was first reduced by incubating with 4 mM TCEP for 30 minutes at 4°C with gentle rotation, and centrifuged at 14,000 rpm for 20 minutes at 4°C to remove any precipitation. Fluorescence dye was added into the protein solution at dye:protein = 3:1 molar ratio and the mixture were incubated for 1 to 2 hours at room temperature with gentle rotation. Unreacted dye was removed by passing through the PD MidiTrap G-25 column (GE Healthcare) three times to reduce background fluorescence.
TolC was labeled with Alexa488 through its primary amine sites. The protein was diluted at a 2 mg/mL concentration in a sodium phosphate buffer (20 mM NaPi pH 8, 150 mM NaCl, 0.9% (w/v) β-OG). We added sodium bicarbonate at a concentration of 1M in a 1:10 (v/v) ratio in order to raise the pH and improve the labeling reaction. We added the dye (stocked at −20°C dissolved in DMSO at 1 mg/mL) in a ratio of 30:1 (mol/mol) dye per protein. The solution was mixed at room temperature during 2.5 hours. The resulting labeled protein was eluted in a gel-filtration column (Econo-Pac® 10DG Columns, Bio-Rad) to remove excess non-bound fluorophores in order to reduce background fluorescence. The labeling ratio was verified by spectrometry and was between 0.7 and 1 dye per protein. Labeled proteins were then dialyzed (150 mM Tris pH 8 300 mM NaCl, 0.9% (w/v) β-OG).
Formation of small liposomes
In case of the protein-free small liposomes the phospholipid mixtures (POPC/POPS/DOPE-Rhodamine 89:10:1 mol%) and the composition of the GUVs with two liquid phases (bottom center in 5) DOPC 40 mol%, DPPC 40 mol% and cholesterol 20 mol%., were initially mixed in glass tubes and dried with nitrogen gas and subsequently under vacuum. The dried lipid film was resuspended in buffer 100 mM NaCl) to a final concentration of 3 mM lipid by vortexing for 15 min to form small liposomes.
The strategy for the formation of small proteo-liposomes depends upon the type of interaction of the protein with the membrane.
The membrane associated protein GL1 is anchored to liposomes via the lipidation reaction described before 27. Briefly, the phospholipid mixture (30 % DOPE, 69.5 % DOPC, 0.5 % DOPE-ATTO647 in mol%) was mixed in a glass tube, dried with nitrogen gas, and subsequently dried under vacuum. Here, DOPE is necessary to for the lipidation process of the protein. The dried lipid film was then resuspended in buffer (100 mM NaCl, 50 mM TrisHCl, pH7.6) to a final concentration of 10 mM lipid and vortexed for 15 min. To anchor the GL1 protein on the lipid, we mixed liposomes (3 mM lipid), 10 μM GL1, 1 mM DTT, 10 μM Atg3, 10 μM Atg7, 1 mM ATP and incubated the mixture for 90 min at 37°C. Residual glycerol was removed from the proteo-liposomes mixture by dialysis through 7 KDa pore membrane overnight at 4°C.
The reconstitution of the t-SNAREs and the TolC, which both are embedded in the membrane via their helical transmembrane domain, into liposomes was basically done as described in 34 with slight modifications. Briefly, lipids (DOPC/DOPS/Atto647-DOPE, 91.2: 8: 0.8 mol%) were dried under nitrogen/ vacuum and then rehydrated with purification buffer containing 0.9 to 1 % octylglucoside and the respective protein. The inclusion of DOPS is a common addition when reconstituting t-SNAREs. After 20 min shaking, rapid dilution was done by addition of reconstitution buffer (t-SNARE: 20 mM Tris, 150 mM NaCl, 1mM DTT, pH 7.4, TolC: 150 mM Tris, 300 mM NaCl, pH 8.0) to dilute the detergent concentration below the critical micellar concentration and mixed for another 20 min. The resulting proteo-liposomes (final lipid concentration: 3 mM) were dialyzed overnight in reconstitution buffer to remove the detergent.
0.2 mM sucrose of lipid were added to the proteo-liposomes to protect the protein during the GUV formation process 17.
GUV formation
Osmotic shock hydration
The coverslip glass was cleaned with isopropanol and a dust free tissue and dried with pressurized nitrogen. A 2 μl drop of small liposomes solution was placed on the coverslip and dried at room temperature under atmospheric pressure. Then, it was rehydrated with 6 μl deionized water, and then dried again under the same conditions. In the next step the lipid film is rehydrated once more with a larger deionized water volume of 10–20 μl and observed under the microscope. GUVs grew instantly from the rehydrated lipid films mainly in the areas with high concentration of lipids (Fig. 2). The two sequential drying-rehydration cycles favor the formation of larger and more uniform GUVs.
Figure 2. Protein-free GUVs preparation from small liposomes.

A 2 μl drop of the 3 mM lipid small liposomes solution in buffer is deposited on a clean coverslip (1) and left to dry at room temperature on a lab bench. After drying, the small liposomes tend to form aggregates (2). A 6 μl water drop is placed on top of the dry sample triggering the formation of small aggregated GUVs by osmotic shock (3). The sample is left to dry a second time (4) before being rehydrated with 10–20 μl of water (5). Larger and a higher number of GUVs are formed. If required the drying/rehydration cycle can be repeated to improve the number and size of the GUVs. The composition of the GUVs with one liquid phases (top and bottom left in 5) was POPC 89 mol%, POPS 10 mol%, DOPE-Rohamine 1 mol%, and the composition of the GUVs with two liquid phases (bottom center in 5) DOPC 40 mol%, DPPC 40 mol% and cholesterol 20 mol%. The images on top in 2, 3, 4 and top left 5 were recorded using transmission microscopy, the images on the bottom in 2, 3, 4 and all the image in 5 except the top left were recorded using confocal microscopy (ATTO647 fluorescence). Scale bars: 10 μm.
The temperature during the hydration steps should be above the highest transition temperature of the dried lipids.
Electroformation
GUVs were grown following the protocol of Angelova et al.35. Briefly, 20 μl of a 1 mM lipid stock solution in chloroform were dried on an ITO plate under vacuum and rehydrated with 2 ml of 200 mM sucrose. GUV formation was performed at 8Hz and 1.1V for 2h.
Unilamellarity
To prove that the giant liposomes produced using our proposed protocol are unilamellar we applied an already published method by Akashi et al. 7 based upon the quantification of the intensity of the incorporated fluorescently labeled lipids in the membrane. In detail, NBD-PE or Atto647-PE is included in the membrane of the protein-free and proteo-GUV at 0.8 to 1 mol% of the lipids. The averaged fluorescence intensity of the membrane of the GUV is measured by confocal microscopy and subtracted by the averaged background signal. The resulting intensities are plotted in dependence of the liposome size and also their occurrence.
Fluorescence microscopy
GUVs were detected using bright field and confocal microscopy at room temperature. The experiments were carried out on a SP5 confocal microscope from Leica equipped with an 10x and a 20x air objective, an Argon (488/543nm) and HeNe (633nm) lasers and a 488/543/633 beam splitter. Image analysis was done with the LAS AF software from Leica.
Micromanipulation and FRAP
Micromanipulation was used to maneuver independent GUVs and keep them static in solution. The internal diameter of the micropipettes was about ~5 μm and to prevent adhesion of the GUV on the glass, the pipette was incubated in a solution of 10% BSA in pure water prior to use for 15 min. The aspiration to maintain the GUVs was controlled by hydrostatic pressure and kept to 20 Pa.
Fluorescent recovering after photobleaching (FRAP) was performed on GUVs to monitor the diffusion coefficient of fluorescent lipid and fluorescent protein on the membrane. The fluorescence recovery curves were then analyzed with Mathematica 10 or Scilab 5.4 to get the Tau coefficient, Tau being the recovery time.
RESULTS
Principles of the osmotic shock method for GUV formation
The osmotic shock method is based upon the assumption that the membranes of the dried small liposomes have to be disrupted in order to form GUVs. This disruption is induced by the generation of an osmotic shock leading to a surface tension larger than the lysis tension (discussed below), thereby destroying the integrity of the membrane: instead of rehydrating with buffer as it is done in other previously published protocols 3, 4, 15, 23, pure water is used to rehydrate. We assume that the osmotic shock spontaneously and synchronously breaks the liposomes, thereby generating floating membranes that merge together and reseal as soon as they can, thus forming bigger membrane surfaces. Together with the lipids the salt of the small liposome buffer is rehydrated too, creating a physiological environment for the GUVs.
The basic protocol is as follows: A 2 μl drop of a 3 mM small liposomes solution is placed on the coverslip and dried at room temperature under atmospheric pressure. It is rehydrated with 6 μl of deionized water and dried again under the same conditions. In the next step, the lipid film is rehydrated once more with a larger deionized water volume of 10–20 μl and observed under the microscope. GUVs grow instantly from the rehydrated lipid films mainly in the areas with high concentration of lipids (Fig. 2). Several drying hydration cycles usually improve the quality and yield of the GUVs. This might be explained by the new reorganization of the lipids during each drying/ hydration cycle resulting in more and larger GUVs.
Conditions for the osmotic shock method
Our proposed GUV formation protocol is based on an osmotic shock which seems to partially break the membrane of the dried liposomes resulting in the formation of GUVs. This new method is applicable under a broad range of conditions. We characterized the properties and yield of protein-free GUVs in regards to the lipid concentration, the volume of liposomes to be dried, the liposome-buffer composition and concentration, the water volume for hydration and the drying procedure (Fig. S1).
The lipid concentration modulates the thickness and density of the dried liposome film. A too high lipid concentration forms a thick layer of liposome aggregates and GUVs only form at the edge of these aggregates whereas a too small concentration of lipids would be washed away during the hydration step. Lipid concentrations in the range of 3–10 mM result in the formation of GUVs at the edge and also in the center of the liposome film. Besides, Aimon et al. 15 also use a 3 mM lipid concentration when drying liposomes prior to electroformation. Therefore, all our experiments were performed at a lipid concentration of 3 and 10 mM and the volume of the deposited drop is 2 μl.
We also tested several liposome-buffer compositions. Tris-HCl (50 mM) with up to 300 mM NaCl or KCl in the liposome buffer lead to reasonable amounts of GUVs, whereas all the buffers made with a concentration of sucrose, glucose, Nycodenz or glycerol above a few tens of mM did not work. We successfully obtained GUVs with up to 4 mM sucrose. Looking at the drying film it seems that these sugar components prevent sufficient drying of the liposome drop and form a thin layer above the lipid film that protects the liposomes leading to an inhibition/reduction of the osmotic shock during the osmotic shock step (Fig. S2). For salt concentrations higher than 500 mM of the liposome buffer, no formation of GUVs can be detected. To keep physiological conditions, we worked with ionic concentrations between 100 mM and 300 mM. The volume of water applied for the rehydration must cover the area of dried liposomes.
Additionally, we found that several consecutive drying and hydrations of the dried liposomes film increase the number and quality of grown GUVs. After the first hydration the GUVs are small (around 5 μm) and only a few can be observed. But after a second drying and sequential hydration step the number of GUVs increases drastically and their diameter is around 40 μm. Depending on the conditions, 1 to 3 consecutive drying and rehydration steps are recommended.
In conclusion, we chose the following parameters to form protein free and proteo-GUVs: 3 mM lipids, 150 mM NaCl salt in tris buffer (pH 7.4), 2 μl drop, two drying-hydration cycles, 6 μl of water at the first hydration then 10 μl.
Formation of Proteo-GUVs by osmotic shock
In this section, we present how the osmotic shock method is used to incorporate proteins in the GUVs. First, small liposomes containing the incorporated protein were produced. Our protocol does not give a generic method to make small proteo-liposomes but a generic method to form proteo-GUVs starting from small proteo-liposomes. Depending on the protein, the method to form small proteo-liposomes is different. In the next paragraphs, we present briefly the proteins we used, GL1, TolC and t-SNARE (Fig. 1), and how they were incorporated into liposomes.
GL1 is a member of the Atg8/LC3 protein family and is active during the autophagy process, an intracellular degradation mechanism. In the autophagic organelles, the autophagosomes, GL1 is responsible for the regulation of membrane dynamics. This peripheral protein is anchored to the membrane of the autophagosome, through the covalent attachment of its C-terminal end to phosphatidylethanolamine (PE) lipid head group via an ubiquitin-like reaction involving the autophagic enzymes Atg7 and Atg3 27. This lipidation can be efficiently recapitulated in vitro on very small liposomes, but is an inherently curvature-sensitive process and thus does not occur on larger flat membrane structures as GUVs. Atg3 proteins need first to bind to the membrane through packing defects before transferring GL1 to a PE lipid. However the low curvature of the GUVs membrane and the low concentration of PE lipids (30 mol%) limit the presence of lipids packing defects and thus the binding of Atg3.
The neuronal t-SNARE is a protein complex with a single alpha-helix transmembrane domain that is involved in membrane fusion. It is composed of Syntaxin 1 and SNAP-25 and is located on the target plasma membrane. Calcium dependent synaptic vesicle fusion in neurons is mediated by the formation of SNARE complexes. During the docking and subsequent fusion process, vesicle and target membrane are brought into close proximity by pairing of vesicle-associated v-SNAREs with cognate t-SNAREs on target membranes 36.
TolC is an integral membrane protein and part of a heterotrimer that together comprise a major multidrug efflux pump in Escherichia coli, AcrA-AcrB-TolC. AcrA and AcrB are embedded in the inner membrane of the bacteria whereas TolC is an outer membrane protein (OMP). TolC itself is composed of 3 monomers of 428 amino acids. The formation of this homotrimer is required for TolC to insert in the membrane, as its transmembrane domain is a beta-barrel constituted of three protomers contributing four beta-strands each. Once the substrate has been recruited by the inner membrane proteins, it is exported through TolC which plays the role of a channel leading to the extracellular space. Its global geometry is a cylinder whose diameter is approximately 40 Å and height 140 Å 37.
In general, it is quite challenging to incorporate membrane proteins which contain one or several transmembrane domains as they span through the whole bilayer. Concerning our biological systems, our studied proteins (t-SNARE and TolC) are complex structures consisting of 2 or more subunits. Therefore, it is quite challenging to keep these units assembled and in their naturally folded state during the drying processes.
Using small proteo-liposomes containing one of the three proteins, we produced proteo-GUVs in the same way as described for the protein-free GUVs (Fig. 2). We observed their presence by labeling the proteins with an Alexa488 fluorophore. In all three cases the proteins were detected to be colocalized with the lipid-fluorophore label of the membrane (Fig. 3).
Figure 3.

Protein-GUVs, formed with the osmotic shock method using GL1, t-SNARE and TolC labeled with Alexa488 (green). The fluorescent lipid is DOPE-Atto 647 (red). Scale bars 10 μm.
Quantification of the protein concentration in the proteo-GUVs
The amount of protein incorporated in the membrane was quantified based on the protocol described by Galush et al.38 using the fluorescent signal of Alexa488 labeled proteins. To the best of our knowledge, no fluorescent lipid with Alexa488 exists to make a fluorescence scale, so we used soluble ATTO488 dye and the DOPE-ATTO488 lipid to set a fluorescence scale. First we established a ratio of fluorescence intensity between those Alexa488 and ATTO488 in bulk. The measurement settings were kept identical and several dye concentrations were measured (Fig. S3.a.). We found a ratio of the fluorescence intensities ATTO488/Alexa488 of 2. Then, GUVs were made with different molar ratios of DOPE-ATTO488 (Fig. S3.b.) to get a scale of fluorescence intensity with the DOPE-ATTO488 percentage in the membrane. Finally, considering the ratio ATTO488/Alexa488, we calculated the amount of incorporated GL1. Knowing that 5% of the GL1 proteins are labeled and using the standard we established, we found a GL1 ratio on the membrane of 1 per 500 lipids, the ratio between the loaded proteins and the lipids is 1 protein per 300 lipids, so 60% of the initially added GL1 protein get incorporated on the GUV membrane. In case of the t-SNAREs, we found that about 90 % of the loaded protein gets incorporated when starting with an initial concentration of 1 protein per 500 lipids (Fig. S3C) of which about 86% were labeled. We are aware that fluorescent intensity of Alexa488 and Atto647 in its soluble form might be slightly different from its bound form. However, this method provides a good approximation of the protein concentration which is what we are looking for.
Activity of incorporated proteins into GUVs
In a next step, we determined if the membrane proteins remain active after their incorporation into the GUVs..
GL1-PE, incorporated into small liposomes or present on natural biological membranes, is a substrate for the protease RavZ, an anti-autophagy factor produced by the bacterium Legionella pneumophila 39. Importantly, neither GL1 alone nor GL1-PE solubilized in detergent are recognized by RavZ, thus RavZ proteolytic activity on GL1-PE can be used as a proxy for assessing whether GL1-PE retains its normal lipidation and membrane incorporation-dependent fold 39. Here, we tested whether our reconstitution approach maintained the folding of membrane-embedded GL1-PE by incubating the proteo-GUVs with 10 nM of recombinant RavZ at room temperature for 40 min. The fluorescent signal of the protein on the proteo-GUV membrane reduced by half, as expected when proteins from the outer leaflet were cleaved while proteins bound to the inner leaflet remained protected (Fig. 4A) To test the functionality of the t-SNARE, proteo-GUVs and protein-free GUVs were incubated with the soluble domain of Alexa647-labeled v-SNAREs (CDV), its cognate binding partner (Fig. 4B and S4) 36. CDV binds specifically to the proteo-GUVs which proves that t-SNAREs are properly folded. It has been reported before that t-SNAREs usually tend to aggregate when reconstituted into GUV losing their activity 40 and therefore usually require more extensive protocols to be reconstituted41. This proof of functionality does not give information about the site fraction of the t-SNAREs bound to the CDV. However, it gives a good estimation of if the t-SNAREs remain active after reconstitution using a simple and fast protocol.
Figure 4. Functionality of the protein after the osmotic shock method.

A. The reconstituted protein GL1-Alexa488 is cleaved from membrane by RavZ (10 nM) after 40 min incubation at room temperature. The gel shows the cleavage of GL1 from the liposome membrane (bottom left). In the first line, GL1 alone was loaded. In the second line, the enzymatic reaction was loaded, so the lipidated GL1 migration is shifted. In the third line, the enzymatic reaction plus RavZ was loaded. For GL1 is evenly distributed between the inner and the outer leaflet of the proteo-GUVs, only half of the protein is accessible to RavZ leading to a 50% reduction of the fluorescence intensity of GL1 (see micormanipulated GUV on the top left and the evaluation of the fluorescence intensity on the bottom left, statistics on 7 GUVs). In conclusion, GL1 remains properly folded after the drying/hydration cycles. B. Resuspended t-GUVs labeled with NBD-PE are incubated in 1 μM Alexa647-CDV for 60 minutes at 37°C. The left panel is shows the lipid-fluorescence of NBD-PE signal (green) of the GUV and the fluorescence intensity of the bound CDV-Alexa647 to the t-SNAREs. The right panel shows the average fluorescence intensity of the Alexa647 signal of the CDV at t- and protein-free GUVs (see figure S3 for picture of protein-free GUVs). Scale bars: 10 μm. The data show that CDV is binding specifically to the GUVs containing t-SNAREs, proving their functionality after reconstitution (15 GUVs were analyzed for each species).
In general, there were no biochemical differences detected when working with micromanipulated or resuspended GUVs. The error bars in the graphs represent the standard deviation from the mean.
The transmembrane part TolC is a 12-stranded right twisted beta-barrel in which each of the monomers take part 37. Incorrect folding of TolC would prevent the correct insertion of the protein as a homotrimer in the membrane. Since we observe that TolC enters and stays within the membrane of the proteo-GUV, its transmembrane part probably remains correctly folded. TolC is a channel protein, so we suppose that, when correctly folded, it remains functional. This is the best functionality test we can easily propose since TolC is part of a 3 protein complex (AcrA-AcrB-TolC) forming an efflux pump embedded in two adjacent bilayers. The outer membrane protein TolC alone is in a closed state, and opens with the help of its inner membrane partner AcrA. Showing that it can function as a channel is therefore very complicated experimentally without reconstituting the whole efflux pump within a two bilayer system.
Unilamellarity
To prove that our proposed protocol produces unilamellar giant liposomes we applied an already published method by Akashi et al. 7 quantifying the intensity of the incorporated fluorescently labeled lipids in the membrane. The measurement results in a distribution of separate groups of fluorescence intensities belonging each to a distinct number of lipid layers in the liposome with unilamellar liposomes exhibiting the lowest intensity. In our case 82% of the analyzed protein free giant liposomes turned out to be unilamellar. In case of the protein reconstituted GUVs 70% of the vesicles containing GL1 were unilamellar, whereas when t-SNAREs are reconstituted the unilamelarity is of the liposomes is about 85% (Fig. S5).
Micromanipulation of proteo-GUVs and diffusion of the protein in the membrane
Proteo-GUVs obtained with the osmotic shock method can easily be separated and micromanipulated: they can be either caught directly from the hydrated lipid/protein film or first be gently detached from the lipid film by generating a flow in the buffer drop and then caught with a pipette (Fig. 5A). To compare the mobility of the lipids (DOPE-NBD) in the GUVs grown by osmotic shock with that in GUVs made by another method like electroformation, we measured their diffusion coefficient in the membrane by FRAP. We found a similar diffusion coefficient of 6.9 ± 1.3 μm2/s for the osmotic shock method and 7.2 ± 0.4 μm2/s for electroformed GUVs. Therefore, the mobility of the lipids in the GUVs formed by our proposed method and electroformation are the same, indicating that both membranes have similar fluidities.
Figure 5. Micromanipulation and FRAP of the proteo-GUVs.

A. A micromanipulated GUV is aspirated by a micropipette. The fluorescent lipid is DOPE-ATTO647. The scale bar equals 10 μm. B. Photobleaching is performed at the top of the GUV on a 10 μm diameter area (left panel). Middle panel: graph and picture of the fluorescence recovery after bleaching, Tau is the recovery time. Right panel: diffusion coefficients (D) of the fluorescent lipid DOPE-NBD (statistics on 5 FRAP measurements on electroformed GUVs and GUVs prepared with the osmotic shock method), GL1 (statistics on 23 FRAP measurements), t-SNARE (statistics on 5 FRAP measurements) and TolC (statistics on 5 FRAP measurements) calculated from the Tau and the diameter of the bleached area (d). The error bars in the graphs represent the standard deviation from the mean measured using multiple GUVs
The diffusion coefficients of the proteins were also measured to check that they were well incorporated in the membrane. The diffusion coefficients were 4.5 ± 0.4 μm2/s for the lipid anchored GL1 protein, 4.6 ± 0.6 μm2/s for the single transmembrane protein t-SNARE and 2.5 ± 0.5 μm2/s for the trimeric protein TolC (Fig. 5B). TolC and t-SNARE diffuse slower than a lipid because of their trans-membrane domains. The difference between both proteins is expected because of the larger trans-membrane domain of TolC. Although GL1 is lipid anchored, it is less mobile than a lipid, possibly because of additional interactions with the membrane.
DISCUSSION
In the upcoming paragraphs, we are going to discuss the role of the osmotic for GUV formation, the conditions for an efficient GUVs growth and how to use the GUVs for micromanipulation.
Mechanism of GUV growth: osmotic shock and drying steps
Liposomes are first formed in a saline buffer and then dried on a glass coverslip under atmospheric pressure. The choice of the drying conditions could be questioned considering that vacuum is the usual way to dry lipids or liposomes. We tested it on liposomes and proteo-liposomes to form GUVs and proteo-GUVs. Vacuum drying of the small liposome for several hours (or overnight) led to a reasonable amount of GUVs but no proteo-GUVs were growing in contrast to drying at atmospheric pressure. We assume that the slow drying under atmospheric pressure avoids a complete destruction of the membrane and the bilayer remains almost intact trapping salt inside the liposomes. Our finding of partially drying the sample and thereby protecting the protein has been suggested before23.
The initial osmolarity of solution to be dried is around 350 mOsm corresponding to an osmotic pressure of ΔP=106 Pa at room temperature. Hence adding water on 400 nm liposomes induces surface tension γ = (radius.ΔP)/2 ~ 0.1 N/m, largely above the lysis tension of 10−2 N/m 13, 42. Therefore, hydration with water triggers an osmotic shock that breaks the cohesion of the membrane at a certain point. We assume that this is the reason why the membrane immediately merges with other nearby pieces of bilayers and reseals to produce larger liposomes. When the liposomes are dried a second time, their large structure is preserved like during the first drying step. Therefore, during the second hydration the liposomes grow from larger liposomes than at the first cycle and become even larger. After the first cycle the osmolarity inside the liposomes is reduced three to five times depending on the rehydration volume, so the surface tension decreases but it stay higher than the lysis tension. Several drying/hydration cycles can improve the GUV yield and quality, possibly through reorganization of the lipids during each cycle
Lipid concentration, drop size, buffer concentration and rehydration volume
The lipid concentration, drop size, buffer concentration and rehydration volume are crucial parameters that have to be adjusted to the respective experimental setup to obtain a large amount of functional proteo-GUVs. Here, we discuss how the formation of the GUVs varies in function of these parameters. Lipid concentrations in the range of 3–10 mM give a reasonable amount of GUVs. For higher concentrations the layer of dried liposomes is too thick and the liposomes are trapped in the layer and form fewer GUVs. The size of the liposome drop to dry is another way to adjust the lipid concentration. Indeed, an increase in the volume of the liposome drop increases the density of liposomes in this area. The buffer concentration and volume of water during the rehydration are two bound parameters. The volume of added water does not affect the growth but only the buffer dilution. The buffer of the small liposomes should be in the range of 100–300 mM, enough to get an osmotic shock. For 500 mM NaCl we could not see any GUVs forming in contrast to other published protocols. Those two parameters should be adjusted in function of each other. The buffer concentration should be close to physiological conditions to support the reconstituted proteins. If a large final volume is needed but still with a high ionic concentration, for the micromanipulation experiment for instance, we recommend to do the last hydration with a small volume around 10 μl and, afterwards carefully add more buffer with the correct dilution. In this last step caution is needed. Because when the diluted buffer is added, it triggers a flow inside the drop that partially detaches the GUVs from the coverslip. To limit this effect, the additional buffer can be added in form of a ring around the initial drop which are simultaneously merging together (Fig. S6) leading to an increase of the initial drop with limited impact on the GUVs.
We assume that the buffer composition inside the GUV matches closely the one outside. The GUVs appear to be completely spherical, meaning they are under tension and their osmolarity is larger than the outer solution. However, a change of more than 30 mOsm (i.e. 10% of a physiological outer medium) would not allow the formation of largest GUVs we observe (up to 100 μm).
Protein addition: GUV growth and sucrose
The addition of proteins in the protocol clearly reduces the amount and the size of GUVs. Those observations could be explained in different ways. The incorporated protein may increase the lysis tension of the membrane 43, which would reduce the breaking of the membrane. It could also affect the viscosity of the membrane and prevent the reformation of the membrane after the osmotic shock. In order to protect the protein during the GUV growth, the addition of sucrose in the μM range has been recommended17. Sucrose is stabilizing proteins via hydrogen bonding during the drying process17,44. Additionally, it is also stabilizing the membrane during the drying process by being a substitute for the missing water molecules. This way the gap between the lipid head groups remains filled preventing its transition to the rigid gel phase during the drying process17. A too high concentration generates a layer of sucrose above the liposomes and proteo-liposomes inhibiting the GUV formation. The additional sucrose does not interfere with the process of proteo-GUV formation. Here, sucrose was used in case of the t-SNARE and TolC reconstitution, which are known to denature easily.
Using our method it is not possible to control the orientation of the incorporated protein. This is a standard issue and there has been an elegant way recently proposed to make asymmetric GUVs using transmembrane proteins in detergent 16. We tried this method on TolC. It worked but the density of incorporated proteins was low (at least 10 times less than the osmotic shock method). This method was not suitable for GL1 because it has to be lipidated on small liposomes. We did not try it on t-SNARE.
Conclusion/summary
In this article we show a new way to form proteo-GUVs. The method is based upon the generation of an osmotic shock when hydrating a lipid film made of dried small liposomes. We have successfully inserted three different kinds of proteins: A membrane associated protein (GL1), a membrane protein with one helical transmembrane domain (t-SNARE) as well as a channel protein (TolC). We have explored the most favorable conditions. In all cases an initial lipid concentration of 3 mM and a salt concentration between 100 to 300 mM have given the highest yield of GUVs. Furthermore, several consecutive drying and hydrations can improve the amount and size of the GUVs.
We verified the unilamellarity of the GUVs as well as the amount protein incorporated. Additionally, we were able to demonstrate that the reconstituted proteins keep their physiological functionality and are mobile in the membrane of the GUVs.
As an application, we show that the GUVs can be micromanipulated.
To conclude, our results show that using our newly developed method different kinds of proteins can be integrated and still keep their natural function and diffusive behavior.
Supplementary Material
Acknowledgments
This work was supported by a Partner University Funds exchange grant between the Yale and Ecole Normale Supérieure laboratories and by the ANR-13-BSV5-0004 grant. We thank Prof. T. Melia for many helpful discussions, Dr. D. Tareste and N. Rodriguez for their comments on the manuscript, Dr. L. Monlezun, Dr. M. Picard and A. Verchère for purifying the TolC protein.
Footnotes
Figure S1 shows the study of the growth parameter for the GUV formation process. Figure S2 shows the characteristics of a drying liposome film with high and low sucrose content. S3 depicts the calibration curves for the estimation of protein reconstituted into the GUVs. Figure S4 shows a picture of the control, proteinfree GUVs incubated with CDV, for the functionality test of the t-SNAREs in the GUVs. Figure S5 shows the measurement of the unilamellarity of the produced GUVs with and without protein. Figure S6 demonstrates how additional buffer can be added without disturbing the grown of the GUVs.
This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Chan YH, Boxer SG. Model membrane systems and their applications. Current opinion in chemical biology. 2007;11(6):581–7. doi: 10.1016/j.cbpa.2007.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Angelova M, Dimitrov DS. A mechanism of liposome electroformation. Faraday Discuss Chem Soc. 1986;81:303–311. [Google Scholar]
- 3.Pott T, Bouvrais H, Meleard P. Giant unilamellar vesicle formation under physiologically relevant conditions. Chem Phys Lipids. 2008;154(2):115–9. doi: 10.1016/j.chemphyslip.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 4.Montes LR, Alonso A, Goni FM, Bagatolli LA. Giant unilamellar vesicles electroformed from native membranes and organic lipid mixtures under physiological conditions. Biophys J. 2007;93(10):3548–54. doi: 10.1529/biophysj.107.116228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Estes DJ, Mayer M. Giant liposomes in physiological buffer using electroformation in a flow chamber. Biochim Biophys Acta. 2005;1712(2):152–60. doi: 10.1016/j.bbamem.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 6.Reeves JP, Dowben RM. Formation and properties of thin-walled phospholipid vesicles. Journal of cellular physiology. 1969;73(1):49–60. doi: 10.1002/jcp.1040730108. [DOI] [PubMed] [Google Scholar]
- 7.Akashi K, Miyata H, Itoh H, Kinosita K., Jr Preparation of giant liposomes in physiological conditions and their characterization under an optical microscope. Biophys J. 1996;71(6):3242–50. doi: 10.1016/S0006-3495(96)79517-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horger KS, Estes DJ, Capone R, Mayer M. Films of agarose enable rapid formation of giant liposomes in solutions of physiologic ionic strength. J Am Chem Soc. 2009;131(5):1810–9. doi: 10.1021/ja805625u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weinberger A, Tsai FC, Koenderink GH, Schmidt TF, Itri R, Meier W, Schmatko T, Schroder A, Marques C. Gel-assisted formation of giant unilamellar vesicles. Biophys J. 2013;105(1):154–64. doi: 10.1016/j.bpj.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.CB, Jeffries GDM, Orwar O, Jesorka A. Formation of giant unilamellar vesicles from spin-coated lipid films by localized IR heating. Soft Matter (Communication) 2012;8:10823–10826. [Google Scholar]
- 11.Buboltz JT, Feigenson GW. A novel strategy for the preparation of liposomes: rapid solvent exchange. Biochim Biophys Acta. 1999;1417(2):232–45. doi: 10.1016/s0005-2736(99)00006-1. [DOI] [PubMed] [Google Scholar]
- 12.Moscho A, Orwar O, Chiu DT, Modi BP, Zare RN. Rapid preparation of giant unilamellar vesicles. Proc Natl Acad Sci U S A. 1996;93(21):11443–7. doi: 10.1073/pnas.93.21.11443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abkarian M, Loiseau E, Massiera G. Continuous droplet interface crossing encapsulation (cDICE) for high throughput monodisperse vesicle design. Soft Matter. 2011;7:4610–4614. [Google Scholar]
- 14.Pautot S, Frisken BJ, Weitz DA. Production of Unilamellar Vesicles Using an Inverted Emulsion. Langmuir. 2003;19(7):2870–2879. [Google Scholar]
- 15.Aimon S, Manzi J, Schmidt D, Poveda Larrosa JA, Bassereau P, Toombes GE. Functional reconstitution of a voltage-gated potassium channel in giant unilamellar vesicles. PLoS One. 2011;6(10):e25529. doi: 10.1371/journal.pone.0025529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dezi M, Di Cicco A, Bassereau P, Levy D. Detergent-mediated incorporation of transmembrane proteins in giant unilamellar vesicles with controlled physiological contents. Proc Natl Acad Sci U S A. 2013;110(18):7276–81. doi: 10.1073/pnas.1303857110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doeven MK, Folgering JH, Krasnikov V, Geertsma ER, van den Bogaart G, Poolman B. Distribution, lateral mobility and function of membrane proteins incorporated into giant unilamellar vesicles. Biophys J. 2005;88(2):1134–42. doi: 10.1529/biophysj.104.053413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kahya N, Pecheur EI, de Boeij WP, Wiersma DA, Hoekstra D. Reconstitution of membrane proteins into giant unilamellar vesicles via peptide-induced fusion. Biophys J. 2001;81(3):1464–74. doi: 10.1016/S0006-3495(01)75801-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu YJ, Hansen GP, Venancio-Marques A, Baigl D. Cell-free preparation of functional and triggerable giant proteoliposomes. Chembiochem : a European journal of chemical biology. 2013;14(17):2243–7. doi: 10.1002/cbic.201300501. [DOI] [PubMed] [Google Scholar]
- 20.Meleard P, Bagatolli LA, Pott T. Giant unilamellar vesicle electroformation from lipid mixtures to native membranes under physiological conditions. Methods in enzymology. 2009;465:161–76. doi: 10.1016/S0076-6879(09)65009-6. [DOI] [PubMed] [Google Scholar]
- 21.Varnier A, Kermarrec F, Blesneac I, Moreau C, Liguori L, Lenormand JL, Picollet-D’hahan N. A simple method for the reconstitution of membrane proteins into giant unilamellar vesicles. J Membr Biol. 2010;233(1–3):85–92. doi: 10.1007/s00232-010-9227-8. [DOI] [PubMed] [Google Scholar]
- 22.Horger KS, Liu H, Rao DK, Shukla S, Sept D, Ambudkar SV, Mayer M. Hydrogel-assisted functional reconstitution of human P-glycoprotein (ABCB1) in giant liposomes. Biochim Biophys Acta. 2015;1848(2):643–53. doi: 10.1016/j.bbamem.2014.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Girard P, Pecreaux J, Lenoir G, Falson P, Rigaud JL, Bassereau P. A new method for the reconstitution of membrane proteins into giant unilamellar vesicles. Biophys J. 2004;87(1):419–29. doi: 10.1529/biophysj.104.040360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shaklee PM, Semrau S, Malkus M, Kubick S, Dogterom M, Schmidt T. Protein incorporation in giant lipid vesicles under physiological conditions. Chembiochem : a European journal of chemical biology. 2010;11(2):175–9. doi: 10.1002/cbic.200900669. [DOI] [PubMed] [Google Scholar]
- 25.Merkle D, Kahya N, Schwille P. Reconstitution and anchoring of cytoskeleton inside giant unilamellar vesicles. Chembiochem : a European journal of chemical biology. 2008;9(16):2673–81. doi: 10.1002/cbic.200800340. [DOI] [PubMed] [Google Scholar]
- 26.Osawa M, Anderson DE, Erickson HP. Reconstitution of contractile FtsZ rings in liposomes. Science (New York, NY. 2008;320(5877):792–4. doi: 10.1126/science.1154520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nath S, Dancourt J, Shteyn V, Puente G, Fong WM, Nag S, Bewersdorf J, Yamamoto A, Antonny B, Melia TJ. Lipidation of the LC3/GABARAP family of autophagy proteins relies on a membrane-curvature-sensing domain in Atg3. Nat Cell Biol. 2014;16(5):415–24. doi: 10.1038/ncb2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parlati F, Weber T, McNew JA, Westermann B, Sollner TH, Rothman JE. Rapid and efficient fusion of phospholipid vesicles by the alpha-helical core of a SNARE complex in the absence of an N-terminal regulatory domain. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(22):12565–70. doi: 10.1073/pnas.96.22.12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li F, Kummel D, Coleman J, Reinisch KM, Rothman JE, Pincet F. A half-zippered SNARE complex represents a functional intermediate in membrane fusion. J Am Chem Soc. 2014;136(9):3456–64. doi: 10.1021/ja410690m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li F, Pincet F, Perez E, Giraudo CG, Tareste D, Rothman JE. Complexin activates and clamps SNAREpins by a common mechanism involving an intermediate energetic state. Nature structural & molecular biology. 2011;18(8):941–6. doi: 10.1038/nsmb.2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kummel D, Krishnakumar SS, Radoff DT, Li F, Giraudo CG, Pincet F, Rothman JE, Reinisch KM. Complexin cross-links prefusion SNAREs into a zigzag array. Nature structural & molecular biology. 2011;18(8):927–33. doi: 10.1038/nsmb.2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li F, Pincet F, Perez E, Eng WS, Melia TJ, Rothman JE, Tareste D. Energetics and dynamics of SNAREpin folding across lipid bilayers. Nature structural & molecular biology. 2007;14(10):890–6. doi: 10.1038/nsmb1310. [DOI] [PubMed] [Google Scholar]
- 33.Broutin I, Benabdelhak H, Moreel X, Lascombe MB, Lerouge D, Ducruix A. Expression, purification, crystallization and preliminary X-ray studies of the outer membrane efflux proteins OprM and OprN from Pseudomonas aeruginosa. Acta crystallographica. Section F, Structural biology and crystallization communications. 2005;61(Pt 3):315–8. doi: 10.1107/S1744309105005014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scott BL, Van Komen JS, Liu S, Weber T, Melia TJ, McNew JA. Liposome fusion assay to monitor intracellular membrane fusion machines. Methods in enzymology. 2003;372:274–300. doi: 10.1016/S0076-6879(03)72016-3. [DOI] [PubMed] [Google Scholar]
- 35.Angelova MI, Soleau S, Meleard P, Faucon JF, Bothorel P. Preparation of Giant Vesicles by External Ac Electric-Fields - Kinetics and Applications. Prog Coll Pol Sci S. 1992;89:127–131. [Google Scholar]
- 36.Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Sollner TH, Rothman JE. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92(6):759–72. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- 37.Koronakis V, Sharff A, Koronakis E, Luisi B, Hughes C. Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature. 2000;405(6789):914–9. doi: 10.1038/35016007. [DOI] [PubMed] [Google Scholar]
- 38.Galush WJ, Nye JA, Groves JT. Quantitative fluorescence microscopy using supported lipid bilayer standards. Biophys J. 2008;95(5):2512–9. doi: 10.1529/biophysj.108.131540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choy A, Dancourt J, Mugo B, O’Connor TJ, Isberg RR, Melia TJ, Roy CR. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science (New York, NY. 2012;338(6110):1072–6. doi: 10.1126/science.1227026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tareste D, Shen J, Melia TJ, Rothman JE. SNAREpin/Munc18 promotes adhesion and fusion of large vesicles to giant membranes. Proc Natl Acad Sci U S A. 2008;105(7):2380–5. doi: 10.1073/pnas.0712125105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malsam J, Parisotto D, Bharat TA, Scheutzow A, Krause JM, Briggs JA, Sollner TH. Complexin arrests a pool of docked vesicles for fast Ca2+-dependent release. EMBO J. 2012;31(15):3270–81. doi: 10.1038/emboj.2012.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Portet T, Dimova R. A new method for measuring edge tensions and stability of lipid bilayers: effect of membrane composition. Biophys J. 2010;99(10):3264–73. doi: 10.1016/j.bpj.2010.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim DH, Frangos JA. Effects of amyloid beta-peptides on the lysis tension of lipid bilayer vesicles containing oxysterols. Biophys J. 2008;95(2):620–8. doi: 10.1529/biophysj.107.114983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crowe JH, Crowe LM, Carpenter JF, Rudolph AS, Wistrom CA, Spargo BJ, Anchordoguy TJ. Interactions of sugars with membranes. Biochim Biophys Acta. 1988;947(2):367–84. doi: 10.1016/0304-4157(88)90015-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.