

ABSTRACT
During embryogenesis, the epicardium undergoes proliferation, migration, and differentiation into several cardiac cell types which contribute to the coronary vessels. The type III transforming growth factor-β receptor (TGFβR3) is required for epicardial cell invasion and development of coronary vasculature in vivo. Bone Morphogenic Protein-2 (BMP2) is a driver of epicardial cell migration. Utilizing a primary epicardial cell line derived from Tgfbr3+/+ and Tgfbr3−/− mouse embryos, we show that Tgfbr3−/− epicardial cells are deficient in BMP2 mRNA expression. Tgfbr3−/− epicardial cells are deficient in 2-dimensional migration relative to Tgfbr3+/+ cells; BMP2 induces cellular migration to Tgfbr3+/+ levels without affecting proliferation. We further demonstrate that Src kinase activity is required for BMP2 driven Tgfbr3−/− migration. BMP2 also requires Src for filamentous actin polymerization in Tgfbr3−/− epicardial cells. Taken together, our data identifies a novel pathway in epicardial cell migration required for development of the coronary vessels.
KEYWORDS: BMP2, epicardium, migration, Src, TGFβR3
Introduction
The coronary vasculature is required for proper development and function of the heart. In embryonic development, the coronary vessels arise from a tissue known as the proepicardium.1 Cells from the proepicardium are transferred to the surface of the heart to form the epicardium that contribute to the coronary vessels. The epicardium undergoes migration, differentiation and cell invasion into the primitive myocardium. The epicardium forms an epithelial sheet that covers the myocardium, and secreted growth factors from the myocardium stimulate synthesis of extracellular matrix molecules including hyaluronan (HA) in the subepicardial space.2,3 Epicardially derived cells differentiate into cardiac fibroblasts resident in the myocardium that contribute to repair, and vascular smooth muscle cells that constitute the mature coronary vessels.4 As cell migration is required for heart development, perturbations in this process can lead to congenital defects or adult coronary artery disease, the leading cause of lethality in the United States.5
The TGFβ family of growth factors and receptors are characterized in a wide range of cellular processes including regulating proliferation, migration and cell differentiation in both cardiovascular development and disease.6,7 TGFβ2 binds TGFβR3 at picomolar affinity,28 and TGFβR3 required for TGFβ2 and TGFβ1 induced epicardial cell invasion.19 The Type III TGFβ receptor (TGFβR3) has no intrinsic catalytic activity and is most well characterized to function in TGFβ ligand presentation to Type I and Type II TGFβ receptors which drive epicardial cell invasion.8,9 Upon TGFβR3 ligand binding, Type I and II receptors engage in canonical (Smad-dependent) signal transduction, but also activate several non-canonical (Smad-independent) signaling pathways such as MAP kinase, Rho GTPase, and Src tyrosine kinase cascades.10 Tgfbr3−/− phenotype is lethal at E14.5 as a result of inhibited development of the coronary vasculature.11 Tgfbr3−/− epicardial cells retain the ability to migrate to and attach to the myocardium, but fail to undergo cell invasion, leading to inhibited coronary vessel formation. In vitro, Tgfbr3−/− epicardial cells fail to undergo cellular invasion (migration into, and residence in a 3D matrix), and exhibit delayed migration (migration in 2-dimensions) relative to wild-type.19
The Bone Morphogenic Proteins (BMPs) are members of the TGFβ super family of ligands and receptors. BMPs have a wide variety of roles in embryonic tissues including inducing proliferation, migration and differentiation of developing tissues in organogenesis.12-14 The role of BMPs in endocardial cell transformation have been heavily investigated revealing a requirement for TGFβR3 in valve development.15-17 TGFβ1, BMP7 and BMP2, directly bind to TGFβR3.34 TGFβR3 is required for BMP2 activation of Smad1 (canonical signaling) and BMP2-dependent endocardial cell transformation.34 It has been shown that BMPs function as directional signals in the attachment of the epicardium to the myocardium.18 In this regard, Tgfbr3−/− epicardial cells fail to undergo 3-dimensional cell invasion in vitro when stimulated with BMP2.8 Tgfbr3−/− cells exhibit deficient 2-dimensional migration relative to Tgfbr3+/+ under unstimulated conditions in a wound healing model of cell motility.19
Although, BMP2 and TGFβ receptor signaling in epicardial cells requires the canonical TGFβ signaling effector Smad4,8 the requirement of non-canonical BMP effectors are not well studied in the epicardium. Src is a ubiquitously expressed non-receptor tyrosine kinase that is characterized as a driver of cell motility in many cell systems.20 Src is activated via Type I TGFβ receptor dependent pathway by TGFβ1 and BMP2.21,22 The Type III TGFβ receptor is directly upstream of Src activity 23 BMP2 is a modulator of migration in many cell systems,24,25 whether BMP2 and Src are required for epicardial cell migration is unknown.
In order to investigate BMP2 in coronary progenitor cell biology, we used Tgfbr3−/− epicardial cells. These cells fail to express BMP2, and as such provides a unique cell model to decipher the function of BMP2 and TGFβR3. The addition of exogenous BMP2 to Tgfbr3−/− cells rescues deficient cell migration in the wound healing assay. We revealed that TGFβR3 is not required for BMP2-induced migration. Finally, BMP2 stimulation induces filamentous actin polymerization in epicardial cells, and that Src kinase is required for BMP2 induced filamentous actin polymerization and epicardial cell migration.
Results
Differential expression of pro-migration genes in Tgfbr3+/+ and Tgfbr3−/− cells
Detection of select genes known to promote epicardial cell invasion and migration was performed by RT-PCR analysis with RNA isolated from Tgfbr3+/+ and Tgfbr3−/− epicardial cells. An up-regulation of molecules related to the Type III TGFβ receptor was detected in Tgfbr3−/− epicardial cells relative to Tgfbr3+/+ expression (Figure 1). TGFβ2 binds TGFβR3 at picomolar affinity, making TGFβ2 its highest affinity ligand binding partner.28 Expression of TGFβ2, but not TGFβ1, is significantly up-regulated in Tgfbr3−/− cells relative to Tgfbr3+/+ ; enhanced expression of TGFβ2 in vitro is likely compensatory for loss of TGFβR3, though not significant to direct cell behavior. Hyaluronan Synthase 2 (Has2) synthesizes the glycosaminoglycan hyaluronan (HA), an important component of the extracellular matrix required for cardiac development and epicardial migration and invasion.29,30 mRNA expression of Has2 is 4.5-fold higher in Tgfbr3−/− cells relative to Tgfbr3+/+. This further outlines TGFβR3 related compensation, as TGFβR3 is required for both TGFβ2 and HMWHA stimulated epicardial cell invasion.31 Markers of cell differentiation, mesenchymal (vimentin) and vascular smooth muscle (SM22α) are both significantly reduced in Tgfbr3−/− cells. BMP2 mRNA is not detected in Tgfbr3−/− epicardial cells, though it is not known if TGFβR3 drives BMP2 expression in epicardial cells. Deficient expression of these genes (SM22α, vimentin, and BMP2) could pre-dispose Tgfbr3−/− epicardial cells to be less sensitive to differentiation and cell migration. Since Tgfbr3−/− epicardial cells lack BMP2, we wanted to understand the role of BMP2 in 2-dimensional epicardial cell migration
Figure 1.
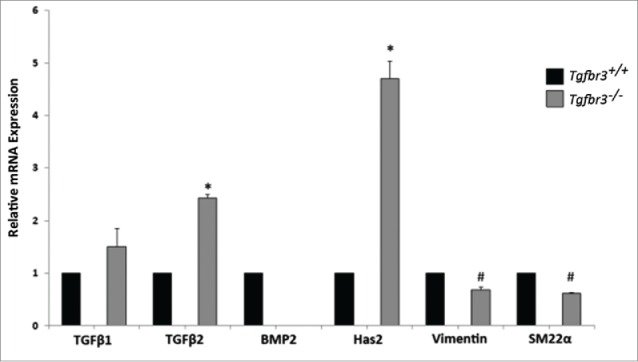
TGFβ2, BMP2, and Has2 are differentially expressed in Tgfbr3−/− cells. RNA isolated from Wild-type (black bars) and Tgfbr3−/− (gray bars) epicardial cells was analyzed by RT-PCR for mRNA expression of molecules known to drive migration. (#=p < 0.005, *=p < 0.0005).
BMP2 rescues Tgfbr3−/− deficient migration
Tgfbr3−/− epicardial cells are non-invasive in response to BMP2 in a 3 dimensional invasion assay,8 we evaluated the effect of BMP2 on 2-dimensional migration. Tgfbr3+/+ and Tgfbr3−/− cells grown to confluence were subjected to the wound healing assay for 24 hours in the presence, or absence of BMP2. Unstimulated Tgfbr3+/+ epicardial cells at 24 hours fill in the wound by 82% (Figure 2 A-B, I). In contrast, there is only 55% wound closure by Tgfbr3−/− epicardial cells at 24 hours (Figure 2C-D, I). Thus, Tgfbr3−/− epicardial cells are deficient in 2-dimensional cell migration as observed in the wound healing assay consistent with previously reported results.19 As BMP2 is shown to stimulate expansion and directional 2-dimensional migration of the avian epicardium,18 and Tgfbr3−/− epicardial cells lack BMP2 mRNA, we investigated whether the addition of exogenous recombinant BMP2 could rescue deficient Tgfbr3−/− epicardial cell migration. BMP2 did not superimpose an increased migration effect in Tgfbr3+/+ cells (Figure 2E-F, I). In contrast, BMP2-induced migration of Tgfbr3−/− epicardial cells to a level comparable to unstimulated Tgfbr3+/+ epicardial cells (Figure 2G-H, I). Migration of Tgfbr3−/− epicardial cells were not statistically significantly different from that of unstimulated or BMP2-stimulated Tgfbr3+/+ epicardial cells (Figure 2I). Thus, TGFβR3 is required for epicardial migration in the wound healing model of motility, and BMP2 is sufficient to rescue the Tgfbr3−/− deficit.
Figure 2.
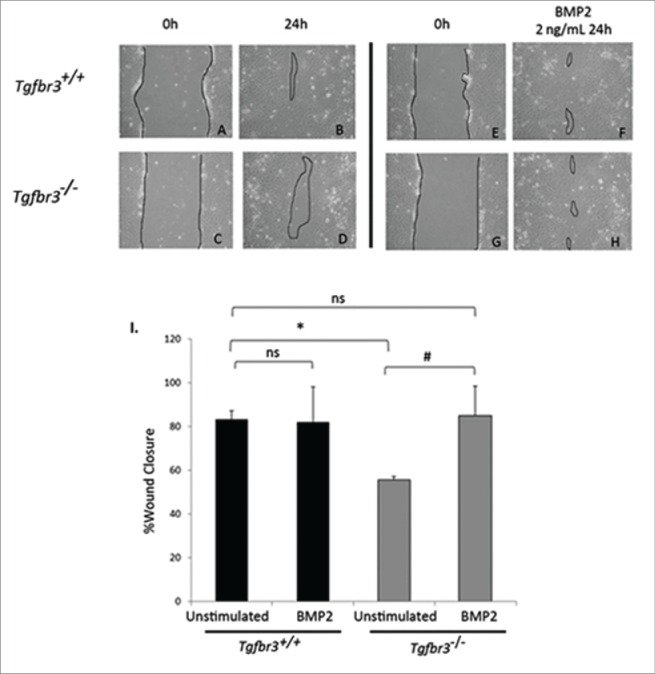
BMP2 rescues Tgfbr3−/− deficient cell migration. Wild-type (A-B, E-F) and Tgfbr3−/− (C-D, G-H) epicardial cells allowed to migrate for 24 hours in the presence (E-H) or absence (A-D) of BMP2 (2 ng/mL) in the wound healing assay. (*=p < 0.0005, #=p < 0.05).
Epicardially secreted BMPs do not drive migration
We have shown that BMP2 can restore Tgfbr3−/− epicardial cell migration in the wound healing assay, which lack endogenous BMP2 expression. A role for epicardially secreted BMPs in driving epicardial wound healing response in Tgfbr3+/+ cells was tested by neutralizing the action of epicardially secreted BMPs. Tgfbr3+/+ and Tgfbr3−/− epicardial cells were grown to confluence, and subjected to the wound healing assay for 24 hours in the presence or absence of 200 ng/mL Noggin, a potent sequester and inactivator of BMP ligands.32 As previously shown, unstimulated Tgfbr3−/− cells have a 20% deficiency in cell migration relative to Tgfbr3+/+ (Figure 3A-D, I). Inactivation of BMP ligands by Noggin in this assay has no statistically significant effect on migration of Tgfbr3+/+ or Tgfbr3−/− epicardial cells (Figure 3E–I). Thus, epicardially secreted BMPs in vitro do not contribute to epicardial migration.
Figure 3.
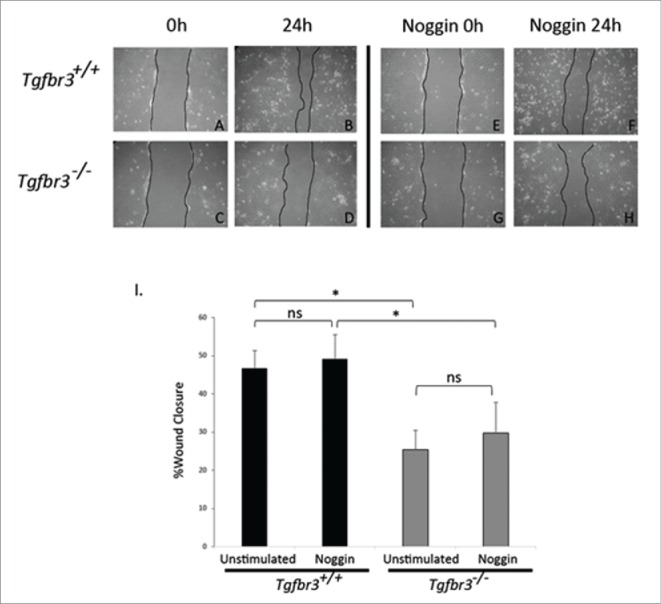
Endogenous BMPs are not required for epicardial cell migration. Wild-type (A-B, E-F) and Tgfbr3−/− (C-D, G-H) epicardial cells were subjected to the wound healing assay, and allowed to migrate for 24 hours in the presence (E-H) or absence (A-D) of Noggin (200 ng/mL). (*=p < 0.05).
Src is required for BMP2 induction of Tgfbr3−/− epicardial cell migration
In order to evaluate the role of Src kinase in BMP2-stimulated Tgfbr3−/− cell migration, an inhibitor of Src kinase activity, PP2, was used in the wound healing assay. Tgfbr3−/− epicardial cells were pre-incubated with PP2, the wound was made and cells were subsequently stimulated with BMP2 (2 ng/mL) for 24 hours in the continued presence of PP2. Unstimulated Tgfbr3−/− epicardial cells fill 40% of the wound area (Figs. 4A, B, I). Tgfbr3−/− cells stimulated with BMP2 increase 2-dimensional migration to fill 60% of the wound area (Figs. 4C, D, I). In the presence of PP2, BMP2-stimulated migration of Tgfbr3−/− cells is completely blocked (Figs. 4G, H, I). Migration of Tgfbr3−/− cells in the presence of PP2 is slightly reduced from that of unstimulated control. These results indicate BMP2-stimulated epicardial cell migration is dependent on Src.
Figure 4.
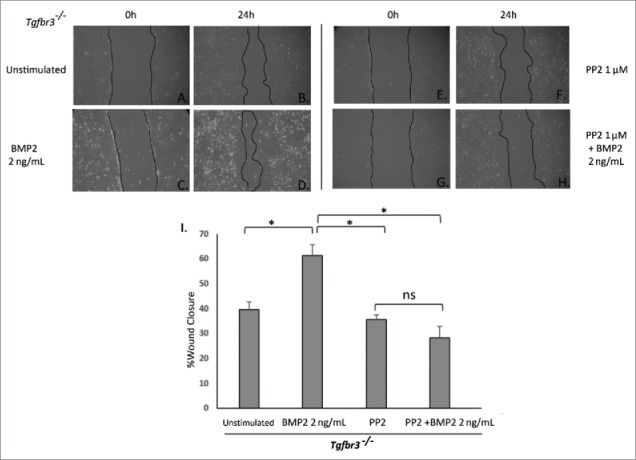
Src is required for BMP2-stimulated Tgbr3−/− epicardial cell migration. Tgfbr3−/− epicardial cells were subjected to the wound healing assay and allowed to close the wound for 24 hours in the presence (C-D) or absence (A-B) of BMP2 (2 ng/mL). Tgfbr3−/− epicardial cells were subjected to the wound healing assay and allowed to migrate for 24 hours in the presence of 1 µM PP2 with (G-H) or without (E-F) BMP2 (2 ng/mL).( *=p < 0.05).
Src is required for BMP2 induction of filamentous actin polymerization
We investigated the roles of BMP2 and Src kinase functioning in filamentous actin dynamics required for cell migration.33 Tgfbr3+/+ and Tgfbr3−/− epicardial cells were stimulated with BMP2 (2 ng/mL) in the presence or absence of PP2 for 1 hour. Phalloidin staining to visualize actin fibers was performed, and the f/g actin assay for relative quantification of filamentous actin polymerization in whole cell lysates. Tgfbr3+/+ epicardial cells stimulated with BMP2 induce filamentous actin polymerization, and f/g actin ratios of 2.1 relative to untreated control (Figure 5A-B, I). Tgfbr3−/− cells under unstimulated conditions have a high basal levels of filamentous actin polymerization (Figure 5C-D), and under BMP2 stimulation induce polymerization of f-actin stress fibers and f/g ratios of 2.1 relative to untreated control (Figure 5I). When stimulated with BMP2 in the presence of PP2, formation of filamentous actin in Tgfbr3+/+ and Tgfbr3−/− epicardial cells is severely blocked (Fig 5F, H, I). f/g actin ratios of cells stimulated with BMP2 and PP2 were reduced in Tgfbr3+/+ (0.87) and Tgfbr3−/− (1.47) epicardial cells. Tgfbr3−/− cells are less affected, but this may be due to high basal f-actin levels in cells. These data show BMP2 is an inducer of f-actin polymerization in epicardial cells, independent of TGFβR3 and dependent on Src kinase activity.
Figure 5.
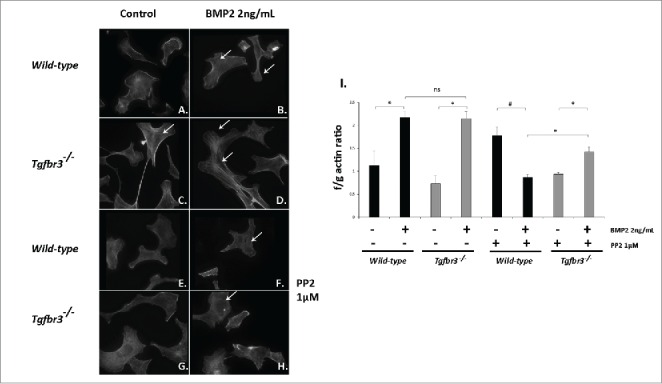
BMP2 induces filamentous actin polymerization in Tgfbr3−/− epicardial cells and requires Src. (A-H) Wild-type and Tgfbr3−/− epicardial cells were stimulated with BMP2 (2 ng/mL) in the presence or absence of 1µM PP2 and subject to phalloidin staining (A-H) and the f/g actin assay (I). #=p < 0.005, *=p < 0.05.
BMP2 does not affect epicardial cell proliferation
BMP2 is sufficient to drive migration in a wound healing model of cell motility in Tgfbr3−/−, but not Tgfbr3+/+ epicardial cells (Figure 2). In order to assess whether this was an artifact of enhanced cell proliferation rather than purely an effect of enhanced migration, the MTT proliferation assay was performed under identical experimental conditions as wound healing assays. Tgfbr3−/− epicardial cells are hypo-proliferative relative to Tgfbr3+/+ cells (Figure 6). Addition of BMP2 for 24 hours did not enhance or inhibit cell proliferation in Tgfbr3+/+ or Tgfbr3−/− epicardial cells. Proliferation rate of Tgfbr3−/− cells in the presence of BMP2 was not statistically significantly different from that of untreated Tgfbr3−/− epicardial cells. This highlights an important aspect of this study in that BMP2 is a pro-migratory signal in mouse epicardial cells that has no effect on proliferation. Thus, we can say with confidence that BMP2 rescue of Tgfbr3−/− deficient migration is not the result of enhanced proliferation.
Figure 6.
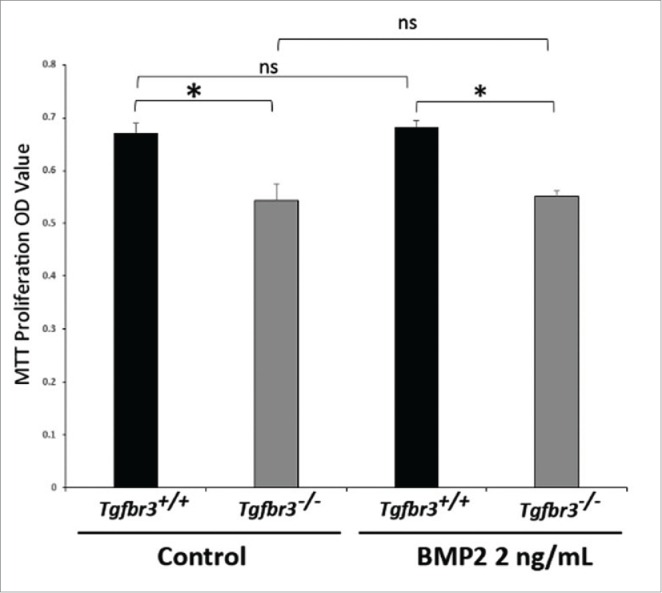
BMP2 does not affect proliferation of epicardial cells. Wild-type and Tgfbr3−/− epicardial cells were stimulated with BMP2 (2 ng/mL) for 24 hours. MTT Proliferation Assay was performed (*=p < 0.005).
Discussion
The development of the coronary vasculature requires migration, invasion, and differentiation of the epicardium. This process is tightly regulated by growth factors influences from the neighboring myocardium to initiate epicardial contact and migration over the developing heart. In this study, we evaluate the role of TGFβR3, BMP2, and Src kinase as a novel pathway in epicardial cell migration required for early developmental events of coronary genesis.
The Tgfbr3−/− phenotype is lethal due to decreased epicardially derived cells resident in the myocardium and therefore inhibiting proper coronary vessel formation.11 In culture, Tgfbr3−/− cells are deficient in in vitro 3-dimensional cell invasion in the presence of BMP2 relative to Tgfbr3+/+. We demonstrate Tgfbr3−/− cells execute 2-dimensional migration in a wound healing model of motility to levels of Tgfbr3+/+ cells when stimulated with BMP2 without affecting cell proliferation (Figs. 2, 6). Though delayed of unstimulated migration of Tgfbr3−/− cells may partly be due to low proliferation, enhancing migration of Tgfbr3−/− cells by BMP2 in vitro is not through increased proliferation (Figs. 2 and 6). Tgfbr3+/+ epicardial migration is not enhanced by addition of BMP2. We postulate TGFβR3 dependent signal transduction pathways required for the wound healing response are saturated in Tgfbr3+/+ cells, and addition of BMP2 would not enhance this response. Conversely, Tgfbr3−/− cells are deficient in these TGFβR3 specific wound healing signaling pathways, but addition of BMP rescues this deficiency independent of TGFβR3. This is reflective of the in vivo phenotype in that the epicardium still migrates to, attaches, and covers the myocardium in Tgfbr3−/− embryos. Thus, BMP2 secretion from the myocardium is likely to be retained in Tgfbr3−/− embryos, and be adequate to instruct epicardial migration, but not cell invasion, to Tgfbr3−/− epicardial cells. Analysis of mRNA expression of Tgfbr3+/+ and Tgfbr3−/− epicardial cells demonstrate that types I and II TGFβ receptors, ALKs 1,2,3,4,5,6, expression is maintained at the same levels in Tgfbr3−/− cells relative to Tgfbr3+/+.8 Retention of these BMP2 responsive TGFβ superfamily receptors must be sufficient to drive epicardial migration in a TGFβR3-independent fashion. This highlights a non-redundant role for Tgfbr3−/− in directing cell invasion, but not migration, in coronary vessel development.
Exploring the role of BMP2 in epicardial cell migration was a logical avenue to address, as Tgfbr3−/− cells lack BMP2 mRNA (Figure 1). Tgfbr3−/− cells also revealed increased expression of TGFβ2 and Has2 relative to wild-type, inducers of epicardial cell invasion in a TGFβR3 dependent fashion. Furthermore, Tgfbr3−/− cells and decreased unstimulated SM22α and Vimentin expression. The importance of these expression disparities in Tgfbr3−/− cell behavior are unclear, and global gene expression analysis is required to fully understand this phenotype. Tgfbr3−/− cells are deficient in 2-dimensional migration relative to Tgfbr3+/+ in a wound healing model of cell motility (Figure 2). BMP2 directs epicardial cell motility toward the myocardium 18 and direct epicardial cell invasion in a TGFβR3 dependent manner.8 We asked whether or not BMPs secreted from epicardial cells in vitro signal in an autocrine manner were required for unstimulated migration of Wild-type cells. Using Noggin to sequester and inhibit BMPs, we found that there was no change in unstimulated migration of epicardial cells (Figure 3). Noggin can sequester other BMP ligands including BMP4, 5, 7, 13, and 14,32 if these BMPs were involved, then an effect would have been observed. This is not all together surprising, as the myocardium is the major source of BMPs secreted to the epicardium in vivo, rather than the epicardium.18 Therefore, this further confirms no role for the action of autocrine BMP signaling in the epicardium. Although exogenous BMP2 rescues Tgfbr3−/− deficient migration, down-regulated BMP2 expression in Tgfbr3−/− cells is not sufficient to fully explain the in vivo phenotype, and further study of disparately expressed genes is required.
Although some previously described non-canonical BMP signaling pathways such as PI3-kinase and cdc42 have been investigated in BMP2-dependent cell migration,25 we assessed the role of Src kinase in BMP2-stimulated 2-dimensional cell migration in the wound healing assay. Y416 phosphorylation of Src is robustly induced by BMP2 in both Tgfbr3−/− and Wild-type epicardial cells (Supplemental Figure 1). Blocking Src attenuates BMP2-stimulated migration in Tgfbr3−/− epicardial cells in the wound healing assay (Figure 4).
BMP2 is known to stimulate cell migration and reorganization of the actin cytoskeleton, though a connection to Src kinase in this process has not yet been described. BMP2-induced f-actin polymerization in Wild-type and Tgfbr3−/− epicardial cells (Figure 5) is blocked by PP2. It appears that BMP2 has a more profound effect on formation of dense f-actin structures in Tgfbr3−/− cells than Wild-type as visualized by phalloidin staining (Figure 5), although f/g actin ratios are not different between BMP2-stimulated Wild-type and Tgfbr3−/− cells. This would suggest that TGFβR3 is not required for BMP2 induced filamentous actin polymerization, and that this occurs in a Src dependent manner through other receptors.
This study highlights a unique signaling pathway required for migration of epicardial cells in coronary vessel development. We show that TGFβR3 is required for epicardial cell migration, but not BMP2-stimulated migration, in a Src dependent manner.
Materials and methods
Cell lines and culture conditions
Conditionally immortal murine Epicardial cells were originally provided by Dr. Joey Barnett (Vanderbilt Medical University). Cell culture conditions were used as previously described.26 Epicardial cells used in this study were isolated from wild-type mouse embryos (Wild-type) and embryos lacking TGFβR3 (Tgfbr3−/−) as previously described.8 Recombinant human BMP2 and mouse Noggin was purchased from R&D Systems (#355-BM-10, #719-NG-050). The Src kinase inhibitor PP2 was purchased from EMD Millipore (#529573).
RT-PCR
RNA from Wild-type and Tgfbr3−/− epicardial cells was isolated and purified using Trizol RNA isolation reagent according to manufacturer's instructions (Invitrogen). cDNA was generated using first strand cDNA synthesis kit (Roche) from 1 μg RNA isolated from each experimental condition. Real-time PCR was performed as previously described using TaqMan Master primer-probe system (Roche). Genes and primer sets in Table 1.
Table 1.
Genes and primer sets.
| RPS7 (f) | 5′-AGCACGTGGTCTTCATTGCT-3′ |
| RPS7 (r) | 5′-CTGTCAGGGTACGGCTTCTG-3′ |
| Bmp2 (f) | 5′-CGGACTGCGGTCTCCTAA-3′ |
| Bmp2 (r) | 5′-GGGGAAGCAGCAACACTAGA-3′ |
| Has2 (f) | 5′-GGCGGAGGACGAGTCTATG-3′ |
| Has2 (r) | 5′-ACACATAGAAACCTCTCACAATGC-3′ |
| Tgfb1 (f) | 5′-TGGAGCAACATGTGGAACTC-3′ |
| Tgfb1 (r) | 5′-GTCAGCAGCCGGTTACCA-3′ |
| Tgfb2 (f) | 5′-TGGAGTTCAGACACTCAACACA-3′ |
| Tgfb2 (r) | 5′-AAGCTTCGGGATTTATGGTGT-3′ |
| Sm22α (f) | 5′-CCTTCCAGTCCACAAACGAC-3′ |
| Sm22α (r) | 5′-GTAGGATGGACCCTTGTTGG-3′ |
| Vim (f) | 5′-TGCGCCAGCAGTATGAAA-3′ |
| Vim (r) | 5′-GCCTCAGAGAGGTCAGCAA-3′ |
Wound healing assay
Wound healing assays were performed in accordance with previously described methods.27 Wild-type and Tgfbr3−/− epicardial cells were seeded at 100,000 cells per well in a 12-well tissue culture dish and grown to confluence. Cells were serum starved for 4 hours (DMEM 0%FBS). A wound was made through the epithelial monolayer with a 200 µL pipette tip and washed twice with 1X phosphate buffered saline. Cells were then stimulated with 2 ng/mL BMP2 and allowed to undergo migration for 24 hours. For experiments using 1 µM PP2 or 200 ng/mL Noggin, cells were pretreated for one hour prior to wound, and replaced with media containing 1 µM PP2 or 200 ng/mL Noggin in the presence or absence of 2 ng/mL BMP2. 0 hour images were taken immediately after the wound was made and immediately prior to BMP2 stimulation. Additional images were taken at 24 hours after BMP2 stimulation and compared to initial wound (0 hour) images for each condition. Image J was used to assess extent of wound healing by comparing the wound area at time 0 and 24 hours, and % closure of wound area was observed and reported. Each condition was performed in duplicate and repeated at least 3 times. Images were acquired using a Canon Powershot G5 and an Olympus CKX41 inverted microscope.
Phalloidin staining
Wild-type and Tgfbr3−/− epicardial cells were seeded on glass coverslips at equal and sub-confluent density (75,000 cells/coverslip in 12-well tissue culture plate) and allowed to adhere overnight. Cells were subject to overnight serum starvation (DMEM, 0% FBS), and stimulated with 2 ng/mL BMP2 for 60 minutes. Immunofluorescence visualization of filamentous actin was accomplished using AlexaFlour594 phalloidin (Life Technologies A12381) and a Leica DMLB microscope. Images were documented using a Retiga 200R camera and ImagePro Plus 5.1 software.
Filamentous actin assay
Wild-type and Tgfbr3−/− epicardial cells were seeded at 800,000 cells/10cm dish. After attachment, cells were serum starved overnight (DMEM, 0% FBS), subsequently stimulated with 2 ng/mL BMP2 for 60 minutes, and subjected to the G-Actin/F-Actin In Vivo Bioassay Biochem Kit (f/g actin assay) according to the manufacturer's instructions (Cytoskeleton BK037). Briefly, whole cell lysates were ultra centrifuged (100,000xg) to fractionate filamentous (f) and globular (g) actin fractions. f/g ratios under stimulated conditions were plotted relative to unstimulated f/g ratios from Wild-type conditions. f/g ratio is used as a read out of relative amount of filamentous actin in whole cell lysates.
Proliferation assay
Wild-type and Tgfbr3−/− epicardial cells were seeded at 10,000 cell/mL in a 96 well tissue culture dish and allowed to adhere overnight. Cells were serum starved (DMEM 0% FBS) for 4 hours, and subsequently stimulated with BMP2 (2 ng/mL) for 24 hours. Vybrant MTT Proliferation Assay (Promega) was performed according to manufacturer's instructions.
Statistical analysis
All graphs represent mean values, all error bars represent the standard deviation of the mean, all experiments were performed in duplicate repeated at least 3 times. Statistical significance was assessed using a 2-way student's t-test, with p values below 0.05 considered significant.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Joey V. Barnett for providing the conditionally immortal murine epicardial cells used in this study.
References
- [1].Mu H, Ohashi R, Lin P, Yao Q, Chen C. Cellular and molecular mechanisms of coronary vessel development. Vasc Med 2005; 10(1):37-44; http://dx.doi.org/ 10.1191/1358863x05vm584ra [DOI] [PubMed] [Google Scholar]
- [2].Kalman F, Viragh S, Modis L. Cell surface glycoconjugates and the extracellular matrix of the developing mouse embryo epicardium. Anat Embryol (Berl) 1995; 191(5):451-64; http://dx.doi.org/ 10.1007/BF00304430 [DOI] [PubMed] [Google Scholar]
- [3].Munoz-Chapuli R, Macias D, Gonzalez-Iriarte M, Carmona R, Atencia G, Perez-Pomares JM. The epicardium and epicardial-derived cells: multiple functions in cardiac development. Rev Esp Cardiol 2002; 55(10):1070-82; http://dx.doi.org/ 10.1016/S0300-8932(02)76758-4 [DOI] [PubMed] [Google Scholar]
- [4].Perez-Pomares JM, de la Pompa JL. Signaling during epicardium and coronary vessel development. Circ Res 2011; 109(12):1429-42; http://dx.doi.org/ 10.1161/CIRCRESAHA.111.245589 [DOI] [PubMed] [Google Scholar]
- [5].Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, et al.. Heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation 2013; 127(1):e6-e245; PMID:23239837; http://dx.doi.org/ 10.1161/CIR.0b013e31828124ad [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Azhar M, Schultz Jel J, Grupp I, Dorn GW 2nd, Meneton P, Molin DG, Gittenberger-de Groot AC, Doetschman T. Transforming growth factor beta in cardiovascular development and function. Cytokine Growth Factor Rev 2003; 14(5):391-407; PMID:12948523; http://dx.doi.org/ 10.1016/S1359-6101(03)00044-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Doetschman T, Barnett JV, Runyan RB, Camenisch TD, Heimark RL, Granzier HL, Conway SJ, Azhar M. Transforming growth factor beta signaling in adult cardiovascular diseases and repair. Cell Tissue Res 2012; 347(1):203-23; PMID:21953136; http://dx.doi.org/ 10.1007/s00441-011-1241-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hill CR, Sanchez NS, Love JD, Arrieta JA, Hong CC, Brown CB, Austin AF, Barnett JV. BMP2 signals loss of epithelial character in epicardial cells but requires the Type III TGFbeta receptor to promote invasion. Cell Signal 2012; 24(5):1012-22; PMID:22237159; http://dx.doi.org/ 10.1016/j.cellsig.2011.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lopez-Casillas F, Wrana JL, Massague J. Betaglycan presents ligand to the TGF beta signaling receptor. Cell 1993; 73(7):1435-44; PMID:8391934; http://dx.doi.org/ 10.1016/0092-8674(93)90368-Z [DOI] [PubMed] [Google Scholar]
- [10].Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res 2009; 19(1):128-39; http://dx.doi.org/ 10.1038/cr.2008.328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Compton LA, Potash DA, Brown CB, Barnett JV. Coronary vessel development is dependent on the type III transforming growth factor beta receptor. Circ Res 2007; 101(8):784-91;http://dx.doi.org/ 10.1161/CIRCRESAHA.107.152082 [DOI] [PubMed] [Google Scholar]
- [12].Shin D, Shin CH, Tucker J, Ober EA, Rentzsch F, Poss KD, Hammerschmidt M, Mullins MC, Stainier DY. Bmp and Fgf signaling are essential for liver specification in zebrafish. Development 2007; 134(11):2041-50; PMID:17507405; http://dx.doi.org/ 10.1242/dev.000281 [DOI] [PubMed] [Google Scholar]
- [13].Weber H, Symes CE, Walmsley ME, Rodaway AR, Patient RK. A role for GATA5 in Xenopus endoderm specification. Development 2000; 127(20):4345-60 [DOI] [PubMed] [Google Scholar]
- [14].Dale L, Jones CM. BMP signalling in early Xenopus development. Bioessays 1999; 21(9):751-60; http://dx.doi.org/ 10.1002/(SICI)1521-1878(199909)21:9%3c751::AID-BIES6%3e3.0.CO;2-I [DOI] [PubMed] [Google Scholar]
- [15].Nakajima Y, Yamagishi T, Hokari S, Nakamura H. Mechanisms involved in valvuloseptal endocardial cushion formation in early cardiogenesis: roles of transforming growth factor (TGF)-beta and bone morphogenetic protein (BMP). Anat Rec 2000; 258(2):119-27;http://dx.doi.org/ 10.1002/(SICI)1097-0185(20000201)258:2%3c119::AID-AR1%3e3.0.CO;2-U [DOI] [PubMed] [Google Scholar]
- [16].Delot EC. Control of endocardial cushion and cardiac valve maturation by BMP signaling pathways. Mol Genet Metab 2003; 80(1-2):27-35; http://dx.doi.org/ 10.1016/j.ymgme.2003.07.004 [DOI] [PubMed] [Google Scholar]
- [17].Townsend TA, Robinson JY, Deig CR, Hill CR, Misfeldt A, Blobe GC, et al.. BMP-2 and TGFbeta2 shared pathways regulate endocardial cell transformation. Cells Tissues Organs 2011; 194(1):1-12; PMID:21212630; http://dx.doi.org/ 10.1159/000322035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ishii Y, Garriock RJ, Navetta AM, Coughlin LE, Mikawa T. BMP signals promote proepicardial protrusion necessary for recruitment of coronary vessel and epicardial progenitors to the heart. Dev Cell 2010; 19(2):307-16; http://dx.doi.org/ 10.1016/j.devcel.2010.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sanchez NS, Hill CR, Love JD, Soslow JH, Craig E, Austin AF, Brown CB, Czirok A, Camenisch TD, Barnett JV. The cytoplasmic domain of TGFbetaR3 through its interaction with the scaffolding protein, GIPC, directs epicardial cell behavior. Dev Biol 2011; 358(2):331-43; PMID:21871877; http://dx.doi.org/ 10.1016/j.ydbio.2011.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bjorge JD, Jakymiw A, Fujita DJ. Selected glimpses into the activation and function of Src kinase. Oncogene 2000. November 20; 19(49):5620-35; http://dx.doi.org/ 10.1038/sj.onc.1203923 [DOI] [PubMed] [Google Scholar]
- [21].Hutcheson JD, Ryzhova LM, Setola V, Merryman WD. 5-HT(2B) antagonism arrests non-canonical TGF-beta1-induced valvular myofibroblast differentiation. J Mol Cell Cardiol 2012. November; 53(5):707-14; http://dx.doi.org/ 10.1016/j.yjmcc.2012.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wong WK, Knowles JA, Morse JH. Bone morphogenetic protein receptor type II C-terminus interacts with c-Src: implication for a role in pulmonary arterial hypertension. Am J Respir Cell Mol Biol 2005. November; 33(5):438-46; http://dx.doi.org/ 10.1165/rcmb.2005-0103OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Allison P, Espiritu D, Barnett JV, Camenisch TD. Type III TGFbeta receptor and Src direct hyaluronan-mediated invasive cell motility. Cell Signal 2015. Mar; 27(3):453-9; PMID:25499979; http://dx.doi.org/ 10.1016/j.cellsig.2014.11.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jin H, Pi J, Huang X, Huang F, Shao W, Li S, Chen Y, Cai J. BMP2 promotes migration and invasion of breast cancer cells via cytoskeletal reorganization and adhesion decrease: an AFM investigation. Appl Microbiol Biotechnol 2012; 93(4):1715-23; PMID:22270235; http://dx.doi.org/ 10.1007/s00253-011-3865-3 [DOI] [PubMed] [Google Scholar]
- [25].Gamell C, Osses N, Bartrons R, Ruckle T, Camps M, Rosa JL, Ventura F. BMP2 induction of actin cytoskeleton reorganization and cell migration requires PI3-kinase and Cdc42 activity. J Cell Sci 2008; 121(Pt 23):3960-70; PMID:19001503; http://dx.doi.org/ 10.1242/jcs.031286 [DOI] [PubMed] [Google Scholar]
- [26].Austin AF, Compton LA, Love JD, Brown CB, Barnett JV. Primary and immortalized mouse epicardial cells undergo differentiation in response to TGFbeta. Dev Dyn 2008; 237(2):366-76; http://dx.doi.org/ 10.1002/dvdy.21421 [DOI] [PubMed] [Google Scholar]
- [27].Sollome JJ, Thavathiru E, Camenisch TD, Vaillancourt RR. HER2/HER3 regulates extracellular acidification and cell migration through MTK1 (MEKK4). Cell Signal 2014; 26(1):70-82; http://dx.doi.org/ 10.1016/j.cellsig.2013.08.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lopez-Casillas F, Cheifetz S, Doody J, Andres JL, Lane WS, Massague J. Structure and expression of the membrane proteoglycan betaglycan, a component of the TGF-beta receptor system. Cell 1991; 67(4):785-95; http://dx.doi.org/ 10.1016/0092-8674(91)90073-8 [DOI] [PubMed] [Google Scholar]
- [29].Camenisch TD, Spicer AP, Brehm-Gibson T, Biesterfeldt J, Augustine ML, Calabro A Jr, Kubalak S, Klewer SE, McDonald JA. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J Clin Invest 2000; 106(3):349-60; PMID:10930438; http://dx.doi.org/ 10.1172/JCI10272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Craig EA, Austin AF, Vaillancourt RR, Barnett JV, Camenisch TD. TGFbeta2-mediated production of hyaluronan is important for the induction of epicardial cell differentiation and invasion. Exp Cell Res 2010; 316(20):3397-405; http://dx.doi.org/ 10.1016/j.yexcr.2010.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sanchez NS, Hill CR, Love JD, Soslow JH, Craig E, Austin AF, Brown CB, Czirok A, Camenisch TD, Barnett JV. The cytoplasmic domain of TGFbetaR3 through its interaction with the scaffolding protein, GIPC, directs epicardial cell behavior. Dev Biol 2011; 358(2):331-43; PMID:21871877; http://dx.doi.org/ 10.1016/j.ydbio.2011.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Krause C, Guzman A, Knaus P. Noggin. Int J Biochem Cell Biol 2011; 43(4):478-81; http://dx.doi.org/ 10.1016/j.biocel.2011.01.007 [DOI] [PubMed] [Google Scholar]
- [33].Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell 2003; 112(4):453-65; http://dx.doi.org/ 10.1016/S0092-8674(03)00120-X [DOI] [PubMed] [Google Scholar]
- [34].Kirkbride KC, Townsend TA, Bruinsma MW, Barnett JV, Blobe GC. Bone Morphogenetic Proteins Signal through the Transforming Growth Factor-β Type III Receptor. J Biol Chem 2008; (283), 7628-7637; PMID:18184661; http://dx.doi.org/ 10.1074/jbc.M704883200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.