

Abstract
DC vaccination with autologous tumor lysate has demonstrated promising results for the treatment of glioblastoma (GBM) in preclinical and clinical studies. While the vaccine appears capable of inducing T cell infiltration into tumors, the effectiveness of active vaccination in progressively growing tumors is less profound. In parallel, a number of studies have identified negative costimulatory pathways, such as programmed death 1/programmed death ligand 1 (PD-1/PD-L1), as relevant mediators of the intratumoral immune responses. Clinical responses to PD-1 pathway inhibition, however, have also been varied. To evaluate the relevance to established glioma, the effects of PD-1 blockade following DC vaccination were tested in intracranial (i.c.) glioma tumor–bearing mice. Treatment with both DC vaccination and PD-1 mAb blockade resulted in long-term survival, while neither agent alone induced a survival benefit in animals with larger, established tumors. This survival benefit was completely dependent on CD8+ T cells. Additionally, DC vaccine plus PD-1 mAb blockade resulted in the upregulation of integrin homing and immunologic memory markers on tumor-infiltrating lymphocytes (TILs). In clinical samples, DC vaccination in GBM patients was associated with upregulation of PD-1 expression in vivo, while ex vivo blockade of PD-1 on freshly isolated TILs dramatically enhanced autologous tumor cell cytolysis. These findings strongly suggest that the PD-1/PD-L1 pathway plays an important role in the adaptive immune resistance of established GBM in response to antitumor active vaccination and provide us with a rationale for the clinical translation of this combination therapy.
The PD-1/PD-L1 immunoregulatory pathway suppresses DC vaccine–induced T cell responses in the setting of established gliomas.
Introduction
Glioblastoma (GBM) is a devastating disease, for which the diagnosis is associated with an extremely poor prognosis and median survival of 14 months following surgery, radiation, and chemotherapy (1–3). Our group and others have pioneered a DC vaccine–based immunotherapy platform, the results of which have suggested benefit in early-phase trials by promoting an endogenous antitumoral immune response (4–7). An ongoing randomized, placebo-controlled phase III clinical trial is now underway based on these results. However, survival in DC vaccine-treated GBM patients has been varied (5). While increased T cell infiltration correlates with survival benefit across subjects, the ability to generate and sustain this response appears to be dependent on factors such as active tumor progression and GBM subtype (4, 8). These findings emphasize the need to more clearly understand the cellular mechanisms by which DC vaccination induces effective tumor-specific immune responses.
A possible explanation for the variability of vaccine efficacy is that the tumor and its microenvironment can adapt to suppress an immune response directed against them. Studies in various cancer models have suggested that checkpoint mechanisms, which exist to promote self-tolerance and protect against autoimmunity, can develop in the tumor microenvironment (9–14). PD-1/PD-L1 (programmed death 1/programmed death ligand 1) has been shown to induce functional anergy and limit activation of cytotoxic T cells during long-term exposure to antigen, a phenomenon associated with neoplastic disease (9, 15–17). The upregulation of inhibitory PD-L1 in tumor cells appears to be associated with increased tumor-infiltrating lymphocytes (TILs), a phenomenon readily noted in immunogenic cancers with an endogenous immune infiltrate (18, 19). Studies in melanoma have frequently shown robust antitumor responses in response to PD-1 mAb blockade (20–22). It was first shown that inhibition of PD-1/PD-L1 promotes the antitumoral activity of TILs present in B16 melanoma models (23–27). This blockade was dependent on the presence of an infiltrating CD8+ population (21). PD-1/PD-L1–mediated suppression was noted in a glioma model, as well. Adjuvant PD-1 mAb blockade combined with external beam ionizing radiation promoted long-term survival in mice when compared with mice that only received radiation alone (28).
Unlike melanoma, however, GBM are not inherently immunogenic, and active vaccination is necessary to first generate an intratumoral immune response. In this study, we demonstrated that PD-1/PD-L1 modulates adaptive immune resistance to tumor lysate–pulsed DC vaccine treatment in our murine glioma model. Specifically, we show that this negative costimulatory ligand plays a role in suppressing TIL activation, trafficking, and memory responses and that blocking PD-1 can reverse this suppression. Finally, we recapitulated these findings in our patient-derived GBM tissue by a series of ex vivo studies, further documenting the clinical relevance of the PD-1 mechanism. Thus, these findings suggest that the combination of DC vaccine with PD-1 mAb blockade in human GBM provides a clinically translatable means of promoting an antitumoral immune response and attenuating immune suppression.
Results
DC vaccination promotes an antitumor, infiltrating T cell response but is ineffective in established intracranial glioma.
We previously noted that established tumor burden and immunosuppressive cytokine levels negatively correlated with immune responsiveness and patient survival after DC vaccination (4). As such, we were interested to examine the critical mechanisms of resistance to vaccine treatment in the face of established tumor burden, despite T cell infiltration. To evaluate this question, mice were implanted with 2 × 104 murine GL261 glioma cells in the brain and treated with tumor lysate–pulsed DC vaccination. Mice that began receiving DC vaccine on the day of tumor implant, prior to establishment, showed significant survival benefit (Figure 1A). Mice that did not begin receiving DC vaccine treatments until 3 days after tumor implantation or later did not demonstrate increased survival when compared with nontreated controls (Figure 1B). Thus, we showed that DC vaccination was ineffective when there is a larger, established tumor burden.
Figure 1. DC vaccination promotes an antitumor but ineffective infiltrating immune response in the established setting.

Mice intracranially (i.c.) implanted with GL261 were randomly assigned to receive DC vaccine treatments (A) at time of implant (low tumor burden) or (B) once tumors became established (elevated tumor burden). Data shown are representative of 1 experiment repeated 2 times with similar findings. (C–F) IHC staining with anti-CD3 antibody (red) on brains harvested from these mice (×20 magnification). (G) CD3+Thy1.2+ cells were FACS sorted from i.c. GL261 gliomas or spleens and plated with GL261 cells for in vitro xCelligence cytotoxicity assay (n = 3) (****P < 0.0001). Data shown are representative of 1 experiment repeated 2 times with similar findings. Each point represents 1 subject (A and B) or the average of biological replicate (n = 4) (G). Survival differences were calculated using log-rank statistical tests and graphed using the method of Kaplan-Meier (A and B), and a Student’s t test was used to calculate statistical significance at individual time points (G).
To determine whether vaccination promoted a local immune response in these established tumors, explanted brains were analyzed by IHC staining. DC vaccine–treated tumors showed a robust CD3+ infiltrating population (Figure 1, C–F) similar to what was seen in patients’ tumors (4), suggesting that DC vaccine treatment elicited a significant intratumoral immune response compared with nontreated controls, despite being ineffective at promoting long-term survival in mice with established tumors. To assess whether this tumor-associated lymphocyte population was glioma reactive, we evaluated the ability of purified TILs to lyse cultured GL261 glioma cells in vitro. In a real-time cytotoxicity assay, CD3+ TILs from DC vaccine–treated mice showed 50% cytotoxicity against cultured GL261 glioma cells over 20 hours, whereas splenic lymphocyte controls demonstrated little or no cytotoxicity (Figure 1G). In whole, these findings demonstrated that, despite a lack of survival benefit in mice with established tumor burden, DC vaccination induced a significant antitumor immune infiltrate.
Adjuvant PD-1 mAb blockade in the setting of established tumor burden enhances the therapeutic benefit of DC vaccination.
We hypothesized that local immune suppression in the intracranial (i.c.) tumor microenvironment prevented an effective antitumor T cell response following DC vaccination. Recent studies have described PD-1–mediated suppression of T cell responses (9, 21). In our mouse glioma model, we found relevant increases in the expression of PD-1 on TILs following DC vaccination in larger, established tumors (Figure 2, A and B). There was also a significant increase in PD-1 expression on TILs compared with splenic lymphocytes in both control nontreatment and DC-vaccinated mice (Figure 2B). In order to gain a more complete understanding of the genetic changes of tumor infiltrating lymphocytes in our mouse model, RNA was isolated from CD3+ TILs of nontreated control and DC vaccine–treated mice, and quantitative transcriptional changes were assessed. We noted a significant upregulation in PD-1, confirming the FACS data (Supplemental Figure 1A; supplemental material available online with this article; doi:10.1172/jci.insight.87059DS1). Other T cell markers of immune inhibition, including CTLA-4, TIGIT, ICOS, BTLA, and TGF-β, were downregulated on TILs following DC vaccine treatment (Supplemental Figure 1, B–F).
Figure 2. PD-1 blockade rescues the survival benefit after DC vaccination in mice with established tumor burden.

(A) Splenic and tumor-infiltrating lymphocytes (TILs) were gated for CD3 expression and (B) median fluorescence intensity (MFI) of PD-1 expression on CD3+ cells quantified between control nontreatment (No Tx) and DC vaccine treatment groups (n = 4/group). Data shown are representative of 1 experiment repeated 2 times with similar findings. FACS-sorted CD3+Thy1.2+ TILs from DC vaccine–treated tumor-bearing mice were cocultured with i.c.-derived tumor bulk with or without ex vivo PD-1 mAb, and tumor cytolysis (C) at 4 hours was quantified (*P < 0.05) (n = 4/group). (D) Mice were randomized into control (tumor-bearing, no treatment), PD-1 mAb, DC vaccine, and DC vaccine plus PD-1 mAb treatment groups. Graphs show evaluation of survival (n = 6/group) (***P < 0.001). Data shown are representative of 1 experiment repeated 4 times with similar findings. Each point represents 1 cell (A) or 1 subject (B–D). Box-and-whisker plots are used to graphically represent the median (line within box), upper- and lower- quartile (bounds of box), and maximum and minimum values (bars) (B and C). A Student’s t test was used to calculate statistical significance (B and C), and survival differences were calculated using log rank statistical tests and graphed using the method of Kaplan-Meier (D).
Because PD-1 was the only inhibitory marker elevated on TILs following DC vaccination in murine samples, we hypothesized that the PD-1/PD-L1 pathway mediated immune suppression of the DC vaccine–induced immune response in established tumors. We noted that GL261 glioma cells expressed PD-L1 (Supplemental Figure 2). To evaluate the relevance of this finding in our murine model, we first cocultured TILs harvested from tumor-bearing mice treated with DC vaccination together with i.c. derived GL261 tumor bulk cells and found that ex vivo PD-1 mAb blockade significantly increased GL261 glioma cell cytolysis over control cocultures of TILs with tumor bulk alone (Figure 2C and Supplemental Figure 3). We then performed in vivo PD-1 mAb blockade together with tumor lysate–pulsed DC vaccination in mice with established i.c. GL261 gliomas. When PD-1 mAb blockade was administered concomitantly with DC vaccination, a highly significant survival benefit resulted. Approximately 40% long-term survival was observed selectively in the combination treatment group (Figure 2D). As we described before, there was no therapeutic benefit with DC vaccination alone in these large, established tumors. Similarly, there was no survival benefit noted with PD-1 mAb blockade alone. As such, this data suggested that antibody blockade of PD-1 recovered the therapeutic benefit of DC vaccination in the established i.c. glioma setting.
PD-1 mAb blockade enhances the functional intratumoral CD8+ T cell response.
To understand this PD-1 regulatory mechanism and the immune cell subsets critical for the survival benefit seen with the combinatorial treatment, we depleted mice of either CD4+ or CD8+ cells. As depicted in Figure 3, A and B, the clinical benefit of a combined DC vaccine and PD-1 mAb blockade was completely dependent on CD8+ cells and not on CD4+ cells. DC vaccination induced a highly significant increase in the tumor infiltrating CD3+ T cell population, which was also maintained in the combination DC vaccine plus PD-1 mAb setting (Figure 3C). Of note, there was no change in the numbers of tumor-infiltrating helper T cell (CD4+) or Treg (CD4+CD25+FoxP3+) populations across treatment groups (Supplemental Figure 4, A and B). A significant proportion of tumor-infiltrating CD3+ cells from DC vaccine– and DC vaccine plus PD-1 mAb–treated mice was composed of CD8+ T cells. Thus, we evaluated differences in the activation pattern of these T cells between DC vaccine– and DC vaccine plus PD-1 mAb–treated mice. Expression of CD25, the high-affinity subunit of IL-2R, was evaluated. Even though there was an elevation in the percentage of activated CD8+CD25+ T cells in PD-1 mAb–treated mice, only a minimal number of infiltrating CD8+ T cells could be isolated in the absence of a DC vaccine. In contrast, we observed a significant number of infiltrating T cells in the presence of a DC vaccine; however, without PD-1 mAb treatment, there was not a significant proportion of activated cells. Only in mice treated with both DC vaccination and PD-1 mAb blockade was there a significant increase in both the proportion and number of activated CD8+CD25+ T cells (Figure 3D). Thus, our findings suggest that the addition of PD-1 mAb blockade to a DC vaccine enhances the migration and activation of T cell responses in a CD8-dependent fashion.
Figure 3. PD-1 mAb blockade enhances the intratumoral CD8+ T cell response.

Survival of mice from control (tumor bearing, no treatment), PD-1 mAb, DC vaccine, and DC vaccine plus PD-1 mAb treatment groups when (A) CD4+ cells or (B) CD8+ cells were depleted is shown (n = 6/group). (C) An absolute number T lymphocytes was isolated from tumor-bearing cerebral hemispheres and cells were then analyzed by flow cytometry (***P < 0.001). (D) The absolute CD8+ count was plotted against the percentage of CD25+CD8+ activated lymphocytes and compared across different treatment groups (n = 4/group) (**P<0.01). Data shown are representative of 1 experiment repeated 4 times with similar endings. Each point represents 1 subject (A–D). Box-and-whisker plots were used to graphically represent the median (line within box), upper- and lower- quartile (bounds of box), and maximum and minimum values (bars) (C). Survival differences were calculated using log-rank statistical tests and graphed using the method of Kaplan-Meier (A and B), and the Student’s t test was used to calculate statistical significance at individual time points.
To obtain a more complete understanding of the gene expression differences between TIL isolated from DC vaccine– versus DC vaccine plus PD-1 mAb–treated animals, we performed an unbiased transcriptional screen of purified TILs. In purified TIL populations isolated from mice treated with DC vaccination and PD-1 mAb blockade, there was increased transcription of the late-activation marker IL-2Rα, as well as genes associated with activation, homing, and survival (Supplemental Figure 5, A and B). Furthermore, cell-free supernatants from ex vivo cocultures of TILs and GL261 tumor cells had significantly elevated concentrations of IL-2 and IFN-γ in the presence of PD-1 mAb (Supplemental Figure 6, A and B). These findings suggested that suppression of the PD-1/PD-L1 signaling mechanism via PD-1 mAb blockade activates CD8+ TILs.
PD-1 mAb blockade enhances the trafficking of a tumor-infiltrating memory CD8+ T cell population into i.c. gliomas.
Previous work has suggested that expression of the α4, β1 integrin was critical for the trafficking of tumor-specific T cells to glioma (29, 30). Additionally, the development of a memory response with inhibition of PD-1 has been previously described (31, 32). To evaluate this in our model, we compared the pattern of memory and integrin homing markers from splenic T cells and TILs harvested from our DC vaccine– and DC vaccine plus PD-1 mAb–treated mice. In both treatment groups, there was a population of memory T cells (CD8+CD44+) in the spleen (Figure 4A) that was not different. However, a significant increase in the percentage of tumor-infiltrating memory T cells was observed with adjuvant PD-1 blockade, such that there was an approximate 2-fold increase across treatment groups (Figure 4, B and C). We further interrogated CD8+CD44+ memory T cells for the expression of CD62L (L-selectin), a lymph node homing receptor expressed on central memory T cells. There was a significantly elevated proportion of CD8+CD44+ memory T cells in the spleen from DC vaccine plus PD-1 mAb–treated mice that expressed CD62L when compared with mice that only received DC vaccination (Figure 4D). This difference was also significant when we looked specifically at tumor-infiltrating cells (Figure 4E). Previous work has demonstrated the role of tumor homing signals, such as the integrin α-4 chain (CD49d), to promote T cell trafficking to CNS tumors (33). In our model, we noted that the concomitant use of PD-1 mAb blockade together with DC vaccination was associated with a statistically elevated proportion of CD8+CD44+ memory T cells that expressed CD49d (Figure 4, F and G). To evaluate how such T cell populations influenced immune memory, we challenged surviving mice from the DC vaccine plus PD-1 mAb treatment cohort 60 days after the initial tumor inoculation with GL261 glioma cells in the contralateral hemisphere. These mice did not receive any additional treatments. When compared with naive control mice, a significant survival benefit was noted in the long-term survivors from previous DC vaccine plus PD-1 mAb therapy (Figure 4H). Such findings suggested that the addition of PD-1 mAb blockade to tumor lysate–pulsed DC vaccination promoted selective i.c. glioma trafficking and immune memory.
Figure 4. PD-1 mAb blockade increases the population of T cells expressing memory and tumor homing markers.

(A–C) Splenic lymphocytes and tumor-infiltrating lymphocytes (TILs) from DC vaccine with or without PD-1 mAb were stained with CD8+CD44+ cells and analyzed by flow cytometry (n = 4/group) (**P < 0.01). (D and E) CD62L+ cells (gated from CD8+CD44+ cells) were analyzed by flow cytometry (n = 4/group) (*P < 0.05, ***P < 0.001). (F and G) CD49d+ cells (gated from CD8+CD44+ cells) were analyzed by flow cytometry (n = 4/group) (*P < 0.05). Data shown are representative of 1 experiment repeated 2 times with similar findings. (H) Survival of long-term DC vaccine plus PD-1 mAb survivors challenged with GL261 glioma cells in the contralateral brain was monitored and compared with naive control mice (control, n = 6; DC vaccine plus PD-1 mAb, n = 5) (****P < 0.0001). Data shown are representative of 1 experiment repeated 3 times with similar findings. Each point represents 1 subject (A, B, and D–G). Box-and-whisker plots were used to graphically represent the median (line within box), upper- and lower- quartile (bounds of box), and maximum and minimum values (bars) (A, B, and D–G). Statistical analyses were performed by a Student’s t test (A, B, and D–G) and Kaplan-Meier method (H).
Ex vivo PD-1 mAb blockade enhances TIL cytotoxicity against patient GBM cells.
To evaluate whether similar biological principles exist in human GBM patients, we interrogated our clinical trial patient samples using a quantitative multiplex immunofluorescence staining panel. There was marked PD-L1 expression in the GBM tumor microenvironment (Supplemental Figure 7, A–F). Additionally, the percentage of CD8+ TILs that dually expressed PD-1 was elevated following DC vaccine treatment compared with the percentage seen in pretreatment samples (Figure 5, A–H). We noted that this difference was significant across multiple patient samples (Figure 5I). Having confirmed upregulated PD-1 expression on our patient TILs, we next evaluated whether ex vivo PD-1 mAb blockade enhanced TIL function in purified, live GBM TILs. To test this, we obtained fresh GBM tissue samples from patients undergoing surgical resection to remove their tumors and harvested CD3+ TILs and autologous tumor cells. When these TILs were cocultured with the original GBM tumor, we noted some cytotoxicity (Figure 5J). However, when a blocking PD-1 mAb was added to the cocultures ex vivo, there was a significant increase in cytotoxicity across samples. This demonstrated that the PD-1/PD-L1 signaling mechanism suppressed the cytotoxicity of TILs. We confirmed that TILs exerted a tumor-specific immune response, as there was minimal tumor cytolysis when TILs from 1 patient were cocultured with GBM from another patient (Supplemental Figure 8). Together, these findings suggested that DC vaccination results in upregulated PD-1 expression in GBM patients and, when blocked ex vivo, could restore function in tumor-infiltrating T cells.
Figure 5. Ex vivo PD-1 blockade enhances TIL cytotoxicity against human GBM.

(A and B) 4’,6-diamidino-2-phenylindole (DAPI), (C and D) CD8, (E and F) PD-1, and (G and H) CD8 and PD-1 costaining is shown across pre- and post-DC vaccine samples from a representative glioblastoma (GBM) patient (40× magnification). (I) Percent PD-1 expression on CD8+ TILs across pre- and post-DC vaccine treatment patient samples was quantified (n = 6) (**P<0.01). (J) TIL cytotoxicity against human GBM at 15 hours with and without PD-1 mAb blockade is shown (n = 4). Each point represents 1 subject (I and J). Box-and-whisker plots were used to graphically represent the median (line within box), upper- and lower- quartile (bounds of box), and maximum and minimum values (bars) (I). Statistical analyses were performed using the Student’s t test (I).
Discussion
In immunogenic cancers, such as melanoma, biologic therapies that incorporate PD-1 blocking antibodies (nivolumab or pembrolizumab) have resulted in extended patient survival in randomized controlled trials (34–42). However, as suggested by Tumeh et al., the significant survival benefit in melanoma patients was dependent on a preexisting infiltrating population of cytotoxic T cells (21). A study in GBM patients noted that increased T cell infiltrates at the time of resection were predictive of increased survival (43). Based on our previous work, such T cell infiltration is not consistent across GBM subtypes, with nonmesenchymal subtypes exhibiting a lower endogenous lymphocyte infiltration when compared with mesenchymal GBM (8). Although results of ongoing trials examining GBM treatment with PD-1 mAb alone have not yet been published, we hypothesize that clinical efficacy may be varied and dependent on a preexisting TIL population for PD-1 mAb treatment to work.
Our group previously demonstrated that, despite promoting a T cell infiltrating response, the ability of active vaccination to initiate an immune response and improve survival is less consistent in patients with large, progressive disease (4). Thus, we were particularly interested to understand the mechanisms that would prevent some patients with established and progressive recurrent disease from showing survival benefits. In this study, we effectively recapitulated these clinical findings in our mouse model by vaccinating mice bearing large established i.c. gliomas. We demonstrated that, despite generating a significant localized CD8-dominant tumor-specific T cell response with vaccination, there was no significant survival benefit when mice were vaccinated in the setting of large, progressive i.c. tumors. Importantly, additional vaccine treatments did not provide therapeutic benefit for mice bearing these larger tumors (data not shown). The corresponding elevation of PD-1 expression with increasing TILs following vaccination suggested that the PD-1/PD-L1 negative costimulatory mechanism of adaptive immune resistance becomes functionally relevant in an environment of elevated tumor burden (44). As such, while the T cell response appears to be crippled by these regulatory mechanisms, it is significantly functionally activated when the PD-1/PD-L1 pathway is inhibited. Thus, by generating an immune response in a nonimmunogenic, established tumor — and by activating it with PD-1 mAb blockade — we are able to both generate and maintain effective immunity with therapeutic benefit (Figure 6).
Figure 6. Therapeutic benefit for i.c. glioma is dependent on TIL infiltration and activation.
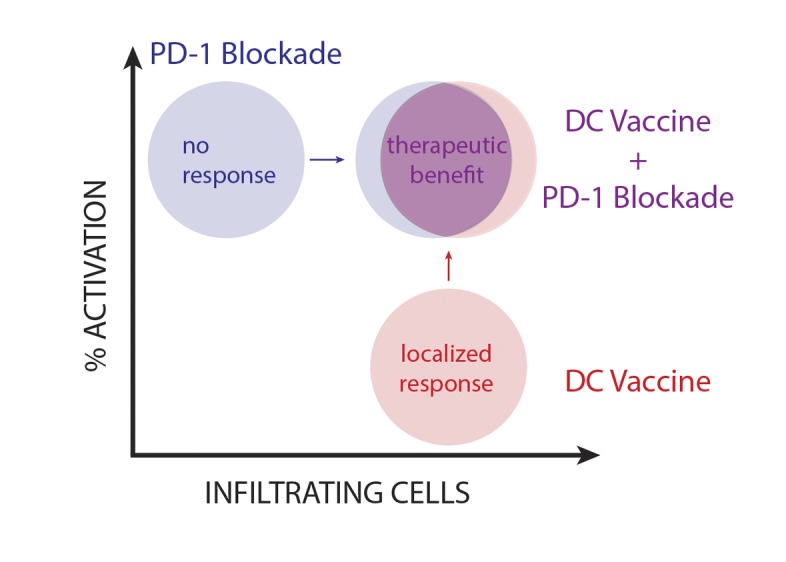
DC vaccination promotes activation of a significant tumor-infiltrating lymphocyte (TIL) population, which is then further activated in the presence of PD-1 mAb blockade.
Mice that had been treated with DC vaccine and PD-1 mAb were able to reject a second tumor inoculation without additional vaccination or PD-1 mAb treatments. The relevance of PD-1/PD-L1 to memory T cell generation in the tumor environment has been described previously (31, 45). Interestingly, an elevated proportion of our central memory T cell population in mice vaccinated with adjuvant PD-1 mAb blockade expressed elevated tumor homing markers CD62L and CD49d. These findings suggest that this population may represent a subset of memory T cells that are recent emigrants into the tumor. As they mature to become effector cells, they may downregulate these homing markers. Another possibility is that these non-CD62L– and CD49d-expressing memory T cells may represent a resident memory T cell population analogous to those present in barrier tissues such as the epithelium of skin, lung, and gastrointestinal tract (46, 47). These resident cells likewise express memory cell markers, but they are generally thought to be unable to recirculate from tissue to lymph and then back again, an important aspect of the homing phenotype (47). Together, these findings suggest that, despite a large, established nonimmunogenic tumor, we are able to generate and maintain an immune response that both targets existing tumors and prevents tumor recurrence: a hallmark of GBM. It is possible that, in patients, dual vaccination and PD-1 mAb blockade will not only enhance survival benefit with established tumors, but also decrease the likelihood of GBM recurring.
The role of helper T cells and Tregs in tumor-mediated immune suppression has been examined (48–51). Although we cannot rule out that PD-1 mAb blockade does not affect the function of Tregs, we did note in our studies that CD8+ T cells largely mediated our antitumoral immune response with no significant changes in CD4+ and CD4+ Treg populations across treatment groups. Depletion of CD4+ T cells did not provide any added benefit compared with DC vaccine alone. Importantly, the DC vaccine plus PD-1 mAb treatment was not improved with the depletion of CD4+ T cells. Our flow cytometry data on harvested cells demonstrated that there was no increase in the tumor-infiltrating helper T cell and Treg populations with DC vaccine treatment, and adjuvant PD-1 mAb did not reduce their proportion in the tumor environment. Therefore, while it is possible that CD4+ cells play some role in the PD-1/PD-L1 mechanism on a broader scale, our results suggest that this is noncontributory in our DC vaccine plus PD-1 mAb treatment model.
In conclusion, we demonstrated that the PD-1/ PD-L1 negative costimulatory system mediates adaptive immune resistance to the vaccine-generated tumor infiltrating T cell response in GBM. Specifically, we described the effect of PD-1 blockade on intratumoral effector and memory T cells. We demonstrated drastically improved survival in the in vivo setting following both treatments. Furthermore, mice that survived the initial tumor challenge inoculation following DC vaccine plus PD-1 mAb treatment sustained survival benefit over naive controls when rechallenged with tumors contralaterally. With this study, we also demonstrated the necessity of a vaccine-generated TIL population in order for the PD-1 pathway to be able to exert an effect within GBM tumors. Although there is probably a broad spectrum of tumor environment–mediated adaptive inhibition, PD-1 is a likely dominant regulatory mechanism in vaccine-induced immune suppression in our model. Future studies are needed to better understand what is likely a heterogeneous array of mechanisms utilized to prevent antitumor activity. The interplay of these mechanisms may provide the key to successfully treating GBM using endogenous immune mechanisms.
Methods
Human tissue samples.
Paraffin-embedded human GBM tissues from pre- and post-DC vaccine treatments were obtained through the Brain Tumor Translation Resource (BTTR) at UCLA and used for histology and IHC. GBM tissue from freshly resected patient samples was placed in collagenase overnight for digestion and then passed through a Percoll gradient to separate TIL and tumor cell layers; it was then used for functional ex vivo assays.
GL261 lysate preparation.
GL261 glioma cells, obtained from Henry Brem (Johns Hopkins University) were cultured in complete DMEM media. Cells were then harvested and subjected to several freeze-thaw cycles. The suspension was then filtered, and the concentration of the lysate obtained was then quantified using a Bradford protein assay.
Cell lines.
Cells were maintained in complete DMEM (Mediatech), supplemented with 10% FBS (Gemini Bio Products), and 1% (v/v) penicillin and streptomycin (Mediatech Cellgro) and cultured in a humidified atmosphere of 5% CO2 at 37°C.
Murine Model.
Female C57BL/6 mice, age 6–8 weeks, were obtained from our institutional breeding colony and kept under defined-flora pathogen-free conditions at the Association for Assessment and Accreditation of Laboratory Animal Care–approved animal facility of the Division of Experimental Radiation Oncology at UCLA.
i.c. glioma implantation.
Female C57BL/6 mice (6–8 weeks old) were anaesthetized, and GL261 glioma cells (2 × 104 in 2 μl PBS) were stereotactically injected at 2.5 mm lateral to bregma at a depth of 3.5 mm below the dura mater with a sterile Hamilton syringe fitted with a 26-gauge needle. The i.c. injection was performed over a 2-minute period. Following i.c. tumor implantation, mice were randomized into treatment groups (n = 6–16 mice/group).
BM-derived DC and vaccination model.
DCs were prepared from murine BM progenitor cells using a method previously described (52). Mice were treated with s.c. injections of lysate-pulsed DC vaccination (1 × 106 cells/mouse) on days 3 and 13 after implantation (elevated tumor burden) or days 0, 7, and 14 (low tumor burden).
In vivo antibody treatments and cell depletions.
PD-1 mAb (clone RMP1-14, BioXCell) was administered i.p. at 250 mg/kg daily on days 3–5 and 13–15. CD4 (clone GK1.5, BioXCell) and CD8 (clone YTS 169.4, BioXCell) depleting antibodies were administered i.p. at 200 mg/kg every other day starting 4 days prior to tumor implantation.
Tissue harvests, IHC, and flow cytometry.
Spleens, lymph nodes, and tumor-bearing brain hemispheres were harvested from mice on day 16, 72 hours following the second DC vaccination. In cases where sectioning and IHC were required, tissue was placed in Zinc Fixative (BD Biosciences) for 24 hours and then transferred to 70% ethanol before being embedded in paraffin wax. Murine tissue was stained via IHC methods with the assistance of the UCLA Translational Pathology Core Laboratory (TPCL) for CD3. Immunofluorescent staining was performed on formalin-fixed paraffin embedded (FFPE) 5-μm patient-derived tissue sections as described (21) using CD8 (clone C8/144B, DAKO, 1/5,000), PD-1 (clone EPR4877, abcam, 1/50,000), and PD-L1 (clone 1-111A, eBioscience, 1:4,000). Cy3 and FITC TSA kits (Invitrogen) were used to visualize primary antibody, and nuclei were visualized with 49,6-diamidino-2-phenylindole (DAPI; Invitrogen). Analyses for all tissue was performed using the Vectra inForm analysis software to select and quantify staining and colocalization of markers of interest.
For flow cytometric staining, tumor-bearing brain hemisphere was placed into tissue collagenase overnight. Lymphocytes were isolated from the tumor suspension the following day using 30%–70% Percoll gradient or from spleens using a 70-μm mesh cell strainer. Remaining tumor burden was maintained in DMEM media supplemented with 10% FBS and 1% (v/v) penicillin and streptomycin. In experiments requiring analysis of isolated lymphocytes, cells were stained with fluorochrome conjugated antibodies to CD3 (clone 17A2), CD4 (clone 4SM95), CD8 (clone 53–6.7), CD25 (clone PC61.5), FoxP3 (clone FJK-16s), Thy1.2 (clone 30-H12), and PD-1 (clone RMP1-14) obtained from eBioscience All FACS analysis was performed with the use of an LSRII (BD Biosciences). Gates were set based on isotype-specific control antibodies (data not shown). Data was analyzed using FlowJo software. TILs used in in vitro assays were FACS sorted for CD3+Thy1.2+ using a FACSAria (BD Biosciences).
nCounter GX NanoString Analysis.
TILs were harvested and then FACS sorted for Thy1.2+CD11b– T cells. Sorted cells were then lysed in RLT buffer (Qiagen, RNEasy Kit). The gene expression (2,500–10,000 cells/ sample) of approximately 770 genes was quantified via direct binding to tagged probes using the nString NanoString system.
Multianalyte Luminex immunoassay.
FACS-sorted TILs were placed into culture with GL261 tumor with or without 10 μg/ml PD-1 mAb (murine, clone RMP1-14; human, clone J110, BioXCell). After 24 hours, supernatant was collected and then processed using the multianalyte Luminex immunoassay.
Real-time cytotoxicity assay.
Cytotoxic killing of tumor cells was assessed using the xCELLigence Real-Time Cell Analyzer System (Acea Biotechnology). In mice, T cells were initially harvested from tumor-bearing brain hemispheres or spleens and then purified via Percoll gradient and FACS sorting (CD11b–, CD3+). In patient samples, T cells were harvested from freshly resected tumor tissue and then purified. After overnight tumor culture to allow adherence of the cells to the well bottom, T cells were added to tumor cell cultures at a 10:1 T cell/tumor ratio. Cell index values (relative cell impedance) were collected over 15 hours and normalized to the maximal cell index value immediately prior to effector cell plating using RTCA Software 2.0 (Acea Biotechnology) as described previously (53). PD-1 mAb was added to the T cell cultures prior to coculture at 10 μg/ml where necessary.
Statistics.
The 2-tailed Student’s t test was used to compare continuous variables. Data that necessitated the comparison of more than 2 groups were analyzed using ANOVA followed by Kruskal-Wallis statistics. Data and data bars in figures represent mean ± SEM. Values were considered significant at P < 0.05. In the cases of survival studies, median survival times were graphed using Kaplan-Meier plots and analyzed using log-rank survival statistics.
Study approval.
For human studies, patients provided informed consent for their tissue to be used for research purposes, which was approved by UCLA Medical IRB. For murine studies, mice were handled in accordance with the UCLA animal care policy and approved animal protocols.
Author contributions
JPA, HS, RGE, LML, and RMP designed the study. JPA, HS, RGE, JO, NS, and SS conducted experiments. JPA, HS, RGE, JO, NS, SS, and GL analyzed the data. GL performed statistical analyses. RGE, DM, WHY, and TFC contributed technical and scientific advice. WHY and TFC provided patient samples. The paper was written by JPA, with input from HS, RGE, DM, LML, and RMP. All authors read and approved the manuscript prior to submission.
Supplementary Material
Acknowledgments
This work was supported in part by NIH/NCI grants R21-CA186004 and R01CA154256 (R.M. Prins), R01 CA125244 (L.M. Liau), and R25 NS079198 (R.M. Prins), R.G. Everson, and L.M. Liau); the Isabel Neidorf Foundation (R.M. Prins, L.M. Liau); the Musella Foundation for Brain Tumor Research (R.M. Prins); and the UCLA Medical Scientist Training Program (MSTP) (J.P. Antonios). We would like to thank the Center for Systems Biomedicine (Integrated Molecular Technologies Core) for their technical assistance with the Nanostring studies, which is supported by CURE/P30DK41301-26. We also thank the UCLA JCCC and Center for AIDS Research Flow Cytometry Core Facility that is supported by NIH awards P30 CA016042 and 5P30 AI028697. Finally, we would like to thank the UCLA BTTR and the TPCL for their assistance with paraffin embedding and histology.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:JCI Insight. 2016;1(10):e87059. doi:10.1172/jci.insight.87059.
Contributor Information
Horacio Soto, Email: hsoto@mednet.ucla.edu.
Joey Orpilla, Email: JOrpilla@mednet.ucla.edu.
Diana Moughon, Email: DMoughon@mednet.ucla.edu.
Namjo Shin, Email: NShin@mednet.ucla.edu.
Shaina Sedighim, Email: SSedighim@mednet.ucla.edu.
Gang Li, Email: vli@ucla.edu.
References
- 1.Crespo I, et al. Molecular and Genomic Alterations in Glioblastoma Multiforme. Am J Pathol. 2015;185(7):1820–1833. doi: 10.1016/j.ajpath.2015.02.023. [DOI] [PubMed] [Google Scholar]
- 2.Johnson DR, O’Neill BP. Glioblastoma survival in the United States before and during the temozolomide era. J Neurooncol. 2012;107(2):359–364. doi: 10.1007/s11060-011-0749-4. [DOI] [PubMed] [Google Scholar]
- 3.Chandana SR, Movva S, Arora M. Primary brain tumors in adults. Am Fam Physician. 2008;77(10):1423–1430. [PubMed] [Google Scholar]
- 4.Liau LM, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res. 2005;11(15):5515–5525. doi: 10.1158/1078-0432.CCR-05-0464. [DOI] [PubMed] [Google Scholar]
- 5.Prins RM, et al. Comparison of glioma-associated antigen peptide-loaded versus autologous tumor lysate-loaded dendritic cell vaccination in malignant glioma patients. J Immunother. 2013;36(2):152–157. doi: 10.1097/CJI.0b013e3182811ae4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heimberger AB, et al. Tumor-specific peptide vaccination in newly-diagnosed patients with GBM. J Clin Oncol. 2006;24(18):e87059 [Google Scholar]
- 7.Kanaly CW, Ding D, Heimberger AB, Sampson JH. Clinical applications of a peptide-based vaccine for glioblastoma. Neurosurg Clin N Am. 2010;21(1):95–109. doi: 10.1016/j.nec.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prins RM, et al. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Cancer Res. 2011;17(6):1603–1615. doi: 10.1158/1078-0432.CCR-10-2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fife BT, et al. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol. 2009;10(11):1185–1192. doi: 10.1038/ni.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi L, Chen S, Yang L, Li Y. The role of PD-1 and PD-L1 in T-cell immune suppression in patients with hematological malignancies. J Hematol Oncol. 2013;6(1):e87059. doi: 10.1186/1756-8722-6-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duraiswamy J, Freeman GJ, Coukos G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res. 2013;73(23):6900–6912. doi: 10.1158/0008-5472.CAN-13-1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamid O, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134–144. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahn BJ, Pollack IF, Okada H. Immune-checkpoint blockade and active immunotherapy for glioma. Cancers (Basel) 2013;5(4):1379–1412. doi: 10.3390/cancers5041379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cojocaru G, et al. S9. Proffered paper: Identification of novel immune checkpoints as targets for cancer immunotherapy. J Immunother Cancer. 2014;2(suppl 2):e87059. doi: 10.1186/2051-1426-2-S2-I5. [DOI] [Google Scholar]
- 15.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27(1):111–122. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ribas A. Tumor immunotherapy directed at PD-1. N Engl J Med. 2012;366(26):2517–2519. doi: 10.1056/NEJMe1205943. [DOI] [PubMed] [Google Scholar]
- 17.Agata Y, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. 1996;8(5):765–772. doi: 10.1093/intimm/8.5.765. [DOI] [PubMed] [Google Scholar]
- 18.Topalian SL, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Topalian SL, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32(10):1020–1030. doi: 10.1200/JCO.2013.53.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamid O, et al. Safety and tumor responses with lambrolizumab (anti\textendash PD-1) in melanoma. N Engl J Med. 2013;369(2):134–144. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tumeh PC, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ribas A, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16(8):908–918. doi: 10.1016/S1470-2045(15)00083-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107(9):4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 2013;73(12):3591–3603. doi: 10.1158/0008-5472.CAN-12-4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pilon-Thomas S, Mackay A, Vohra N, Mulé JJ. Blockade of programmed death ligand 1 enhances the therapeutic efficacy of combination immunotherapy against melanoma. J Immunol. 2010;184(7):3442–3449. doi: 10.4049/jimmunol.0904114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rajani K, et al. Combination Therapy With Reovirus and Anti-PD-1 Blockade Controls Tumor Growth Through Innate and Adaptive Immune Responses. Mol Ther. 2016;24(1):166–174. doi: 10.1038/mt.2015.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Binder DC, et al. Antigen-specific bacterial vaccine combined with anti-PD-L1 rescues dysfunctional endogenous T cells to reject long-established cancer. Cancer Immunol Res. 2013;1(2):123–133. doi: 10.1158/2326-6066.CIR-13-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeng J, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013;86(2):343–349. doi: 10.1016/j.ijrobp.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masson F, Calzascia T, Di Berardino-Besson W, de Tribolet N, Dietrich PY, Walker PR. Brain microenvironment promotes the final functional maturation of tumor-specific effector CD8+ T cells. J Immunol. 2007;179(2):845–853. doi: 10.4049/jimmunol.179.2.845. [DOI] [PubMed] [Google Scholar]
- 30.Sasaki K, et al. Stat6 signaling suppresses VLA-4 expression by CD8+ T cells and limits their ability to infiltrate tumor lesions in vivo. J Immunol. 2008;181(1):104–108. doi: 10.4049/jimmunol.181.1.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song MY, Park SH, Nam HJ, Choi DH, Sung YC. Enhancement of vaccine-induced primary and memory CD8(+) T-cell responses by soluble PD-1. J Immunother. 2011;34(3):297–306. doi: 10.1097/CJI.0b013e318210ed0e. [DOI] [PubMed] [Google Scholar]
- 32.Ribas A, et al. PD-1 blockade expands intratumoral memory T cells. Cancer Immunol Res. 2016;4(3):194–203. doi: 10.1158/2326-6066.CIR-15-0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sasaki K, et al. Preferential expression of very late antigen-4 on type 1 CTL cells plays a critical role in trafficking into central nervous system tumors. Cancer Res. 2007;67(13):6451–6458. doi: 10.1158/0008-5472.CAN-06-3280. [DOI] [PubMed] [Google Scholar]
- 34.Robert C, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014;384(9948):1109–1117. doi: 10.1016/S0140-6736(14)60958-2. [DOI] [PubMed] [Google Scholar]
- 35.Garon EB, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372(21):2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 36.Robert C, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015;372(26):2521–2532. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 37.McDermott DF, et al. Survival, durable response, and long-term safety in patients with previously treated advanced renal cell carcinoma receiving Nivolumab. J Clin Oncol. 2015;33(18):2013–2020. doi: 10.1200/JCO.2014.58.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robert C, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320–330. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 39.Gettinger SN, et al. Overall Survival and Long-Term Safety of Nivolumab (Anti-Programmed Death 1 Antibody, BMS-936558, ONO-4538) in Patients With Previously Treated Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 2015;33(18):2004–2012. doi: 10.1200/JCO.2014.58.3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Motzer RJ, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373(19):1803–1813. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Borghaei H, et al. Nivolumab versus Docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373(17):1627–1639. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burki TK. Pembrolizumab for patients with advanced melanoma. Lancet Oncol. 2015;16(6):e87059. doi: 10.1016/S1470-2045(15)70193-2. [DOI] [PubMed] [Google Scholar]
- 43.Yang I, et al. CD8+ T-cell infiltrate in newly diagnosed glioblastoma is associated with long-term survival. J Clin Neurosci. 2010;17(11):1381–1385. doi: 10.1016/j.jocn.2010.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taube JM, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4(127):e87059. doi: 10.1126/scitranslmed.3003689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ge Y, Xi H, Ju S, Zhang X. Blockade of PD-1/PD-L1 immune checkpoint during DC vaccination induces potent protective immunity against breast cancer in hu-SCID mice. Cancer Lett. 2013;336(2):253–259. doi: 10.1016/j.canlet.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 46.Wakim LM, et al. The molecular signature of tissue resident memory CD8 T cells isolated from the brain. J Immunol. 2012;189(7):3462–3471. doi: 10.4049/jimmunol.1201305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park CO, Kupper TS. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat Med. 2015;21(7):688–697. doi: 10.1038/nm.3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.El Andaloussi A, Lesniak MS. An increase in CD4+CD25+FOXP3+ regulatory T cells in tumor-infiltrating lymphocytes of human glioblastoma multiforme. Neuro-oncology. 2006;8(3):234–243. doi: 10.1215/15228517-2006-006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang W, Lau R, Yu D, Zhu W, Korman A, Weber J. PD1 blockade reverses the suppression of melanoma antigen-specific CTL by CD4+ CD25(Hi) regulatory T cells. Int Immunol. 2009;21(9):1065–1077. doi: 10.1093/intimm/dxp072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Siddiqui SA, et al. Tumor-infiltrating Foxp3–CD4+CD25+ T cells predict poor survival in renal cell carcinoma. Clin Cancer Res. 2007;13(7):2075–2081. doi: 10.1158/1078-0432.CCR-06-2139. [DOI] [PubMed] [Google Scholar]
- 51.Grauer OM, et al. CD4+FoxP3+ regulatory T cells gradually accumulate in gliomas during tumor growth and efficiently suppress antiglioma immune responses in vivo. Int J Cancer. 2007;121(1):95–105. doi: 10.1002/ijc.22607. [DOI] [PubMed] [Google Scholar]
- 52.Prins RM, Odesa SK, Liau LM. Immunotherapeutic targeting of shared melanoma-associated antigens in a murine glioma model. Cancer Res. 2003;63(23):8487–8491. [PubMed] [Google Scholar]
- 53.Ramis G, et al. Optimization of cytotoxicity assay by real-time, impedance-based cell analysis. Biomed Microdevices. 2013;15(6):985–995. doi: 10.1007/s10544-013-9790-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.