

Abstract
Sudden unexplained nocturnal death syndrome (SUNDS) is a perplexing disorder to both forensic pathologists and clinic physicians. Desmoplakin (DSP) gene was the first desmosomal gene linked to arrhythmogenic right ventricular cardiomyopathy (ARVC) which was associated with sudden death. To identify the genetic variants of the DSP gene in SUNDS in the southern Chinese Han population, we genetically screened the DSP gene in 40 sporadic SUNDS victims, 16 Brugada syndrome (BrS) patients and 2 Early Repolarization syndrome (ERS) patients using Next Generation Sequencing (NSG) and direct Sanger sequencing. A total of 10 genetic variants of the DSP gene were detected in 11 cases, comprised of two novel missense mutations (p.I125F and p.D521A) and eight previously reported rare variants. Of eight reported variants, two were previously considered pathogenic (p.Q90R and p.R2639Q), three were predicted in silico to bepathogenic (p.R315C, p.E1357D and p.D2579H), and the rest three were predicted to be benign (p.N1234S, p.R1308Q and p.T2267S). This is the first report of DSP genetic screening in Chinese SUNDS and Brugada syndrome. Our results implies that DSP mutations contribute to the genetic cause of some SUNDS victims and maybe a new susceptible gene for Brugada syndrome.
Keywords: Sudden Unexplained Nocturnal Death Syndrome, Brugada syndrome, DSP gene, Cardiac arrhythmia, Variant
Introduction
Sudden unexplained nocturnal death syndrome (SUNDS) is a perplexing disorder often presenting to forensic pathologists [1–3]. SUNDS prevails in Southeast Asia and has some common clinical features: (1) the majority of the decedents are male, apparently healthy, 20 to 50 years old; (2) the death mostly occurred at night during sleep or at rest; (3) no evident histopathology changes explain the cause of death. Lethal cardiac arrhythmias were assumed to be associated with the pathogenesis of sudden cardiac death (SCD) in the young, such as congenital Long-QT syndrome (LQTS), Brugada syndrome (BrS), catecholaminergic polymorphic ventricular tachycardia (CPVT), and arrhythmogenic right ventricular cardiomyopathy (ARVC) [4].
Desmosomes are intercellular junctions that tightly link adjacent cells. Desmoplakin (DSP) is essential for normal desmosomal adhesion; it links the desmosomes and intermediate filaments to maintain mechanical integrity of cardiomyocytes [5]. DSP was the first desmosomal gene linked to autosomal dominant ARVC [6]. Almost 100 DSP pathogenic variants have been related to ARVC (type 8) with 40% missense variants, 30% nonsense variants, and the rest being small insertion/deletions or changes that alter splicingsites [7]. Current studies showed that the misregulation of DSP affected the expression and redistribution of Cx43, concomitant with the expression of Nav1.5 [8]. We hypothesized that both DSP mutants associated ARVC and DSP mediated abnormality of Nav1.5 may contribute to the pathogenesis of some SUNDS cases.
We have performed postmortem molecular autopsy in sporadic cases of SUNDS on the lethal cardiac arrhythmia-associated genes (SCN5A, SCN1B-4B, GPD1L, MOG1, KCNQ1, KCNH2, KCNE1, KCNE2, and RyR2) in the southern Chinese Han population and found that inherited cardiacarrhythmias, such as LQTS, BrS, CPVT and ARVC may explain the cause of death in just under 20% of SUNDS [1–3], leaving unclear the genetic etiology of more than 80% of SUNDS. With the goal of identifying additional genes associated with SUNDS, in this study we screened our SUNDS cohort for variants in the DSP gene.
Materials and Methods
Study population
We screened 40 sporadic SUNDS cases referred to the Department of Forensic Pathology, Zhongshan School of Medicine, Sun Yat-sen University, from 2005 to 2014, as defined by the following inclusion criteria [9]: (1) southern Chinese Han male, older than or equal to 18-year-old; (2) in good antemortem health without any significant disease relating to the death; (3) sudden unexpected death during sleep or rest; (4) and a negative comprehensive forensic autopsy. In addition to SUNDS cases, 16 patients with BrS, and 2 patients with Early Repolarization Syndrome (ERS) were also screened in this study. Genetic screening controls were collected from 110 unrelated apparently healthy males in southern China, provided by the First Affiliated Hospital of Sun Yat-sen University. All participants gave informed consent and the principles outlined in the Declaration of Helsinki were followed. The project was approved for human research by the ethics committee of Sun Yat-sen University.
Genetic variants analysis
The genomics DNA was extracted from the dried-blood spot using the Magen Bind Blood and Tissue kit (Magen, Guangzhou, China) and was quantified with Qubit® 2.0 Fluorometer (Qubit® dsDNA HS Assay Kit). Library preparation was performed by the standard Illumina protocols [10] and then the prepared library were hybridized to the customized probe following standard SureSelectQXT Target Enrichment for Illumina Multiplexed Sequencing protocol (Agilent, USA). At last the final captured DNA libraries were sequenced using the Illumina HiSeq2500 Analyzers. Image analysis and base calling were performed using the Illumine pipeline. After the entire run was completed, primary reads were generated. The average depth of the target regions of all samples is over 200×, and over 98% of the bases were covered >50×. Variants with a depth of coverage <30× or a heterozygous reads ratio <20% was excluded.
Unqualified reads were filtered out using the SOAPnuke software (http://soap.genomics.org.cn/) developed by BGI. Aligned the resulting reads to the hg19 reference genome with BWA version 0.7.10-r789 [11], marked duplications by Picard tools (http://picard.sourceforge.net), applied GATK[12]base quality score recalibration, indel realignment, duplicate removal, and performed SNP and INDEL discovery and genotyping across all samples simultaneously using standard hard filtering parameters or variant quality score recalibration according to GATK Best Practices recommendations [13,14]. Finally, variant calls were annotated with NCBI database (Build 37).
A total of 80 genes associated with inherited cardiac arrhythmia (Table 1) were investigated using target captured next generation sequencing technology.
Table 1.
List of the 80 sudden cardiac arrhythmia related genes included in our panel.
| Sudden cardiac arrhythmia | Genes |
|---|---|
| Long QT Syndrome (LQTS) | KCNQ1, KCNH2, SCN5A, ANK2, KCNE1, KCNE2, KCNJ2, CACNA1C, CAV3, SCN4B, AKAP9, SNTA1, KCNJ5 |
| Brugada Syndrome (BrS) | SCN5A, GPD1L, CACNA1C, CACNB2, SCN1B, KCNE3, SCN3B, KCND3, KCNJ8 |
| Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT) | RYR2, CASQ2, ANK2, KCNJ2, TRDN |
| WPW Syndrome | PRKAG2 |
| Short QT Syndrome (SQTS) | KCNH2, KCNJ2, CACNA1C, CACNB2, KCNQ1 |
| Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) | DSC2, DSG2, TMEM43, PKP2, DSP, RYR2, JUP, RPSA, TGFB3 |
| Sick sinus syndrome (SSS) | SCN5A, HCN4 |
| Cardiac conduction disease (CCD) | SCN5A |
| Hypertrophic Cardiomyopathy (HCM) | MYH7, MYBPC3, TNNT2, TNNI3, TPM1, ACTC1, MYL2, MYL3, MYH6, LAMP2, PRKAG2, CSRP3, GLA, VCL, TNNC1, TTR, TTN, TCAP, MYLK2, MYOZ2, MYO6 |
| Dilated cardiomyopathy (DCM) | DES, TNNT2, MYH7, CSRP3, PLN, TCAP, ABCC9, LMNA, DMD, TAZ, TTN, VCL, EYA4, EMD, ACTN2, SGCD, ACTC1, TNNI3, TPM1, MYBPC3, LDB3, MYH6, CTF1, ANKRD1 |
| Dilated cardiomyopathy with Cardiac conduction defect (DCM+CCD) | SCN5A, LMNA |
| Left ventricular noncompaction (LVNC) | LDB3, TAZ, MYH7, MYBPC3, ACTC1, TNNT2, SCN5A, LMNA, DMPK, DTNA, AMPD1, PMP22, LMX1B, YWHAE |
| Restrictive cardiomyopathy (RCM) | MYH7, TNNI3, TNNT2 |
| Atrial Fbrillation (AF) | KCNQ1, KCNE2, KCNJ2, KCNH2, SCN5A, KCNA5, NPPA, NUP155, GJA5, ABCC9, GATA4, GATA5, GATA6, KCNE1L |
Different types of sudden cardiac arrhythmia share the same pathogenic gene.
All the variants were filtered for candidate mutations based on: allele frequency in 1000G database; the functional mutation types; the predictive programs (SIFT, Polyphen2 and CONDEL software); related published literature. Briefly, allele frequency above 1% was considered to be a sequence polymorphisms and nonsense, frame shift, splice-site mutations were considered to be candidate pathogenic mutations unless identified as a polymorphism. All the filtered variants were then classified to five categories based on their clinical significance (pathogenic, likely pathogenic, uncertain, likely benign, and benign) according the ACMG Guidelines Revisions 2007 [15].
All the pathogenic and likely pathogenic mutation and some rare genetic variants in DSP gene were confirmed by Sanger Direct sequencing (the primer sequences and PCR conditions are in the table in Electronic Supplementary Material on line). The sequencing data were compared with the corresponding reference cDNA sequence of DSP gene (NM_004415.2) using SeqMan™II sequence analysis software (DNASTAR, Inc., Madison, WI, USA). These detected variants were also screened in 110 unrelated ethnically matched healthy controls.
Results
The investigation of the 80 candidate genes in both SUNDS victims and clinical patients yielded rich novel or reported mutations, rare variants, and polymorphisms. The additional cases, and the detailed epidemiological, morphological and clinical data are being accumulated to address the molecular pathological spectrum of Chinese SUNDS for the first time. Considering the independent role of DSP gene in SUNDS and the uncertain delay time of the whole study, we just aimed to report the new findings of DSP gene screening in SUNDS in this paper.
Among the total 58 cases, 10 DSP gene missense variants were identified in 11 cases (Fig 1 and Table 2), these variants were all absent in 110 healthy controls. Of the 10 variants, 7 were identified in SUNDS, 2 were identified in BrS and 1 in ERS. In these 11 cases with DSP variants, there were no reported pathogenic mutations detected in known LQTS, BrS, SQTS, CPVT and ARVC associated genes (Table 1). There were common polymorphisms found in these 11 cases in the screened 80 genes (which will be reported separately). Eight of these 10 variants were previously catalogued in international databases (NCBI and database of BGI) and 2 were novel (p.I125F and p.D521A). Of eight reported variants, two were previously considered pathogenic (p.Q90R and p.R2639Q), three were predicted in silico as pathogenic (p.R315C, p.E1357D and p.D2579H), and 3 were predicted as benign (p.N1234S, p.R1308Q and p.T2267S).
Figure 1. Structure of DSP gene and localization of variants.
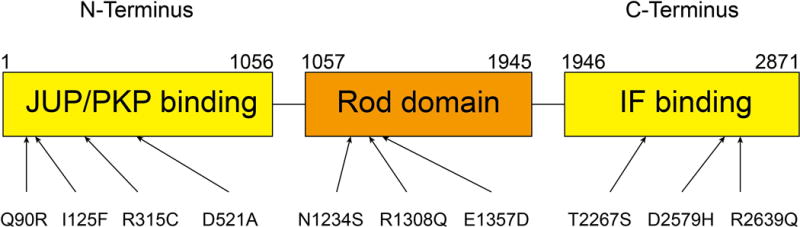
DSP is composed of 3 functional domains (JUP/PKP binding domain, rod domain and intermediate filament [IF] binding domain). Arrows indicate the positions of variants identified.
Table 2.
Variants of DSP gene in SUNDS, BrS and ERS
| Case no. | Exon | Loci* | Amino acid change | Disease | Age | Identity in dbSNP** | PVFD MAF*** | Prediction**** |
|---|---|---|---|---|---|---|---|---|
| E128 | 2 | c.269A>G | Q90R | SUNDS | 24 | rs188516326 | 0.0049 | Malignant |
| E111 | 3 | c.373A>T | I125F | SUNDS | 24 | novel | novel | Malignant |
| IPS | 8 | c.943C>T | R315C | BrS | 56 | rs200476515 | novel | Malignant |
| E5 | 12 | c.1562A>C | D521A | SUNDS | N/A | novel | novel | Benign |
| ZS105 | 23 | c.3701A>G | N1234S | BrS | 60 | rs185367490 | 0.0031 | Benign |
| E144 | 23 | c.3923G>A | R1308Q | SUNDS | 20 | rs184154918 | 0.0089 | Benign |
| E145 | 23 | c.4071G>C | E1357D | SUNDS | 33 | rs569786610 | 0.0004 | Malignant |
| ZS113 | 24 | c.6799A>T | T2267S | ERS | 44 | rs181378432 | 0.0027 | Benign |
| E100 | 24 | c.7735G>C | D2579H | SUNDS | 37 | novel | 0.0004 | Malignant |
| E101 | 24 | c.7916G>A | R2639Q | SUNDS | 31 | rs116888866 | 0.0018 | Malignant |
| A7600 | 24 | c.7916G>A | R2639Q | SUNDS | 33 | rs116888866 | 0.0018 | Malignant |
Nucleotide number relative to the translation start site.
Single Nucleotide Polymorphism database.
Minimum Allele Frequency in database of BGI
Prediction by SIFT, Polyphen2 and CONDEL.
One of the two pathogenic mutations, p.Q90R (rs188516326) is located in exon 5 of the DSP gene in SUNDS case E128. The substitution c.269A>G leads to a replacement of glutamine by arginine. The minimum allele frequency (MAF) was 0.01/0.005 (Han Chinese in Beijing/Southern Han Chinese, respectively, data from 1000 Genomes Project). The other variant p.R2639Q (rs116888866) is located in exon 24 of the DSP gene in SUNDS case E101 and SUNDS case A7600. The substitution c.7916G>A leads to a replacement of arginine by glutamine. The MAF was 0.015/0.019 (Han Chinese in Beijing/Southern Han Chinese, respectively, data from 1000 Genomes Project).
Six more variants were reported previously in public databases but with unknown disease effect. The first three were p.N1234S (rs185367490), p.R1308Q (rs185367490) and p.E1357D (rs569786610), the three variants all located in exon 23 with c.3701A>G, c.3923G>A and c.4071G>C substitution respectively. The variants p.N1234S and p.R1308Q were predicted in silico to be benign and p.E1357D was predicted to be malignant.
The other three DSP variants were p.R315C (rs200476515), p.T2267S (rs181378432) and p.D2579H. The p.R315C variant has a nucleotide change of C-T in the position 943 and is located in exon 8. The p.T2267S and p.D2579H variant had nucleotide changes of A-T in the position 6679 and G-C in the position of 7735 respectively, both located in exon 24. The variants p.R315C and p. D2579H were predicted to be malignant and p.T2267S was predicted to be benign. Interestingly, p.R315C (rs200476515) was novel in database of BGI while p.D2579H with MAF 0.0004 in the database of BGI was absent in dbSNP, neither was considered to be novel in this study.
Finally, two novel variants p.I125F and p.D521A were located in exon 3 and exon 12 respectively. The variant p.I125F has an A-T nucleotide change in the position 373 and was predicted in silico to be malignant. The variant p.D521A has an A-C nucleotide change in the position of 1562 and was predicted to be benign.
Discussion
In this study, we focused our genetic analysis on the DSP gene coding desmoplakin in the desmosome. Desmosomes are transmembrane multiprotein complexes that provide important intercellular structural integrity. Desmoplakin is a component of desmosome that acts as an intracellular link between other desmosomal components and intermediate filaments [16]. Mutations in desmosomal components including DSP are associated with arrhythmogenic cardiomyopathy (AC) [17, 18], an inherited disorder that comprises both an arrhythmia component and structural features.
In case E101and A7600, a rare SNP c.7916G>A at codon 2639 in exon 24 of DSP was identified, leading to replacement of arginine with glutamine (R2639Q). Yu et al. [19] reported the same mutation in a patient who did not at the time meet the diagnostic criteria for ARVC but had sustained ventricular tachycardia on an ECG with the patient experiencing palpitations. They concluded the patient was very likely to have ARVC because of severe dilatation and dysfunction of the right ventricle revealed by the subsequent cardiac MRI. Sato et al. also reported the same mutation in a sudden death case, the decedent, an 18-year-old man with no previous medical history, died suddenly during a class at school [20].
The Q90R variation has been identified in previous studies [21–23]. Yang et al. found the N-terminal missense mutation of DSP Q90R affected the normal localization of DSP to cell membranes in vitro, probably due to loss of binding to the desmosomal protein JUP. In addition, overexpression of the Q90R mutant in embryonic mouse heart lead to embryonic lethality [21].
At autopsy no histopathologic features of AC were detected in our SUNDS cases. In the early stage of AC sudden cardiac death can occur due to ventricular fibrillation, even in the absence of overt structural defect. This early concealed phase of AC is characterized by a propensity to ventricular arrhythmia and SCD in the setting of well-preserved morphology, histology, and ventricular function [16]. Previous findings suggest that Cx43 reduction associated electrophysiological abnormalities may appear prior to fibrosis in the myocardium of AC patients with DSP gene mutations[24], consistent with the finding in the cardiomyocytes from mice with knockout of DSP (DSP-cKO) [25]. These findings suggest that the histopathology changes are not necessary to generate arrhythmia, while less is known about the nature of the arrhythmogenic substrate during the concealed phase of ARVC [25].
The desmosome is a specialized adhesive junction that participates in crosstalk with gap junctions. Mutations in components of the desmosome (including desmoplakin) underlie AC. The gap junction channel is composed of 2 connexons and each connexon consists of 6 connexins. Cx43 is the most abundant connexin isotype in heart. If gap junction channels are not present, normal propagation is disrupted and lethal arrhythmias ensue [26, 27]. Lyon et al. found that in the DSP-cKO mice, the gap junction at the intercalated disks in cardiac ventricular cells is lost with the loss of desmoplakin and dose-dependent loss of desmoplakin directly affects the expression and phosphorylation of Cx43 [25]. Zhang et al. effectively demonstrates that loss of DSP gene leads to the down-expression and redistribution of the Nav1.5 protein and decreased DSP expression results in the decrease of sodium current as well as the slower of conduction velocity in cultured DSP-depleted HL-1 cells [8]. Further, Patel et al. showed that DSP regulates the EB1-base targeted delivery of Cx43 to the membrane, and that this delivery does not occur when DSP gene mutants exist in the N terminus hot-spot [28]. Jansen showed in inducible Cx43 knockout mice with arrhythmia, the heterogeneous expression of Cx43 was concomitant with heterogeneous expression of Nav1.5, and patch-clamp experiments in isolated adult rat ventricular myocytes showed that the loss of Cx43 expression led to a significant reduction in the sodium current amplitude [29]. These studies suggest that the loss of DSP brings out the heterogeneous and combined misregulation of Cx43 and Nav1.5, and lead to slowed and dispersed conduction, which sensitizes the heart for ventricular arrhythmia.
Although the functional effects of the mutations we report here are not yet known, these known effects of loss of DSP function suggest the plausibility or pathogenicity for the clinical syndrome of SUNDS and BrS. Additional data supporting the possible pathogenicity of these mutations include their absence from healthy controls and very low frequency in on-line databases, and some of them have been found to be pathogenic previously. For our forensic investigations, meticulous gross autopsy and pathological detection was important to establish the cohort of negative autopsy cases. However, the absence of clinical data and family history often made it difficult to determine the cause of death. Postmortem genetic analysis can be a powerful technique [30,31] that combined with the traditional forensic investigation may contribute to finding the underlying causes for SUNDS.
Conclusion
In summary, we identified 10 DSP gene variants in 11 cases (8 SUNDS victims, 2 BrS patients, and 1 ERS patient) out of 70 postmortem or clinical cases. No known pathogenic mutation or novel variants associated with LQTS, BrS, SQTS and CPVT were detected in the 11 cases that might otherwise account for their clinical presentation. Considering the low frequency in population, the absence in control, the presence in sudden cardiac death and the known regulation of DSP on Cx43&Nav1.5, we deduced that the variants in DSP gene identified in this study are genetic causes of some SUNDS cases. Ongoing electrophysiological investigations may determine whether they affect function and confirm and support their role as pathogenic variants by establishing the biophysical mechanism by which they cause SUNDS and BrS.
Supplementary Material
Acknowledgments
None.
Sources of funding
This work was supported by the Key Program (81430046), General Program (81172901) from National Natural Science Foundation of China (to JC), and the grants R56 HL71092 &R01HL128076-01from National Institutes of Health of United states of American (to JCM).
Footnotes
Conflict of interest
None.
Contributor Information
Qianhao Zhao, Department of Forensic Pathology, Zhongshan School of Medicine, Sun Yat-sen University, No. 74, Zhongshan 2nd Road, Guangzhou, Guangdong 510080, China. Tel.: +86 20 87330704; fax: +86 20 87334353.
Yili Chen, Department of Cardiology, The First Affiliated Hospital of Sun Yat-Sen University, Guangzhou, 510080, China.
Longlun Peng, Department of Cardiology, The First Affiliated Hospital of Sun Yat-Sen University, Guangzhou, 510080, China.
Rui Gao, BGI-Shenzhen, Shenzhen, 518083, China.
Nian Liu, Department of Cardiology, Beijing Anzhen Hospital, Capital Medical University, Beijing100029, China.
Pingping Jiang, BGI-Shenzhen, Shenzhen, 518083, China.
Chao Liu, BGI-Shenzhen, Shenzhen, 518083, China.
Shuangbo Tang, Department of Forensic Pathology, Zhongshan School of Medicine, Sun Yat-sen University, No. 74, Zhongshan 2nd Road, Guangzhou, Guangdong 510080, China. Tel.: +86 20 87330704; fax: +86 20 87334353.
Li Quan, Email: quanli@mail.sysu.edu.cn, Department of Forensic Pathology, Zhongshan School of Medicine, Sun Yat-sen University, No. 74, Zhongshan 2nd Road, Guangzhou, Guangdong 510080, China. Tel.: +86 20 87330704; fax: +86 20 87334353.
Jonathan C. Makielski, Email: jcm@medicine.wisc.edu, Division of Cardiovascular Medicine, Department of Medicine, University of Wisconsin, Madison, WI 53792, USA.
Jianding Cheng, Email: chengjd@mail.sysu.edu.cn, Department of Forensic Pathology, Zhongshan School of Medicine, Sun Yat-sen University, No. 74, Zhongshan 2nd Road, Guangzhou, Guangdong 510080, China. Tel.: +86 20 87330704; fax: +86 20 87334353.
References
- 1.Liu C, Zhao Q, Su T, Tang S, Lv G, Liu H, et al. Postmortem molecular analysis of KCNQ1, KCNH2, KCNE1 and KCNE2 genes in sudden unexplained nocturnal death syndrome in the Chinese Han population. Forensic Sci Int. 2013;231:82–7. doi: 10.1016/j.forsciint.2013.04.020. [DOI] [PubMed] [Google Scholar]
- 2.Liu C, Tester DJ, Hou Y, Wang W, Lv G, Ackerman MJ, et al. Is sudden unexplained nocturnal death syndrome in Southern China a cardiac sodium channel dysfunction disorder? Forensic Sci Int. 2014;236:38–45. doi: 10.1016/j.forsciint.2013.12.033. [DOI] [PubMed] [Google Scholar]
- 3.Huang L, Liu C, Tang S, Su T, Cheng J. Postmortem genetic screening of SNPs in RyR2 gene in sudden unexplained nocturnal death syndrome in the southern Chinese Han population. Forensic Sci Int. 2014;235:14–8. doi: 10.1016/j.forsciint.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 4.Elger BS, Michaud K, Fellmann F, Mangin P. Sudden death: ethical and legal problems of post-mortem forensic genetic testing for hereditary cardiac diseases. Clin Genet. 2010;77:287–92. doi: 10.1111/j.1399-0004.2009.01293.x. [DOI] [PubMed] [Google Scholar]
- 5.Sheikh F, Ross RS, Chen J. Cell-cell connection to cardiac disease. Trends Cardiovasc Med. 2009;19:182–90. doi: 10.1016/j.tcm.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rampazzo A, Nava A, Malacrida S, Beffagna G, Bauce B, Rossi V, et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2002;71:1200–6. doi: 10.1086/344208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Zwaag PA, Jongbloed JD, van den Berg MP, van der Smagt JJ, Jongbloed R, Bikker H, et al. A genetic variants database for arrhythmogenic right ventricular dysplasia/cardiomyopathy. Hum Mutat. 2009;30:1278–83. doi: 10.1002/humu.21064. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Q, Deng C, Rao F, Modi RM, Zhu J, Liu X, et al. Silencing of desmoplakin decreases connexin43/Nav1.5 expression and sodium current in HL1 cardiomyocytes. Mol Med Rep. 2013;8:780–6. doi: 10.3892/mmr.2013.1594. [DOI] [PubMed] [Google Scholar]
- 9.Cheng J, Makielski JC, Yuan P, Shi N, Zhou F, Ye B, et al. Sudden unexplained nocturnal death syndrome in Southern China: an epidemiological survey and SCN5A gene screening. The American journal of forensic medicine and pathology. 2011;32:359–63. doi: 10.1097/PAF.0b013e3181d03d02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.BioTechniques Protocol Guide. Biotechniques; New York, NY: 2006. Illumina Protocol for Whole Genome Sequencing using SBS Technology. [Google Scholar]
- 11.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;11:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richards CS, Bale S, Bellissimo DB, Das S, Grody WW, Hegde MR, et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med. 2008;10:294–300. doi: 10.1097/GIM.0b013e31816b5cae. [DOI] [PubMed] [Google Scholar]
- 16.Lapouge K, Fontao L, Champliaud MF, Jaunin F, Frias MA, Favre B, et al. New insights into the molecular basis of desmoplakin- and desmin-related cardiomyopathies. J Cell Sci. 2006;119:4974–85. doi: 10.1242/jcs.03255. [DOI] [PubMed] [Google Scholar]
- 17.Delmar M, McKenna WJ. The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res. 2010;107:700–14. doi: 10.1161/CIRCRESAHA.110.223412. [DOI] [PubMed] [Google Scholar]
- 18.Basso C, Bauce B, Corrado D, Thiene G. Pathophysiology of arrhythmogenic cardiomyopathy. Nat Rev Cardiol. 2012;9:223–33. [Google Scholar]
- 19.Yu CC, Yu CH, Hsueh CH, Yang CT, Juang JM, Hwang JJ, et al. Arrhythmogenic right ventricular dysplasia: clinical characteristics and identification of novel desmosome gene mutations. J Formos Med Assoc. 2008;107:548–58. doi: 10.1016/S0929-6646(08)60168-0. [DOI] [PubMed] [Google Scholar]
- 20.Sato T, Nishio H, Suzuki K. Identification of arrhythmogenic right ventricular cardiomyopathy-causing gene mutations in young sudden unexpected death autopsy cases. J Forensic Sci. 2015;60:457–61. doi: 10.1111/1556-4029.12657. [DOI] [PubMed] [Google Scholar]
- 21.Yang Z, Bowles NE, Scherer SE, Taylor MD, Kearney DL, Ge S, et al. Desmosomal dysfunction due to mutations in desmoplakin causes arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Res. 2006;99:646–55. doi: 10.1161/01.RES.0000241482.19382.c6. [DOI] [PubMed] [Google Scholar]
- 22.Xu T, Yang Z, Vatta M, Rampazzo A, Beffagna G, Pilichou K, et al. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2010;55:587–97. doi: 10.1016/j.jacc.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cox MG, van der Zwaag PA, van der Werf C, van der Smagt JJ, Noorman M, Bhuiyan ZA, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: pathogenic desmosome mutations in index-patients predict outcome of family screening: Dutch arrhythmogenic right ventricular dysplasia/cardiomyopathy genotype-phenotype follow-up study. Circulation. 2011;123:2690–700. doi: 10.1161/CIRCULATIONAHA.110.988287. [DOI] [PubMed] [Google Scholar]
- 24.Gomes J, Finlay M, Ahmed AK, Ciaccio EJ, Asimaki A, Saffitz JE, et al. Electrophysiological abnormalities precede overt structural changes in arrhythmogenic right ventricular cardiomyopathy due to mutations in desmoplakin-A combined murine and human study. Eur Heart J. 2012;33:1942–53. doi: 10.1093/eurheartj/ehr472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lyon RC, Mezzano V, Wright AT, Pfeiffer E, Chuang J, Banares K, et al. Connexin defects underlie arrhythmogenic right ventricular cardiomyopathy in a novel mouse model. Hum Mol Genet. 2014;23:1134–50. doi: 10.1093/hmg/ddt508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gutstein DE, Morley GE, Tamaddon H, Vaidya D, Schneider MD, Chen J, et al. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ Res. 2001;88:333–9. doi: 10.1161/01.res.88.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gutstein DE, Morley GE, Vaidya D, Liu F, Chen FL, Stuhlmann H, et al. Heterogeneous expression of Gap junction channels in the heart leads to conduction defects and ventricular dysfunction. Circulation. 2001;104:1194–9. doi: 10.1161/hc3601.093990. [DOI] [PubMed] [Google Scholar]
- 28.Patel DM, Dubash AD, Kreitzer G, Green KJ. Disease mutations in desmoplakin inhibit Cx43 membrane targeting mediated by desmoplakin-EB1 interactions. J Cell Biol. 2014;206:779–97. doi: 10.1083/jcb.201312110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jansen JA, Noorman M, Musa H, Stein M, de Jong S, van der Nagel R, et al. Reduced heterogeneous expression of Cx43 results in decreased Nav1.5 expression and reduced sodium current that accounts for arrhythmia vulnerability in conditional Cx43 knockout mice. Heart Rhythm. 2012;9:600–7. doi: 10.1016/j.hrthm.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hertz CL, Christiansen SL, Ferrero-Miliani L, Dahl M, Weeke PE, LuCamp, et al. Next-generation sequencing of100 candidate genes in young victims of suspected sudden cardiac death withstructural abnormalities of the heart. Int J Legal Med. 2015 doi: 10.1007/s00414-015-1261-8. [DOI] [PubMed] [Google Scholar]
- 31.Alcalde M, Campuzano O, Allegue C, Torres M, Arbelo E, Partemi S, et al. Sequenom MassARRAY approach in thearrhythmogenic right ventricular cardiomyopathy post-mortem setting: clinical andforensic implications. Int J Legal Med. 2015;129:1–10. doi: 10.1007/s00414-014-0996-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.