

Abstract
Successful addiction treatment depends on maintaining long-term abstinence, making relapse prevention an essential therapeutic goal. However, exposure to environmental cues associated with drug use often thwarts abstinence efforts by triggering drug using memories that drive craving and relapse. We sought to develop a dual approach for weakening cocaine memories through phosphoproteomic identification of targets regulated in opposite directions by memory extinction compared with reconsolidation in male Sprague-Dawley rats that had been trained to self-administer cocaine paired with an audiovisual cue. We discovered a novel, inversely regulated, memory-dependent phosphorylation event on calcium-calmodulin-dependent kinase II α (CaMKIIα) at serine (S)331. Correspondingly, extinction-associated S331 phosphorylation inhibited CaMKIIα activity. Intra-basolateral amygdala inhibition of CaMKII promoted memory extinction and disrupted reconsolidation, leading to a reduction in subsequent cue-induced reinstatement. CaMKII inhibition had no effect if the memory was neither retrieved nor extinguished. Therefore, inhibition of CaMKII represents a novel mechanism for memory-based addiction treatment that leverages both extinction enhancement and reconsolidation disruption to reduce relapse-like behavior.
SIGNIFICANCE STATEMENT Preventing relapse to drug use is an important goal for the successful treatment of addictive disorders. Relapse-prevention therapies attempt to interfere with drug-associated memories, but are often hindered by unintentional memory strengthening. In this study, we identify phosphorylation events that are bidirectionally regulated by the reconsolidation versus extinction of a cocaine-associated memory, including a novel site on CaMKIIα. Additionally, using a rodent model of addiction, we show that CaMKII inhibition in the amygdala can reduce relapse-like behavior. Together, our data supports the existence of mechanisms that can be used to enhance current strategies for addiction treatment.
Keywords: CaMKIIa, cocaine, extinction, proteomics, reconsolidation, reinstatement
Introduction
The successful treatment of addictive disorders requires maintaining long-term abstinence from drug use. (Kalivas and Volkow, 2005; Milton and Everitt, 2010). However, individuals frequently encounter environmental cues previously associated with drug use that can increase craving and the likelihood of relapse (Fuchs et al., 2009; Kalivas, 2009). The ability of drug-associated memories to induce relapse is perhaps the greatest obstacle to overcome for the successful treatment of addictive disorders (Torregrossa et al., 2011; Bossert et al., 2013). Thus, reducing the strength of drug-associated memories has therapeutic potential for individuals struggling with addiction.
Behavioral and pharmacological interventions can disrupt or weaken drug-associated memories through two primary mechanisms. The first is to pharmacologically prevent reconsolidation of the memory into long-term storage after the memory is retrieved or reactivated (Taylor et al., 2009; Torregrossa and Taylor, 2013). It is possible to disrupt reconsolidation because when a memory is retrieved it can enter a labile state that requires protein synthesis-dependent restabilization (reconsolidation) for the memory to be maintained in long-term storage. During the period of lability, memories are susceptible to interference, and can be weakened or strengthened with specific pharmacological manipulations (Tronson and Taylor, 2007). The second mechanism for weakening drug-associated memories is through the process of extinction. Extinction involves repeatedly presenting drug-associated cues in the absence of the drug. In this situation, the individual learns that the cues are no longer predictive of drug availability, and thus, subsequent encounters with the cue produce less craving and relapse. Extinction memories undergo their own consolidation and reconsolidation processes that can also be enhanced or inhibited with pharmacological manipulations (Holmes and Quirk, 2010; Nic Dhonnchadha et al., 2010; Vurbic et al., 2011). Disrupting reconsolidation and enhancing extinction have long been proposed as potential strategies to disrupt aversive memories that form the basis of anxiety disorders (Monfils et al., 2009; Agren et al., 2012; Gamache et al., 2012), and these strategies have been extended to the addiction field (Torregrossa et al., 2010; Tronson et al., 2012; Arguello et al., 2014). However, clinical application of extinction- or reconsolidation-based treatments has met with limited success, likely because both processes involve overlapping molecular mechanisms, making selective targeting of one or the other challenging. Indeed, previous attempts to disrupt drug-associated memories using pharmacological agents combined with extinction training may have been unsuccessful due to unintentional memory strengthening (Hofmann et al., 2012; Price et al., 2013).
Therefore, the goal of the present study was to identify cellular signaling events that are regulated in opposition by extinction relative to reconsolidation of a memory associated with self-administered cocaine. Such molecules, once identified, could represent targets for the development of a single medication that can enhance extinction while inhibiting reconsolidation to ensure memory weakening and a reduction in relapse. We used a discovery-based phosphoproteomics strategy to identify proteins that exhibit a divergent pattern of phosphorylation following reconsolidation relative to extinction, as initial memory consolidation events involve the activation of kinase and phosphatase cascades (Sanchez et al., 2010; Merlo et al., 2014). The basolateral amygdala (BLA) was analyzed as it is the locus for associative learning and mediates the encoding of drug-associated memories (Fuchs et al., 2006; Sanchez et al., 2010; Nic Dhonnchadha et al., 2013). The proteomics analysis found a small number of opposing signaling events, including identification of a novel phosphorylation event on calcium-calmodulin-dependent kinase II α (CaMKIIα) on serine (S)331. Although CaMKIIα phosphorylation events on the highly characterized autophosphorylation site at threonine (T)286 have been previously linked to drug-related learning (Easton et al., 2013, 2014; Salling et al., 2016), the role of S331 phosphorylation has not been investigated. Here, we report a functional role for S331 phosphorylation in vitro and describe the behavioral effects of CaMKII inhibition on both the reconsolidation and extinction of a cocaine-associated memory in vivo. Our findings suggest that molecular mechanisms exist that could allow for the combined enhancement of extinction and disruption of reconsolidation.
Materials and Methods
Subjects.
Naive, adult male Sprague-Dawley rats, weighing 275–325 g on arrival, were used in all studies. All rats were housed in a temperature- and humidity-controlled room. Animals were housed in pairs, given ad libitum access to food and water, and maintained on a 12 h light/dark cycle. Rats were given at least 5 d to acclimate to the facility before undergoing surgical procedures. Following surgery, rats were individually housed and given at least 1 week to recover before the start of behavioral training. Rats were food-deprived 24 h before the start of behavioral experiments and maintained at ∼90% of their free-feeding body weight (∼20 g of chow per day) for the duration of testing. All behavioral experiments were run during the light cycle. The experiments used to generate samples for proteomic analysis were conducted at Yale University in the Connecticut Mental Health Center, whereas all subsequent experiments were conducted at the University of Pittsburgh. Sprague-Dawley rats were obtained from Charles River Laboratories at Yale and from Harlan at the University of Pittsburgh. We used different vendors to minimize animal shipping time to both facilities. At Yale, rats were housed in shoebox cages with water bottles on standard racks, whereas at the University of Pittsburgh, rats were housed in auto-ventilated racks with an automated watering system. All other housing and procedural parameters were the same between the two universities, unless otherwise noted. In addition, all procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by each institution's Institutional Animal Care and Use Committee.
Self-administration test chambers.
Rats were trained to self-administer cocaine in standard operant conditioning chambers (MedAssociates). Experiments were counterbalanced across one of two chamber designs, providing two distinct contexts. The first type of chamber consisted of bar floors and two nosepoke apertures. The second type of chamber consisted of grid floors and five nosepoke apertures. All chambers contained two retractable levers on one wall of the chamber, a tone-generator, stimulus light above each lever, house light, and infusion pump. Operant boxes were kept in sound-attenuating chambers equipped with a fan for background noise.
Drugs.
Cocaine hydrochloride (generously provided by the National Institute on Drug Abuse) was dissolved in sterile 0.9% saline (2 mg/ml) and filter-sterilized for self-administration. KN-62 (Tocris Bioscience) was dissolved in 0.9% saline + 62% DMSO. KN-93 (Tocris Bioscience) was dissolved in 1× PBS + 10% DMSO.
Surgical procedures.
Rats were fully anesthetized with ketamine hydrochloride (87.5 mg/kg, i.m.) and xylazine hydrochloride (5 mg/kg, i.m.), and then received an analgesic (Rimadyl, 5 mg/kg, s.c.) and 5 ml of lactated Ringer's (s.c.) before surgery. Betadine and 70% ethanol were applied to all incision sites. All rats were implanted with a chronic indwelling intravenous catheter (CamCaths) into the right jugular vein, as described previously (Torregrossa and Kalivas, 2008; Torregrossa et al., 2010). Catheters were fed subcutaneously to the midscapular region, where they exited through a round incision. For experiments involving intracranial infusions, rats were immediately placed into a stereotaxic instrument (Stoelting), and implanted with bilateral stainless steel guide cannulae (22 gauge; Plastics One) targeting the area just dorsal to the BLA (AP −3.0 mm, ML ±5.3 mm, DV −7.9 mm, relative to bregma; Paxinos and Watson, 2007). Guide cannulae were secured to the skull with three miniature screws and dental acrylic resin. Dummy cannulae were inserted the length of the guide cannulae to maintain patency. Rimadyl (5 mg/kg, s.c.) was administered for the first 2 d after surgery. Catheters were kept patent by daily infusions of 0.1 ml of an antibiotic solution of cefazolin (10.0 mg/ml) dissolved in heparinized saline (30 USP heparin/ml).
Cocaine self-administration procedures.
Rats administered cocaine during daily sessions for 1 h, on a fixed ratio 1 (FR1) schedule of reinforcement with a 10 s timeout. The designated active lever (counterbalanced across left and right levers) produced a cocaine infusion paired with a 10 s tone-light conditioned stimulus (CS). Pump durations were adjusted daily according to body weight to deliver the correct dose of drug (1.0 mg/kg of body weight per infusion). Responses on the other, inactive, lever were recorded, but had no programmed consequences. Rats underwent training for at least 10 d and until they administered at least eight infusions per day over 3 consecutive days. Rats that did not meet acquisition criteria by 14 d were excluded from the study. The program was controlled by and data were collected using MedPC (MedAssociates).
Instrumental lever extinction.
After successful acquisition of self-administration, rats underwent instrumental lever extinction for at least 7 d. During these daily 1 h sessions, responses on both the active and inactive levers were recorded, but had no programmed consequences. Lever extinction continued until extinction criteria had been met (an average of <25 lever presses on the last 2 d of extinction). Throughout lever extinction, rats received no cocaine or cocaine-associated cue reinforcement. Lever extinction was conducted to reduce responding to a stable, low rate to later assess cue-induced reinstatement. In addition, lever extinction reduces the motivational value of other cues in the self-administration context, such as the levers, so that subsequent testing specifically isolates the memory for the discrete cue associated with cocaine infusion.
Identification of cellular signaling events regulated by cocaine-associated memory extinction and reconsolidation
Memory manipulations.
Following cocaine self-administration and instrumental lever extinction, rats were assigned to a memory manipulation group (extinction, memory reactivation, or no manipulation controls) based on a matching procedure that ensured that each group had no statistical differences in their cocaine infusions acquired over days, or differences in lever extinction behavior. All groups were placed in the opposite context from which they received self-administration training (different in flooring texture, shape, and smell), for a 30 min session on 2 consecutive days. During these sessions, rats had no opportunity for instrumental responding, ie, the levers were retracted.
Extinction.
For cue extinction, the cocaine-associated CS was presented for 10 s, 60 times, with each presentation separated by 30 s, on each of the 2 d. Thus, the rats were exposed to a total of 120 CS presentations, which we have previously shown to significantly reduce cue-induced reinstatement on a subsequent test day (Torregrossa et al., 2010).
Memory reactivation.
For cue reactivation, the CS was presented three times at the end of the last session on the second day, with each CS presentation separated by 1 min. Previous work from our laboratory has shown that 3 CS presentations is sufficient to induce memory reactivation and reconsolidation, but is not sufficient to produce extinction (Sanchez et al., 2010; Wan et al., 2014).
No memory manipulation control.
The control group was placed in the operant chambers for the same amount of time as rats in the cue extinction and reactivation groups, but with no CS presentations. Thus, the time spent in the operant boxes and the type of operant box was equivalent between groups before kill.
Tissue collection.
Fifteen minutes following memory manipulations, rats were lightly anesthetized with isofluorane to minimize stress before euthanasia by focused microwave irradiation. Focused microwave irradiation was used to preserve the phosphorylation state of proteins. Importantly, because no group had the opportunity to make instrumental responses before kill, differences in behavioral activity should not substantially affect levels of protein phosphorylation. The brains were immediately dissected and individual brain regions, including the basolateral amygdala complex were obtained and stored at −80°C until processing.
Label-free quantitative proteomics: sample preparation.
Brain regions-of-interest, including the amygdala reported here, were homogenized by sonication in a buffer containing urea (8 m), ammonium bicarbonate (0.4 m), and phosphatase inhibitor cocktails (Sigma-Aldrich). Samples from two rats in each experimental group were randomly pooled to create a total of four to five sample pools per group. The extinction group included one sample “pool” that consisted of just one rat due to an odd number of rats meeting acquisition criteria. Pooled samples were then analyzed by the Yale/NIDA Neuroproteomics Center at Yale. Twenty microliters of 45 mm DTT was added to each sample and incubated at 37°C for 20 min to reduce Cys residues. Samples were cooled and 20 μl of 100 mm iodoacetamide (IAM) was added to each sample and incubated at RT in the dark for 20 min for alkylation of the reactive-free sulfhydro of the reduced Cys. Dual enzymatic digestion was performed by adding 600 μl of dH2O and 30 μl of 1 mg/ml Lys C followed by incubation at 37°C for 4 h, with subsequent digestion by incubation with 30 μl of 1 mg/ml trypsin overnight at 37°C. Samples were macrospin desalted and dried by SpeedVac. Pellets were dissolved in 50 μl of a solution containing 0.5% TFA and 50% acetonitrile. Samples were then subjected to titanium dioxide (TiO2) phosphopeptide enrichment using TopTips (Glygen). The TopTip was washed 3 times at 2000 rpm for 1 min with 40 μl 100% acetonitrile, then 0.2 m sodium phosphate, pH 7.0, 0.5% TFA, and 50% acetonitrile. The acidified digest supernatants were loaded into the TopTip, spun at 1000 rpm for 1 min, and then at 3000 rpm for 2 min. The flow through from these washes (less phosphorylated fraction) was saved for analysis by liquid chromatography tandem mass spectrometry (LC-MS/MS) as described below. Phosphopeptides were eluted from each TopTip by three washes with 30 μl of 28% ammonium hydroxide. Both the flow through and eluted fractions were dried by SpeedVac. Enriched fractions were dissolved in 10 μl of 70% formic acid and 30 μl of 50 mm sodium phosphate. Peptide concentrations were determined by Nanodrop to load 0.3 μg/5 μl of each sample.
LC/MS-MS.
Five microliters of each sample was injected onto a LTQ Orbitrap LC-MS/MS system. Peptide separation was performed on the nanoACQUITY ultra-high pressure liquid chromatography (UPLC) system (Waters), using a Waters Symmetry C18 180 μm × 20 mm trap column and a 1.7 μm, 75 μm × 250 mm nanoACQUITY UPLC column (35°C). Trapping was done at 15 μl/min, with 99% Buffer A (0.1% formic acid in water) for 1 min. Peptide separation was performed over 120 min at a flow rate of 300 nl/min beginning with 95% Buffer A and 5% Buffer B (0.075% formic acid in acetonitrile) to 40% B from 1 to 9 min, to 85% B from 9 to 91 min, held at 85% B from 91 to 95 min, then returned to 5% B from 95 to 96 min. Two washes were made between each sample run to ensure no carry over [(1) 100% acetonitrile, (2) Buffer A]. The LC was in-line with an LTQ-Orbitrap mass spectrometer. MS was acquired in the Orbitrap using 1 microscan, and a maximum inject time of 900 ms followed by three to six data-dependent MS/MS acquisitions in the ion trap (with precursor ion threshold of >3000). The total cycle time for both MS and MS/MS fragmentation by collision-induced dissociation were first isolated with a 2 Da window followed by normalized collision energy of 35%. Dynamic exclusion was activated where former target ions were excluded for 30 s. Three technical replicates were injected for each sample and all samples and replicates were randomized across an entire run time.
Data analyses.
Feature extraction, chromatographic/spectral alignment, data filtering, and statistical analysis used Nonlinear Dynamics Progenesis LC-MS software (www.nonlinear.com). Raw data files were imported into the program and the detected mass spectral features were aligned based on retention time of the detected m/z peaks based on a randomly selected reference run. All other runs were automatically aligned to the reference run to minimize retention time variability between runs. No adjustments were necessary in the m/z dimension due to high mass accuracy of the spectrometer (typically <3 ppm). All runs were selected for detection with an automatic detection limit. Features within retention time ranges of 0–5 min were filtered out, as were features with charge state greater than +6 or singly charged peptides (as no MS/MS fragmentations were taken for these charge states during data collection) for reduction of false-positive peptide assignments. A normalization factor was then calculated for each run to account for differences in sample load between injections. The experimental design grouped multiple injections from each condition. The algorithm then calculated and tabulated raw and normalized abundances, maximum fold-change, and ANOVA p values for each feature in the dataset. Stringent conditions were set in MASCOT to filter out low scoring identified peptides by imposing a confidence probability score (p) of <0.05. Additionally a positively identified protein that was quantified contained at least two unique identified tryptic peptides. The filtered MS/MS spectral features along with their precursor spectra were exported in the form of an .mgf file (Mascot generic file) for database searching using the Mascot algorithm (Hirosawa et al., 1993). The data were searched against the Uniprot (Rattus norvegicus) database. The confidence level was set to 95% within the MASCOT search engine for peptides assigned hits based on randomness. MS/MS analysis was based on the use of trypsin and the following variable modifications: carboamidomethyl (Cys), oxidation (Met), Phospho (Ser, Thr, Tyr). Other search parameters included peptide mass tolerance of ±15 ppm, fragment mass tolerance of ±0.5 Da, and maximum missed cleavages of 3. A decoy search (based on the reverse sequence search) was performed to estimate false discovery rate (FDR), with setting of acceptable protein ID having FDR of 2%. Using the Mascot database search algorithm, a protein is considered identified when Mascot lists it as significant (bold red) and more than two unique peptides match the same protein. The Mascot significance score match is based on a MOWSE score and relies on multiple matches to more than one peptide from the same protein. The Mascot search results were exported to an .xml file using a significance cutoff of <0.05, and ion score cutoff of 28, and a requirement of at least one bold (first time any match to the spectrum has appeared in the report) and red (top scoring peptide match for this spectrum) peptide. The .xml file was then imported into the Progenesis LCMS software, where search hits were assigned to corresponding detected features, identified as described above.
SRM proteomics.
Identified peptides and their modifications were mined to a list of ∼80 unique peptides for quantitative analysis between groups by selective reaction monitoring (SRM). The list was created based on ionization signal quality and to reduce the number of high abundance, highly modified phosphoproteins (eg, neurofilament proteins) and proteins with unclear function. Analyses were performed on a 5500 Q-TRAP instrument coupled online to a Waters nanoACQUITY UPLC system. Four microliters of each sample was loaded onto a 5 μm, 180 μm × 20 mm Symmetry C18 nanoAcquity trapping column with 100% water/0.1% formic acid at a flow rate of 15 μl/min for 1 min. Peptides were then separated on a 1.7 μm, 75 μm × 100 mm BEH130 C18 nanoAcquity column with a 30 min, 2–40% acetonitrile/0.1% formic acid linear gradient at a flow rate of 0.75 μl/min. SRM scanning was conducted using 211 transitions and a cycle time of 2.4 s with a 5 ms dwell time per transition (5 transitions/peptide) in positive polarity. Data were processed using MRMPilot 2.0, Analyst 1.5 with MIDAS, and Multiquant 2.0 software. Peptide identification was further confirmed using MASCOT. Data were analyzed using nested linear models with transition nested within peptide. Interaction effects were included in the model and significant group × transition effects within a peptide were used to identify outlying transitions for exclusion from further analysis. Statistics were calculated using SRMstats with restricted scope (freely available in R; Chang et al., 2012). We report phosphopeptides with significantly different abundances in either the extinction or reactivation group relative to control with p < 0.05 considered significant after correcting for multiple comparisons. The statistical methods used here, as described by Chang et al. (2012), are robust in detecting statistical differences from SRM experiments, particularly when there is little underlying biological variability in the protein abundance. This statistical method was chosen for hypothesis generation, as the primary aim of the present study was to identify phosphorylation events that are bidirectionally regulated by the extinction versus reconsolidation of cocaine-associated memories. The analysis was therefore biased toward potentially identifying false-positives rather than creating false-negatives. We also include analytical results using a more conservative linear mixed effects model for comparison in Table 1. Within the table, the first column lists the Uniprot defined protein abbreviations (not gene names), the second column shows the peptide sequence identified with lower case “p”s indicating sites of phosphorylation and “ox” indicating oxidation of Methionine. The next set of six columns give the results from the SRM stats analysis and the linear mixed effects model analysis. Each analysis includes the unadjusted p value, the estimated fold-change from controls, and the p value after correcting for multiple comparisons. The results from the extinction group relative to control, and reactivation group relative to control, are labeled in the Protein column.
Table 1.
Results from SRM proteomics analysis
| Protein | Peptide sequence + modification(s) | SRM stats restricted scope |
Linear mixed-effects model |
||||
|---|---|---|---|---|---|---|---|
| Non-adjusted p value | Fold-change estimate | Adjusted p value | Non-adjusted p value | Fold-change estimate | Adjusted p value | ||
| Extinction versus control | |||||||
| ADCY9 | GQGTASpPGSpVSDLAQTVK | 0.2413 | 0.3017 | 0.3475 | 0.5004 | 0.3017 | 0.7506 |
| AKA12 | ALGSpLGGSpPSLPDQDK | 0.0172 | −0.5640 | 0.0403 | 0.3275 | −0.5640 | 0.7284 |
| ARHG7 | MSpGFIYQGK | 0.4373 | 0.0582 | 0.5524 | 0.6516 | 0.0582 | 0.8530 |
| BAIP2 | SSSpMAAGLER | 0.7626 | 0.0362 | 0.8133 | 0.8628 | 0.0362 | 0.9557 |
| BSN | SPQVLYSpPVSpPLSPHR | 0.0118 | 0.2460 | 0.0340 | 0.1478 | 0.2460 | 0.7284 |
| CKD18 | RASpLSDIGFGK | 0.0000 | −0.2172 | 0.0000 | 0.4012 | −0.2172 | 0.7284 |
| CSKI1 | KVPLPGPGSpPEVK | 0.0173 | −0.2256 | 0.0403 | 0.1374 | −0.2256 | 0.7284 |
| CTNA1 | SRTpSpVQTpEDDQLIAGQSAR | 0.0144 | −0.2016 | 0.0370 | 0.1565 | −0.2016 | 0.7284 |
| CTNA1 | SRTSpVQTpEDDQLIAGQSAR | 0.0345 | 0.1416 | 0.0653 | 0.3944 | 0.1416 | 0.7284 |
| CTNB1 | RTSMGGTpQQQFVEGVR | 0.2586 | −0.0518 | 0.3581 | 0.8423 | −0.0518 | 0.9557 |
| CTND2 | ALQSpPEHHIDPIYEDR | 0.9798 | −0.0032 | 0.9798 | 0.9794 | −0.0032 | 0.9925 |
| CXA1 | M(ox)GQAGSTISpNSpHAQPFDFPDDNQNAK | 0.3219 | −0.0462 | 0.4292 | 0.8552 | −0.0462 | 0.9557 |
| CXA1 | MGQAGSTISpNSpHAQPFDFPDDNQNAK | 0.0152 | −0.3070 | 0.0377 | 0.3096 | −0.3070 | 0.7284 |
| CXA1 | VAAGHELQPLAIVDQRPSSpRASpSpR | 0.0000 | −0.5198 | 0.0000 | 0.0540 | −0.5198 | 0.7284 |
| DGKB | GAITpPPRSSpPANTCSPEVIHLK | 0.0032 | −0.1344 | 0.0117 | 0.5449 | −0.1344 | 0.7846 |
| EF1D | ATAPQTQHVSpPMR | 0.7508 | −0.1949 | 0.8133 | 0.7508 | −0.1949 | 0.8862 |
| FAK2 | RNSpLPQIPTLNLESR | 0.0000 | 0.2027 | 0.0000 | 0.2270 | 0.2027 | 0.7284 |
| FGF12 | EPSpLHEIGEK | 0.2060 | −0.1028 | 0.3027 | 0.4973 | −0.1028 | 0.7506 |
| GABR1 | HPPTpPPDPSGGLPR | 0.0002 | 0.1330 | 0.0011 | 0.4323 | 0.1330 | 0.7284 |
| GABR1 | RHPPTpPPDPSGGLPR | 0.0556 | 0.1371 | 0.1002 | 0.3379 | 0.1371 | 0.7284 |
| GABR2 | DPIEDINSpPEHIQR | 0.0035 | 0.2341 | 0.0119 | 0.2114 | 0.2341 | 0.7284 |
| GIT1 | HGSpGAESDYENTQSGEPLLGLEGK | 0.0000 | −0.2619 | 0.0002 | 0.0726 | −0.2619 | 0.7284 |
| GIT1 | NQSDLDDQHDYDSpVASpDEDTDQEPLPSAGATR | 0.1288 | 0.0910 | 0.2065 | 0.3636 | 0.0910 | 0.7284 |
| IF3M | LLYLTpSpAK | 0.5401 | 0.2342 | 0.6418 | 0.5401 | 0.2342 | 0.7846 |
| IPP2 | EQESpSpGEEDNDLSPEER | 0.9515 | 0.0051 | 0.9787 | 0.9830 | 0.0051 | 0.9925 |
| KCC2A | ESSESTpNTpTpIEDEDTK | 0.5437 | 0.0698 | 0.6418 | 0.8973 | 0.0698 | 0.9762 |
| KCC2A | ESSpESTNTTIEDEDTK | 0.0000 | 0.3596 | 0.0004 | 0.0737 | 0.3596 | 0.7284 |
| KCNQ2 | HGTSpPVGDHGSLVR | 0.1291 | −0.2891 | 0.2065 | 0.1291 | −0.2891 | 0.7284 |
| KPCB | HPPVLTpPPDQEVIR | 0.0000 | −0.1477 | 0.0002 | 0.5564 | −0.1477 | 0.7855 |
| KPCG | AAPALTpPPDR | 0.5667 | −0.2324 | 0.6535 | 0.5902 | −0.2324 | 0.8058 |
| KPCG | TpFCGTPDYIAPEIIAYQPYGK | 0.0008 | −0.1151 | 0.0034 | 0.4350 | −0.1151 | 0.7284 |
| MARCS | EAAEAEPAEPGSpPSAETEGASASSTSSPK | 0.0000 | 0.2408 | 0.0000 | 0.1891 | 0.2408 | 0.7284 |
| MARCS | GEAAAERPGEAAVASpSPSK | 0.3813 | 0.2147 | 0.4992 | 0.3813 | 0.2147 | 0.7284 |
| MARK2 | DQQNLPFGVTPASpPSGHSQGR | 0.0020 | 0.1821 | 0.0075 | 0.0738 | 0.1821 | 0.7284 |
| MINK1 | SDSVLPASHGHLPQAGSpLER | 0.0001 | 0.1289 | 0.0005 | 0.2101 | 0.1289 | 0.7284 |
| MK01 | VADPDHDHTGFLTpEYpVATR | 0.0002 | 0.1278 | 0.0014 | 0.3636 | 0.1278 | 0.7284 |
| MK10 | TAGTSFMMTpPYpVVTR | 0.0236 | −0.4020 | 0.0499 | 0.4244 | −0.4020 | 0.7284 |
| NBEA | EIEDLSQSQSpPESpETDYPVSTDTR | 0.2533 | 0.0973 | 0.3576 | 0.4144 | 0.0973 | 0.7284 |
| NBEA | TPLENVPGNLSpPIKDPDR | 0.0180 | −0.1595 | 0.0406 | 0.3981 | −0.1595 | 0.7284 |
| NCAM1 | DESpKEPIVEVR | 0.9721 | 0.0015 | 0.9798 | 0.9925 | 0.0015 | 0.9925 |
| NCAM2 | ITNHEDGSpPVNEPNETTPLTEPEK | 0.0423 | 0.1622 | 0.0781 | 0.2935 | 0.1622 | 0.7284 |
| NCAM2 | ITNHEDGSpPVNEPNETTpPLTEPEK | 0.3886 | 0.4858 | 0.4996 | 0.3886 | 0.4858 | 0.7284 |
| NMDE2 | HSQLSDLYpGK | 0.9273 | 0.0241 | 0.9676 | 0.9608 | 0.0241 | 0.9925 |
| PAK1 | TVSETPAVPPVSpEDEDDDDDATPPPVIAPRPEHTK | 0.7681 | 0.0103 | 0.8133 | 0.9220 | 0.0103 | 0.9762 |
| PEA15 | QPSpEEEIIK | 0.7666 | −0.0792 | 0.8133 | 0.8489 | −0.0792 | 0.9557 |
| PLCB1 | SEPSSPDHGSSpAIEQDLAALDAEMTQK | 0.0129 | −0.1209 | 0.0344 | 0.4324 | −0.1209 | 0.7284 |
| PLCB1 | VNLKSpPSpSEEVQGENAGR | 0.1122 | 0.1155 | 0.1878 | 0.3816 | 0.1155 | 0.7284 |
| PP1R7 | HGGGIVADLSpQQSpLK | 0.0211 | 0.1865 | 0.0460 | 0.3519 | 0.1865 | 0.7284 |
| PPR1B | IAESHLQTISNLSENQASpEEEDELGELR | 0.0000 | −0.3538 | 0.0001 | 0.5932 | −0.3538 | 0.8058 |
| PTPRN | LPEEGGSSpRAEDSSpEGHEEEVLGGHGEK | 0.0263 | 0.2670 | 0.0513 | 0.4188 | 0.2670 | 0.7284 |
| RLA1 | KEESpEESpEDDM(ox)GFGLFD | 0.0002 | 0.2178 | 0.0013 | 0.1795 | 0.2178 | 0.7284 |
| RLA2 | KEESpEESpDDDM(ox)GFGLFD | 0.0003 | 0.3184 | 0.0016 | 0.1069 | 0.3184 | 0.7284 |
| RP3A | WHQLQNENHVSSpD | 0.4637 | −0.2760 | 0.5757 | 0.4625 | −0.2760 | 0.7506 |
| SCN2A | RFSSpPHQSpLLSIR | 0.0005 | 0.1191 | 0.0021 | 0.6348 | 0.1191 | 0.8464 |
| SHAN2 | RAPSpPVVSpPTELSK | 0.0002 | 0.2541 | 0.0014 | 0.3608 | 0.2541 | 0.7284 |
| SHAN3 | SRSpPSpPSpPLPSPSPGSGPSAGPR | 0.0263 | 0.2147 | 0.0513 | 0.3748 | 0.2147 | 0.7284 |
| SRBS2 | SESpMGSpLLCDEGSK | 0.0068 | 0.4027 | 0.0204 | 0.1111 | 0.4027 | 0.7284 |
| SRCN1 | DSGSSSVFAESpPGGK | 0.0051 | 0.1440 | 0.0161 | 0.2492 | 0.1440 | 0.7284 |
| SRCN1 | KAESpEELEIQKPQVK | 0.6499 | 0.0178 | 0.7312 | 0.9149 | 0.0178 | 0.9762 |
| SRCN1 | RFSpNVGLVHTSER | 0.0005 | −0.2261 | 0.0021 | 0.0394 | −0.2261 | 0.7284 |
| SRCN1 | RGSpDELTVPR | 0.1518 | −0.0512 | 0.2377 | 0.6823 | −0.0512 | 0.8773 |
| SRCN1 | SSpGATpPVSGPPPPAVSSTPAGQPTAVSR | 0.3208 | 0.1193 | 0.4292 | 0.4850 | 0.1193 | 0.7506 |
| STMN1 | DLSpLEEIQK | 0.2030 | 0.0679 | 0.3027 | 0.7102 | 0.0679 | 0.8862 |
| STMN1 | ESVPEFPLSpPPK | 0.0251 | −0.0784 | 0.0513 | 0.7438 | −0.0784 | 0.8862 |
| STMN1 | RASpGQAFELILSpPR | 0.0123 | 0.2622 | 0.0340 | 0.3793 | 0.2622 | 0.7284 |
| STX1A | HSAILASpPNPDEK | 0.0676 | −0.1172 | 0.1187 | 0.3204 | −0.1172 | 0.7284 |
| STX1A | TAKDSpDDDDDVTVTVDR | 0.0004 | 0.1918 | 0.0020 | 0.1052 | 0.1918 | 0.7284 |
| SYPH | LHQVYpFDAPSCVK | 0.0847 | 0.0850 | 0.1452 | 0.7301 | 0.0850 | 0.8862 |
| SYT1 | DQALKDDDAETGLTpDGEEK | 0.4814 | 0.1493 | 0.5875 | 0.4814 | 0.1493 | 0.7506 |
| TNIK | SEGSpPVLPHEPSK | 0.1999 | −0.0503 | 0.3027 | 0.3956 | −0.0503 | 0.7284 |
| VIAAT | GGAPLPPSGSpK | 0.5718 | −0.0553 | 0.6535 | 0.7363 | −0.0553 | 0.8862 |
| Reactivation versus control | |||||||
| ADCY9 | GQGTASpPGSpVSDLAQTVK | 0.0041 | 0.5934 | 0.0084 | 0.1647 | 0.5934 | 0.4146 |
| AKA12 | ALGSpLGGSpPSLPDQDK | 0.7666 | −0.0604 | 0.8119 | 0.9193 | −0.0604 | 0.9734 |
| ARHG7 | MSpGFIYQGK | 0.0030 | 0.3177 | 0.0065 | 0.1157 | 0.3177 | 0.3783 |
| BAIP2 | SSSpMAAGLER | 0.7281 | 0.0759 | 0.8119 | 0.8649 | 0.0759 | 0.9730 |
| BSN | SPQVLYSpPVSpPLSPHR | 0.0063 | 0.2866 | 0.0120 | 0.0908 | 0.2866 | 0.3783 |
| CKD18 | RASpLSDIGFGK | 0.1156 | −0.0447 | 0.1734 | 0.8288 | −0.0447 | 0.9730 |
| CSKI1 | KVPLPGPGSpPEVK | 0.0305 | −0.3568 | 0.0500 | 0.0260 | −0.3568 | 0.2341 |
| CTNA1 | SRTpSpVQTpEDDQLIAGQSAR | 0.0604 | −0.1133 | 0.0945 | 0.5393 | −0.1133 | 0.7949 |
| CTNA1 | SRTSpVQTpEDDQLIAGQSAR | 0.0000 | 0.2878 | 0.0000 | 0.0124 | 0.2878 | 0.1491 |
| CTNB1 | RTSMGGTpQQQFVEGVR | 0.6712 | 0.0640 | 0.7922 | 0.8436 | 0.0640 | 0.9730 |
| CTND2 | ALQSpPEHHIDPIYEDR | 0.7668 | 0.0369 | 0.8119 | 0.7588 | 0.0369 | 0.9419 |
| CXA1 | M(ox)GQAGSTISpNSpHAQPFDFPDDNQNAK | 0.0000 | 0.3038 | 0.0000 | 0.1234 | 0.3038 | 0.3783 |
| CXA1 | MGQAGSTISpNSpHAQPFDFPDDNQNAK | 0.0000 | 0.3992 | 0.0001 | 0.1918 | 0.3992 | 0.4359 |
| CXA1 | VAAGHELQPLAIVDQRPSSpRASpSpR | 0.0000 | −0.3599 | 0.0000 | 0.0198 | −0.3599 | 0.2037 |
| DGKB | GAITpPPRSSpPANTCSPEVIHLK | 0.9499 | −0.0044 | 0.9738 | 0.9901 | −0.0044 | 0.9901 |
| EF1D | ATAPQTQHVSpPMR | 0.7183 | −0.2315 | 0.8119 | 0.7183 | −0.2315 | 0.9403 |
| FAK2 | RNSpLPQIPTLNLESR | 0.0000 | 0.2167 | 0.0001 | 0.1670 | 0.2167 | 0.4146 |
| FGF12 | EPSpLHEIGEK | 0.0000 | −0.8485 | 0.0000 | 0.2585 | −0.8485 | 0.5444 |
| GABR1 | HPPTpPPDPSGGLPR | 0.5901 | −0.0188 | 0.7201 | 0.9144 | −0.0188 | 0.9734 |
| GABR1 | RHPPTpPPDPSGGLPR | 0.4862 | −0.0397 | 0.6251 | 0.8359 | −0.0397 | 0.9730 |
| GABR2 | DPIEDINSpPEHIQR | 0.0054 | −0.2011 | 0.0105 | 0.3330 | −0.2011 | 0.5994 |
| GIT1 | HGSpGAESDYENTQSGEPLLGLEGK | 0.3029 | −0.0707 | 0.4194 | 0.5410 | −0.0707 | 0.7949 |
| GIT1 | NQSDLDDQHDYDSpVASpDEDTDQEPLPSAGATR | 0.0002 | 0.2143 | 0.0005 | 0.0370 | 0.2143 | 0.2661 |
| IF3M | LLYLTpSpAK | 0.7412 | 0.1111 | 0.8119 | 0.7412 | 0.1111 | 0.9419 |
| IPP2 | EQESpSpGEEDNDLSPEER | 0.0052 | 0.3180 | 0.0104 | 0.2948 | 0.3180 | 0.5736 |
| KCC2A | ESSESTpNTpTpIEDEDTK | 0.0003 | 0.4759 | 0.0008 | 0.0322 | 0.4759 | 0.2578 |
| KCC2A | ESSpESTNTTIEDEDTK | 0.0000 | −0.6186 | 0.0001 | 0.4973 | −0.6186 | 0.7949 |
| KCNQ2 | HGTSpPVGDHGSLVR | 0.1937 | −1.4748 | 0.2847 | 0.1937 | −1.4748 | 0.4359 |
| KPCB | HPPVLTpPPDQEVIR | 0.0000 | −0.1782 | 0.0000 | 0.6023 | −0.1782 | 0.8339 |
| KPCG | AAPALTpPPDR | 0.3605 | −0.3204 | 0.4806 | 0.3961 | −0.3204 | 0.6633 |
| KPCG | TpFCGTPDYIAPEIIAYQPYGK | 0.0000 | −0.2607 | 0.0000 | 0.1253 | −0.2607 | 0.3783 |
| MARCS | EAAEAEPAEPGSpPSAETEGASASSTSSPK | 0.0000 | 0.3923 | 0.0000 | 0.0730 | 0.3923 | 0.3726 |
| MARCS | GEAAAERPGEAAVASpSPSK | 0.2646 | −1.9668 | 0.3736 | 0.2646 | −1.9668 | 0.5444 |
| MARK2 | DQQNLPFGVTPASpPSGHSQGR | 0.0000 | 0.2598 | 0.0001 | 0.0023 | 0.2598 | 0.0559 |
| MINK1 | SDSVLPASHGHLPQAGSpLER | 0.0085 | 0.1011 | 0.0153 | 0.3707 | 0.1011 | 0.6509 |
| MK01 | VADPDHDHTGFLTpEYpVATR | 0.0004 | 0.1374 | 0.0008 | 0.3844 | 0.1374 | 0.6590 |
| MK10 | TAGTSFMMTpPYpVVTR | 0.0002 | 0.7296 | 0.0004 | 0.1492 | 0.7296 | 0.4132 |
| NBEA | EIEDLSQSQSpPESpETDYPVSTDTR | 0.0088 | 0.2889 | 0.0155 | 0.0659 | 0.2889 | 0.3648 |
| NBEA | TPLENVPGNLSpPIKDPDR | 0.0003 | −0.2541 | 0.0006 | 0.1069 | −0.2541 | 0.3783 |
| NCAM1 | DESpKEPIVEVR | 0.4820 | −0.0317 | 0.6251 | 0.8555 | −0.0317 | 0.9730 |
| NCAM2 | ITNHEDGSpPVNEPNETTPLTEPEK | 0.9769 | −0.0021 | 0.9769 | 0.9894 | −0.0021 | 0.9901 |
| NCAM2 | ITNHEDGSpPVNEPNETTpPLTEPEK | 0.5354 | −0.3821 | 0.6763 | 0.5354 | −0.3821 | 0.7949 |
| NMDE2 | HSQLSDLYpGK | 0.9603 | 0.0113 | 0.9738 | 0.9690 | 0.0113 | 0.9901 |
| PAK1 | TVSETPAVPPVSpEDEDDDDDATPPPVIAPRPEHTK | 0.2036 | −0.0521 | 0.2932 | 0.6999 | −0.0521 | 0.9383 |
| PEA15 | QPSpEEEIIK | 0.7288 | 0.0539 | 0.8119 | 0.8991 | 0.0539 | 0.9734 |
| PLCB1 | SEPSSPDHGSSpAIEQDLAALDAEMTQK | 0.0348 | 0.1013 | 0.0558 | 0.7037 | 0.1013 | 0.9383 |
| PLCB1 | VNLKSpPSpSEEVQGENAGR | 0.5520 | −0.1562 | 0.6853 | 0.5302 | −0.1562 | 0.7949 |
| PP1R7 | HGGGIVADLSpQQSpLK | 0.0001 | 0.4104 | 0.0002 | 0.0546 | 0.4104 | 0.3275 |
| PPR1B | IAESHLQTISNLSENQASpEEEDELGELR | 0.0000 | −0.4040 | 0.0000 | 0.5816 | −0.4040 | 0.8211 |
| PTPRN | LPEEGGSSpRAEDSSpEGHEEEVLGGHGEK | 0.0001 | 0.4983 | 0.0003 | 0.1004 | 0.4983 | 0.3783 |
| RLA1 | KEESpEESpEDDM(ox)GFGLFD | 0.0000 | 0.3790 | 0.0000 | 0.0048 | 0.3790 | 0.0868 |
| RLA2 | KEESpEESpDDDM(ox)GFGLFD | 0.0001 | 0.4373 | 0.0003 | 0.0009 | 0.4373 | 0.0559 |
| RP3A | WHQLQNENHVSSpD | 0.0067 | −0.4470 | 0.0124 | 0.5687 | −0.4470 | 0.8189 |
| SCN2A | RFSSpPHQSpLLSIR | 0.0000 | 0.4239 | 0.0000 | 0.0446 | 0.4239 | 0.2922 |
| SHAN2 | RAPSpPVVSpPTELSK | 0.0000 | 0.3424 | 0.0000 | 0.2148 | 0.3424 | 0.4686 |
| SHAN3 | SRSpPSpPSpPLPSPSPGSGPSAGPR | 0.0009 | 0.5152 | 0.0019 | 0.1261 | 0.5152 | 0.3783 |
| SRBS2 | SESpMGSpLLCDEGSK | 0.0002 | 0.6422 | 0.0005 | 0.0801 | 0.6422 | 0.3726 |
| SRCN1 | DSGSSSVFAESpPGGK | 0.0000 | 0.2076 | 0.0001 | 0.1560 | 0.2076 | 0.4146 |
| SRCN1 | KAESpEELEIQKPQVK | 0.0146 | −0.1133 | 0.0250 | 0.4970 | −0.1133 | 0.7949 |
| SRCN1 | RFSpNVGLVHTSER | 0.0000 | −0.3600 | 0.0001 | 0.0083 | −0.3600 | 0.1191 |
| SRCN1 | RGSpDELTVPR | 0.0001 | −0.2053 | 0.0004 | 0.1774 | −0.2053 | 0.4259 |
| SRCN1 | SSpGATpPVSGPPPPAVSSTPAGQPTAVSR | 0.9596 | 0.0141 | 0.9738 | 0.9630 | 0.0141 | 0.9901 |
| STMN1 | DLSpLEEIQK | 0.6268 | 0.0313 | 0.7522 | 0.8983 | 0.0313 | 0.9734 |
| STMN1 | ESVPEFPLSpPPK | 0.3329 | −0.0552 | 0.4523 | 0.7488 | −0.0552 | 0.9419 |
| STMN1 | RASpGQAFELILSpPR | 0.0000 | 0.5433 | 0.0001 | 0.1049 | 0.5433 | 0.3783 |
| STX1A | HSAILASpPNPDEK | 0.0193 | −0.1344 | 0.0323 | 0.3284 | −0.1344 | 0.5994 |
| STX1A | TAKDSpDDDDDVTVTVDR | 0.7533 | −0.0147 | 0.8119 | 0.8233 | −0.0147 | 0.9730 |
| SYPH | LHQVYpFDAPSCVK | 0.0037 | −0.4914 | 0.0079 | 0.2792 | −0.4914 | 0.5583 |
| SYT1 | DQALKDDDAETGLTpDGEEK | 0.0828 | −0.2498 | 0.1269 | 0.0828 | −0.2498 | 0.3726 |
| TNIK | SEGSpPVLPHEPSK | 0.0000 | −0.3677 | 0.0001 | 0.0019 | −0.3677 | 0.0559 |
| VIAAT | GGAPLPPSGSpK | 0.0002 | −0.4927 | 0.0005 | 0.3042 | −0.4927 | 0.5763 |
Assessment of effect of CaMKIIα phosphorylation at S331 on catalytic activity
Site-directed mutagenesis.
CaMKIIα phospho-deficient (S331 to alanine; S331A) and phospho-mimetic (S331 to glutamate; S331E) mutants were created using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies). The PCR mixture contained 50 ng of template plasmid DNA (pCMV6-CaMKIIα-Myc-DDK plasmid; Origene, RR201121), 125 ng of each oligonucleotide (For: GGCGTGAAGGAATCCGCTGAGAGCACCAACACC, Rev: GGTGTTGGTGCTCTCAGCGGATTCCTTCACGCC), 1 μl dNTP mix, 2.5 U PfuUltra DNA polymerase, 5 μl 10× Reaction buffer, and 3 μl Quiksolution reagent in a final volume of 50 μl. PCR was performed under the following conditions; denaturation at 95°C for 50 s, annealing at 60°C for 50 s and extension at 68°C for 1 min/kb. The PCR product was digested with 10 U of DpnI for 1 h at 37°C and then transformed into 45 μl of XL10-Gold Ultracompetent Escherichia coli cells. Presence of the mutation was confirmed by DNA sequencing of the construct.
Cell culture and plasmid transfection.
HEK293T cells were maintained in culture in a humidified 5% CO2 atmosphere at 37°C in medium consisting of DMEM/F12 supplemented with 10% fetal bovine serum, and penicillin-streptomycin. Transfections were performed using the Lipofectamine 2000 Reagent (Invitrogen). HEK293T cells were seeded in 6-well plates at 3 × 105 cells per well, grown for 24 h and then incubated overnight with 2 ml of serum-free medium containing 5 μg wild-type (WT) or mutant pCMV6-CaMKIIα expression vectors, and 7.5 μg Lipofectamine. Control cells were incubated with a plasmid-free mixture of medium and Lipofectamine. After overnight incubation, the medium was replaced by fresh medium supplemented with 500 μg/μl geneticin (Invitrogen) to select for transfected cells. To determine basal levels of CaMKIIα expression in HEK293T cultures, a subset of the control cells did not receive geneticin treatment.
Immunoblot and kinase activity assay.
HEK293T whole-cell lysates were prepared by washing cultures with ice-cold 1× PBS followed by treatment with ice-cold lysis buffer containing protease inhibitors (1:100). Cells were collected and incubated on ice for 30 min, then centrifuged at 16,000 × g for 20 min at 4°C. Supernatants from two 6-well plates were collected and pooled together, and protein content was quantified using the Pierce BCA assay kit. Supernatants with equal amounts of protein (10 μg) were resolved via SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blocked with 5% nonfat dairy milk in PBST (PBS + 0.1% Tween 20). Membranes were then probed with the following primary antibodies: Rabbit Anti-CaMKII (Millipore 07-1496; 1:500) and Mouse Anti-GAPDH (Millipore MAB374; 1:1000). Goat anti-rabbit IgG (Li-Cor 926-32211; 1:5000) and goat anti-mouse IgG (Li-Cor 926-68070; 1:5000) secondary antibodies were used. Odyssey Infrared Imager (Li-Cor) was used for the detection of protein bands. Kinase activity was quantified following the instructions of the ADP-Glo Kinase Assay (Promega) in the presence of CaCl2, MgCl2, calmodulin, and the CaMKII selective substrate, autocamtide-2. For each reaction, relative amounts of protein supernatant (∼2.5 μg) were loaded based on total protein levels in the lysate, corrected for loading of GAPDH. Reactions were initiated by the addition of 250 μm ATP then incubated for 15 min at 30°C. Reactions were terminated and the unconsumed ATP depleted by the addition of ADP-Glo Reagent, then incubated for 40 min at RT. Kinase Detection Reagent was then added to each reaction to convert ADP back to ATP while also introducing luciferase and luciferin to detect ATP. This was followed by another 40 min incubation at RT. Luminescence was measured using the FLx800 Multi-Detection Microplate Reader (BioTek) Using a linear standard curve, the amount of depleted ADP was determined, and these values were converted to specific kinase activity.
CaMKII inhibition and reinstatement of cocaine seeking
Memory manipulations.
Memory manipulations took place as described in the proteomics experiment above with the following differences. Pavlovian cue extinction was conducted in the same context in which rats received self-administration training, and took place for either a single 30 min session (moderate extinction) or a 30 min session on 2 consecutive days (extended extinction). For cue reactivation, rats were placed in operant chambers, in the opposite context from which they were trained, for a single memory reactivation session. During this session, the cocaine-associated cue was presented for 10 s, three times, with each presentation separated by 1 min. Control rats were again placed in the opposite context form which they were trained, but were never presented with the cocaine-associated cues.
Intracranial infusions.
Drugs were administered in a volume of 0.5 μl/hemisphere. KN-62 was given at one of two doses: 340 or 680 ng/side. KN-93 was given at either 5 or 10 μg/side. These doses were based on effective doses in prior publications using intracranial infusions (Rodrigues et al., 2004; Sakurai et al., 2007). Infusions were given immediately following the memory manipulations, by removing dummy cannulae and inserting injection cannulae (28 gauge; Plastics One) that extended 1 mm beyond the guide cannulae. The injectors were connected to Hamilton syringes (Hamilton Robotics) controlled by a syringe pump via polyethylene tubing. Infusions were given over the course of 2 min and injectors were left in the cannulae for an additional 1 min to allow for drug diffusion.
Cue-induced reinstatement.
Twenty-four hours after the final Pavlovian cue extinction session or memory reactivation session, cue-induced reinstatement was assessed during a 1 h session that took place in the original self-administration context. A lever press on the active lever produced a 10 s presentation of the cocaine-associated cue on an FR1 schedule, but no drug reinforcement. Lever presses on the inactive lever were recorded but had no programmed consequences.
Histological analysis.
After the completion of experiments, rats with intracranial cannulae were killed via decapitation. Brains were dissected and placed in 10% formalin for at least 3 d then transferred to 30% sucrose for at least 3 d. Brains were then frozen and sectioned coronally through the BLA on a cryostat. Sections were taken at 50 μm and placed on slides for visualization of infusion placements. The investigator was blind to treatment group when analyzing histology, and animals with infusions outside of the BLA were removed from the main analysis.
Code availability.
All behavioral training and testing was conducted using custom MedPC programs. The computer code used is available to researchers upon request.
Statistical analyses.
Behavioral and kinase activity data were analyzed using GraphPadPrism for Windows. For proteomic data, the normalized average intensity across transitions for each phosphopeptide was computed for each sample and these values were averaged within groups. Group averages were compared statistically using a SRMstats restricted analysis and p values corrected for multiple comparisons. For immunoblot data, intensities of protein bands were quantified using Odyssey Imaging software v3.0. Total CaMKII amount was normalized based on the signal intensity of GAPDH and expressed as %WT. For kinase activity data, samples were run in duplicate. Luminescence was normalized to a “no-substrate control” and converted to kinase specific activity based on a standard conversion curve. Data were analyzed using a one-way ANOVA. Reinstatement tests were analyzed by two-way ANOVA with repeated-measures, with the between-subjects factor being responding on the last day of lever extinction versus reinstatement responding and the within-subjects factor being the dose of drug. For the no reactivation control group, vehicle groups were statistically similar and therefore collapsed across treatment groups. For the anatomical control experiment, data were analyzed using an independent samples t test. For all analyses, significant effects were further analyzed by Bonferroni's post hoc tests, with significance set at p < 0.05. All data were determined to be normally distributed using the Shapiro–Wilk test, and Bartlett's test was used to determine that there were no significant differences in the estimated variance between groups.
Results
Identification of novel signaling events that are regulated by cocaine-cue memory extinction and reconsolidation
To identify candidate signaling events that are regulated in opposition by extinction versus reconsolidation of a memory associated with self-administered cocaine, we used a high resolution tandem mass spectrometry-based phosphoproteomics approach, where we examined differential protein phosphorylation events after memory manipulations as an index of increased or decreased protein activity. Rats were trained to self-administer cocaine paired with an audiovisual cue (CS) for 10 d, followed by 5–7 d of extinction of instrumental lever responding (Fig. 1a). Each rat was assigned to a memory manipulation group in a random manner that ensured no training differences between groups. There were no significant differences or interactions with day of training for infusions earned (Fig. 1b) or active lever presses during extinction (Fig. 1c; two-way repeated-measures ANOVA: p > 0.05, n = 7–10 rats per group). No statistical differences were observed for active lever presses during acquisition or inactive lever presses at any stage (data not shown). Following lever extinction, rats were exposed to one of three memory conditions: reactivation, extinction, or control context exposure. Fifteen minutes following the memory manipulation, rats were euthanized by focused microwave irradiation to maintain protein post-translational modifications, and the brains were dissected for proteomics analysis. Fifteen minutes was chosen as it is thought to represent the time when many of the intracellular signaling cascades regulating the extinction or reconsolidation of a memory would be active. Here we describe results from the analysis of the BLA, as it is the locus for associative learning and mediates the encoding of drug-associated memories (LeDoux, 2000; Schafe and LeDoux, 2000; See, 2005).
Figure 1.

Self-administration and extinction training data from rats used for phosphoproteomic analysis. a, Timeline for proteomics experiments. b, Mean number of cocaine infusions per day during acquisition and (c) mean number of active lever presses per day during instrumental extinction in rats before memory manipulations. There were no differences in acquisition or instrumental extinction between manipulation groups (both p > 0.05, n = 7, 9, and 10 rats, respectively, for control, extinction, and reconsolidation). Data are expressed as mean + SEM.
Total homogenates were enriched for phosphopeptides, and discovery-based, label ad libitum quantitative analysis was used as a first step to identify putative memory-regulated proteins. A large number of phosphopeptides were detected corresponding to 355 unique proteins. From this list of phosphopeptides, in a second step also used for validation, we pseudorandomly chose ∼80 phosphopeptides for quantitative validation by SRM mass spectrometry. We prioritized the phosphopeptides to be chosen by selecting those with robust ionization intensity that were likely to provide clear MS signals and to exclude highly abundant, heavily phosphorylated proteins, such as neurofilaments and microtubule-associated proteins. Phosphorylation of these proteins is potentially relevant to memory processes, but because of their abundance and high degree of phosphorylation, they may be over-represented in the analysis. A total of 72 unique phosphopeptides had sufficient data quality from the SRM experiment for data analysis. Table 1 contains the statistical analysis, as described in the methods, and estimated fold-change in the level of phosphopeptides in the extinction and reactivation groups relative to the control. Figure 2 summarizes the data from all phosphopeptides that showed a significant change from control when analyzed using SRMstats with a significance cutoff of p < 0.05 after correction for multiple comparisons. Each panel is organized to illustrate the phosphopeptides significantly regulated by both extinction and reconsolidation in the opposite direction (Fig. 2a); those significantly regulated in the same direction by both conditions (b); those only significantly regulated in the extinction condition (c); and those only significantly regulated in the reactivation condition (d). Of greatest interest for our study were the five phosphopeptides regulated in the opposite direction by extinction versus reconsolidation, as these could represent targets that can both enhance extinction while inhibiting reconsolidation to ensure sufficient weakening of a drug-associated memory. These phosphopeptides included CaMKIIα (KCC2A, pS331), c-Jun N-terminal kinase 3 (JNK3 or MK10, pT221+Y223), the gap junction protein connexin 43 (CXA1, pS328+pS330), the GABAB receptor subunit 2 (GABR2, pS883), and phospholipase C β 1 (PLCB1, pS988). Interestingly, CaMKIIα, phospholipase C, and GABAB receptors are important regulators of Ca2+ signaling (Chalifoux and Carter, 2011; Lyon and Tesmer, 2013), and JNK3 activation can be regulated by PLC (Buckley et al., 2004), indicating a convergence of opposing calcium-related cellular events dependent on whether a cocaine memory is reconsolidated or extinguished.
Figure 2.

Identification of signaling pathways regulated by cocaine-associated memory extinction and reconsolidation. a–d, Phosphoproteomic analysis from tissue collected 15 min following memory manipulations reveals several phosphopeptides that were significantly regulated (a) in the opposite direction by extinction versus reconsolidation, (b) in the same direction by both memory conditions, (c) by memory extinction only (only red bars significantly different from control), or (d) by memory reconsolidation only (only blue bars significantly different from control). The y-axes list the Uniprot database protein abbreviation. The number in parenthesis following an abbreviation is the ID we assigned when multiple unique phosphopeptides were identified from the same protein. Bars represent the estimated fold-change in phosphopeptide abundance relative to non-memory manipulated controls. Positive values indicate significant increases in the abundance of the phosphopeptide, and negative values decreased abundance. All p < 0.05 relative to control after correction for multiple comparisons using SRMstats for statistical analysis of SRM data (Chang et al., 2012).
Phosphorylation of CaMKII at S331 decreases catalytic activity
CaMKIIα is known to be an important regulator of learning and memory (Coultrap and Bayer, 2012; Sanhueza and Lisman, 2013), and is an abundant protein, thus it is not surprising that CaMKIIα was identified in our analysis. Yet, somewhat surprisingly, the differentially regulated phosphorylation event on CaMKIIα was on residue S331. S331 phosphorylation was significantly increased after memory extinction, but was significantly decreased after memory reactivation (Fig. 2a). To our knowledge, S331 phosphorylation has never been previously reported in relation to learning and memory or any other biological process. However, one recent study has found that S331 is a potential autophosphorylation site (Baucum et al., 2015). Therefore, we proceeded to determine whether phosphorylation of S331 regulates the enzymatic activity of CaMKIIα. Phospho-deficient (S331A) and phospho-mimetic (S331E) forms of CaMKIIα DNA were generated (Fig. 3a) and, along with a WT form, expressed in HEK293T cells. The absence of endogenous CaMKIIα in HEK293T cells, as previously reported (Tsui et al., 2005; Jenkins et al., 2010), was confirmed by immunoblot analysis, while expression of all recombinant forms of CaMKIIα was comparable (Fig. 3b; one-way ANOVA: F(3,28) = 25.84, p < 0.001, n = 8 samples per group). CaMKIIα activity in whole-cell lysates was measured in the presence of Ca2+, calmodulin, ATP, and the CaMKIIα selective substrate, autocamtide-2. Kinase activity differed across plasmid types (Fig. 3c; one-way ANOVA: F(3,28) = 8.714, p < 0.001, n = 8 samples per group). The catalytic activity of phospho-mimetic CaMKIIα (S331E) was significantly lower than that of the S331A mutant enzymes (p < 0.05). The kinase activity of WT CaMKIIα was between the levels observed for S331A- and S331E-mutated lysates. Although the phosphorylation state of the WT CaMKIIα at S331 is unknown, it is likely that the total protein contains a mixture of phosphorylated and non-phosphorylated S331, and as an autophosphorylation site, had the potential to be phosphorylated in the presence of Ca2+, calmodulin, and ATP. However, our results suggest that in HEK293 cells, WT CaMKIIα is predominantly not phosphorylated, as kinase activity more closely mimicked that of the phospho-deficient mutant.
Figure 3.

Phosphorylation of CaMKIIα at S331 inhibits enzyme activity. a, Schematic representation of site-directed mutagenesis of CaMKIIα at S331 to generate phospho-deficient (S331A) or phospho-mimetic (S331E) mutants. b, WT S331A and S331E plasmids were expressed in HEK293T cells. Supernatants were collected and CaMKIIα expression was quantified via immunoblot, normalized to GAPDH, and expressed as %WT. Results indicate a lack of CaMKIIα in the non-transfected control (No Tfx) condition, but similar expression of all recombinant forms of CaMKIIα (n = 8 samples per group). Representative immunoblots are shown in the top. c, Quantification of CaMKII activity from whole-cell lysates. Kinase activity was regulated by the phospho-mimetic mutation of S331 (n = 8 samples per group). Catalytic activity of the CaMKIIα S331E mutant was significantly lower than that of the S331A mutant. Data are expressed as mean + SEM. *p < 0.05.
Inhibition of CaMKII in the BLA enhances drug-cue memory extinction
Based on the apparent increase in an inhibitory phosphorylation event at S331 on CaMKIIα after extinction, and a decrease in phosphorylation after reactivation, we hypothesized that memory reconsolidation must require active CaMKIIα in the BLA, whereas extinction requires a deactivation of CaMKIIα. Therefore, we investigated whether CaMKII inhibition could both inhibit drug-cue memory reconsolidation and enhance extinction to reduce relapse-like cue-induced reinstatement. For this set of experiments, rats again underwent standard cocaine self-administration and lever extinction training before placement into memory manipulation groups. Following memory manipulations rats received immediate infusion of one of two doses of two related CaMKII inhibitors (KN-93 or KN-62) or the vehicle directly into the BLA, and the propensity to reinstate cocaine-seeking in response to the cue was assessed 24 h later (Fig. 4a). For all experiments involving CaMKII inhibition, there were no pre-existing differences between drug-treatment groups in number of reinforcers earned, active lever responding, or inactive lever responding during acquisition or lever extinction (KN-93: Fig. 4b; KN-62: Fig. 4c; all p > 0.05, n = 5–11 rats/group). This ensures that any memory manipulation or drug effect on cue-induced reinstatement was not due to pre-existing differences in propensity to self-administer drug or extinguish drug seeking.
Figure 4.

No differences in training data between groups before memory manipulations/BLA infusions. a, Experimental timeline. Rats received bilateral infusions of one of two CaMKII inhibitors (KN-93 or KN-62) or vehicle into the BLA immediately following memory manipulation sessions. Twenty-four hours later, rats were tested for cue-induced reinstatement under drug-free conditions. Data are displayed separately for groups treated with the CaMKII inhibitor KN-93 (b) or KN-62 (c). Left, Mean number of cocaine infusions per day during acquisition. Center, Mean number of active (solid shapes) and inactive (open shapes) lever presses per day during acquisition. Right, Mean number of active and inactive lever presses per day during instrumental extinction. For simplicity, data are grouped via dose of CaMKII inhibitor, collapsed across memory manipulation. There are no differences, in any measures, during acquisition or instrumental extinction between treatment groups for any of the memory manipulations (all p > 0.05). Data are expressed as mean + SEM.
Pavlovian cue extinction has previously been demonstrated as an effective method for reducing drug-cue motivated behavior (Torregrossa et al., 2010, 2013). To determine whether the effects of cue extinction on reinstatement could be amplified by inhibiting CaMKII, rats were given intra-BLA infusions of a CaMKII inhibitor immediately following two types of extinction training: 60 versus 120 CS presentations. In this manner, we could determine whether CaMKII inhibition could facilitate suboptimal (moderate), as well as extended, extinction training. All cue extinction and reinstatement testing occurred in the same context to specifically understand extinction consolidation processes, rather than contextual encoding or renewal. CaMKII inhibition with either KN-93 or KN-62 following moderate cue extinction reduced subsequent cue-induced reinstatement (Fig. 5a; KN-93: F(2,15) = 6.671, p = 0.009, n = 5–7 rats per group; KN-62: F(2,17) = 5.036, p = 0.019, n = 6–7 rats per group; both two-way ANOVA). These data suggest that CaMKII inhibition after extinction training can enhance the effects of moderate cue extinction training to reduce reinstatement. On the other hand, an extended cue extinction-duration protocol (120 cues) immediately followed by infusion of the CaMKII inhibitor KN-62, failed to reduce reinstatement relative to vehicle-treated controls. As a whole, all rats that experienced extended cue extinction in the self-administration context exhibited the expected extinction-induced reduction in reinstatement that could not be further potentiated by CaMKII inhibition (Fig. 5a; two-way ANOVA: p > 0.05, n = 6–8 rats per group), indicating that 120 CS presentations likely produces a floor effect. These results are similar to previously published work demonstrating that extended cue extinction in the self-administration context could not be potentiated by treatment with d-cycloserine (DCS), even though DCS can promote extinction learning under other conditions (Torregrossa et al., 2010).
Figure 5.

CaMKII inhibition enhances drug-cue memory extinction and disrupts memory reconsolidation to reduce cue-induced reinstatement. a, Mean number of active lever presses during cue-induced reinstatement, 24 h after extinction manipulations. CaMKII inhibition by KN-93 (left) and KN-62 (middle) enhanced the effects of a single (moderate) cue extinction session. Both doses of KN-93 and the 680 ng/side dose of KN-62 significantly decreased reinstatement in the moderate extinction condition. CaMKII inhibition by KN-62 (right) had no effect after extended extinction training (p > 0.05). b, Mean number of active lever presses during cue-induced reinstatement, 24 h after memory reactivation manipulations. CaMKII inhibition by KN-93 (left) and KN-62 (right) disrupted drug-cue memory reconsolidation. Both doses of KN-93, but only the 340 ng/side dose of KN-62 reduced reinstatement. Data are expressed as mean + SEM; number of rats per group indicated by number inside bars. *p < 0.05 versus vehicle.
Inhibition of CaMKII in the BLA interferes with drug-cue memory reconsolidation
We then tested whether CaMKII inhibition in the BLA following reactivation of the cocaine cue memory (3 CS presentations) could disrupt reconsolidation. After cocaine self-administration and lever extinction training, the cocaine-associated cue memory was reactivated in a novel context, followed by immediate infusion of CaMKII inhibitors into the BLA. Rats were then tested for cue-induced reinstatement of cocaine seeking 24 h later. KN-93 at both doses decreased active lever responding during reinstatement relative to VEH-treated controls, whereas only the low dose of KN-62 significantly reduced reinstatement (Fig. 5b; KN-93: main effect: F(2,20) = 3.587, p = 0.047, n = 7–9 rats per group; KN-62: significant interaction between treatments: F(2,23) = 4.413, p = 0.024, n = 6–11 rats per group; both two-way ANOVA). Due to the consistency of effects with both doses of KN-93 and the low dose of KN-62, we suspect that the high dose of KN-62 produced nonspecific effects that occluded any influence of CaMKII inhibition; however, we cannot rule out the possibility that there is a U-shaped dose effect function in the ability of CaMKII inhibition to interfere with memory reconsolidation. Nevertheless, together these data suggest that CaMKII may be important for the reconsolidation (restabilization) of the drug-cue memory following reactivation, and inhibition of CaMKII has the potential to interfere with this reconsolidation.
Intra-BLA CaMKII inhibition does not affect reinstatement in the absence of memory manipulations
Finally, we wanted to confirm that the effects of CaMKII inhibition were selective to memory manipulations rather than nonspecific effects on locomotion or motivation, and that inhibitor actions were mediated within the BLA. First, we determined whether the highest dose of either CaMKII inhibitor could affect cue-induced reinstatement if it was given following placement of rats in a novel operant context without cue presentation or the opportunity to make an instrumental response (ie, a no reactivation control experiment). Post-session treatment with the high dose of either KN-93 or KN-62 had no significant effect on active responding during cue-induced reinstatement, indicating no effect on cocaine seeking in the absence of a memory manipulation (Fig. 6a; two-way ANOVA: p > 0.05, n = 5–9 rats per group). Therefore, effects of CaMKII inhibition on cue-induced reinstatement are likely due to specific actions on extinction and reconsolidation processes. Next, we determined whether infusions of CaMKII inhibitors outside of the BLA had any effect on subsequent cue-induced reinstatement. We analyzed the data from animals whose vehicle or inhibitor infusion locations were found to lie outside of the BLA after histological analysis. We included rats whose infusion locations were dorsal, lateral, and medial to the BLA. We found no significant effect of CaMKII inhibitors infused after extinction or reactivation of the cocaine memory on cue-induced reinstatement if the infusions were outside of the BLA, suggesting that drug spread into other regions does not explain our behavioral results (Fig. 6b,c; t test, p values >0.05, n = 5–8 rats per group). Figure 7 illustrates the intracranial infusion locations that were considered to lie within the BLA. Infusions outside the areas shown by the black dots were considered misses.
Figure 6.

CaMKII inhibition has no effect on cue-induced reinstatement if the memory was not reactivated or infusions occurred outside of the BLA. a, Cue-induced reinstatement in animals that did not receive drug-cue memory extinction or reactivation. For this control, rats were placed back in self-administration chambers but received 0 CS presentations. There were no effects of CaMKII inhibition on active lever presses during the cue-induced reinstatement test. (left bar, vehicle; middle bar, KN-93; right bar, KN-62). b, c, Cue-induced reinstatement in animals with infusions outside of the BLA. There were no effects of CaMKII inhibition on active lever presses during the cue-induced reinstatement test in rats that received infusions outside of the boundaries of the BLA after (b) a single (moderate) cue extinction session or (c) reactivation. Reinstatement data are from rats infused with either the effective dose of KN-93 or KN-62 (5 μg/side and 680 ng/side, respectively). Rats were pooled due to the low numbers of animals in each individual inhibitor condition, and because there was no difference in reinstatement behavior between the compounds. Data are expressed as mean + SEM; number of rats per group indicated by number inside bars.
Figure 7.
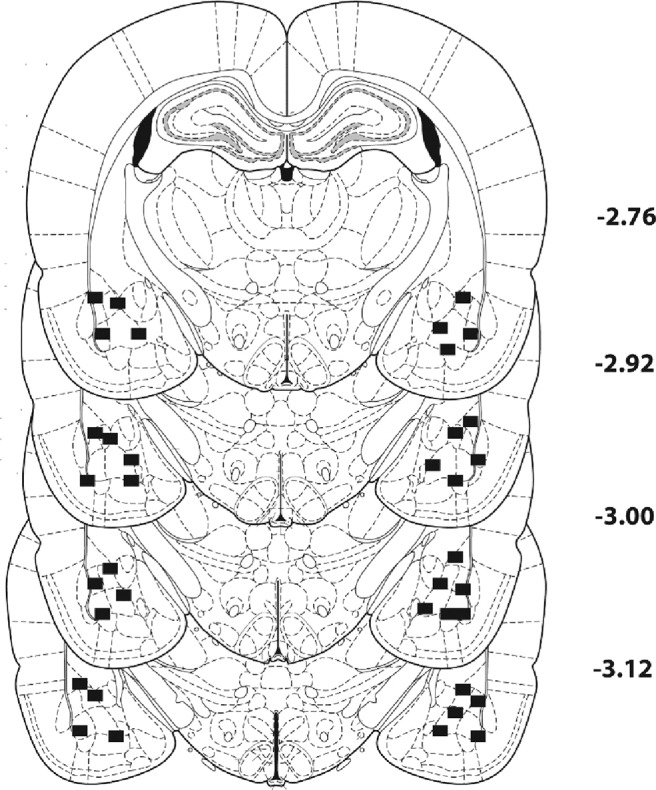
Histological representation of infusion locations in the BLA. The black squares represent the outermost range in the medial-lateral and dorsal-ventral planes that were considered a hit in the BLA, all other placements were located within these bounds. Numbers by each section represent millimeters from bregma.
Discussion
The present study identified brain-region-specific protein phosphorylation events that occur in response to the reactivation or extinction of cocaine-cue-specific memories. Our proteomic analysis indicated that several cellular signaling cascades were activated in the same manner by both extinction and reconsolidation, including increased phosphorylation of extracellular regulated kinase 2 and 60S ribosomal subunits, which regulate protein synthesis. These results are consistent with prior findings in the literature indicating that ERK activity and protein synthesis are required for both the reconsolidation and extinction of memory (Duvarci et al., 2005; Cestari et al., 2006; Tronson and Taylor, 2007). The proteomic analysis also revealed several phosphorylation events that were selective to one memory condition or the other, and importantly, identified five proteins that exhibited opposing directions of phosphorylation under extinction versus reactivation conditions. Thus, we report the identification of several signaling pathways that may be viable targets for developing medications to be used in conjunction with memory-based behavioral interventions. In particular, phospholipase C and c-jun N-terminal kinase 3 are two proteins that have been associated with memory regulation in other paradigms, and deserve further study. It should be noted that it is possible that some of the protein phosphorylation changes observed are not related to memory, but rather could be associated with a change in motivational state. Nonetheless, use of this discovery-based approach represents a novel method for determining signaling events involved in specific memory processes.
Specifically, we found a novel residue on CaMKIIα, S331, where phosphorylation was increased after extinction and decreased after reactivation. Mutagenesis experiments indicated that phosphorylation of S331 reduced enzymatic activity of CaMKIIα. Additionally, we found that pharmacological inhibition of CaMKII within the BLA can lead to a reduction in drug-cue motivated behavior if the inhibition occurs immediately following either memory reactivation or memory extinction. This is consistent with both a partial impairment in the reconsolidation of the original drug-cue memory and an enhancement in the learning/consolidation of the new cue-extinction memory, effectively weakening the drug-associated memory trace.
Targeting memory processes as a therapeutic strategy
Manipulations that inhibit drug-cue memory reconsolidation and promote extinction have high therapeutic potential for relapse prevention in addiction (Torregrossa et al., 2011; Sorg, 2012). Despite a remarkable effort to develop pharmacotherapies for drug addiction, there has been little success in helping addicted individuals maintain long-term abstinence (Koob et al., 2009). Currently, clinical efforts that combine treatment medications with behavioral therapies have demonstrated the highest success rates (Carroll and Onken, 2005). A common behavioral approach involves exposing individuals to multimodal cues in an extinction-based therapy to decrease subsequent craving and relapse when drug-associated cues are re-encountered (Price et al., 2013). Previous research has demonstrated some effectiveness of this approach to inhibit fear-associated memories in rodent models and in humans (Ressler et al., 2004; Monfils et al., 2009; Hofmann et al., 2012). In the present study, we show that a single cue extinction session is mildly effective in reducing responding for cocaine during a cue-induced reinstatement session, but that this reduction is amplified by the addition of a second cue extinction session, consistent with the fact that the number of unreinforced cue presentations is a critical determinant of extinction efficacy (Price et al., 2013; Unrod et al., 2014).
Although cue extinction therapy can have moderate success (Price et al., 2010; Kantak and Nic Dhonnchadha, 2011), developing medications that can be given in conjunction with exposure therapy to augment extinction learning will likely improve treatment outcomes. In particular, intra-BLA CaMKII inhibition immediately following a single cue-extinction session (moderate extinction) potentiated the reduction in cue-induced reinstatement observed 24 h later. CaMKII inhibition also disrupted reconsolidation, indicating that insufficient extinction training would not prevent efficacy, as may be the case for glutamatergic agonists like DCS (Lee et al., 2009; Price et al., 2013). Therefore, inhibiting CaMKII signaling pathways in conjunction with exposure therapy may be a viable treatment strategy. However, CaMKII inhibition had no additional benefit after extended cue-extinction indicating a potential floor effect. Extended extinction alone was sufficient to drastically reduce levels of responding during reinstatement, even in VEH-treated animals, suggesting that sufficient exposure therapy could be beneficial, though the addition of CaMKII inhibition may have been able to inhibit renewal of cocaine seeking after a change of context or reduce spontaneous recovery, which could be tested in future experiments.
Mechanisms of CaMKII action in memory processes
Identifying CaMKIIα as an important component in the maintenance of drug-associated memories is not altogether surprising. It is well established that CaMKII activity is necessary for NMDA-dependent forms of long-term potentiation (LTP) and synaptic modifications (Sanhueza et al., 2011; Coultrap and Bayer, 2012; Lisman et al., 2012). However, the identification of a phosphorylation event that is bidirectionally regulated by reconsolidation relative to extinction of a memory is novel. CaMKII is activated by Ca2+ influx through NMDA receptors or voltage-gated Ca2+ channels, resulting in its autophosphorylation at T286. CaMKII then translocates to synapses, initiating a biochemical cascade, including phosphorylation of glutamatergic receptors that potentiates synaptic transmission (Barria et al., 1997; Bayer et al., 2006; Raveendran et al., 2009; Lu et al., 2010; El Gaamouch et al., 2012; Lemieux et al., 2012; Sanhueza and Lisman, 2013; Coultrap et al., 2014). Moreover, CaMKIIα knock-out mice have deficits in LTP (Silva et al., 1992a), and in learning a hippocampus-dependent spatial task (Silva et al., 1992b), further demonstrating that CaMKIIα is necessary for memory formation. On the other hand, CaMKII activity, including phosphorylation of T286, is also needed for some forms of long-term depression (LTD; Pi et al., 2010). Whereas CaMKII mediates LTP via phosphorylation of GluA1 at S831, LTD is likely evoked by CaMKII-mediated phosphorylation of GluA1 at S567 (Coultrap et al., 2014). Thus, in our study, the effect of CaMKII inhibitors on reconsolidation and extinction might be explained by differential effects on LTP versus LTD, respectively.
Consistent with this possibility, it has been shown that increased activation of CaMKIIα can impair an NMDA-dependent form of synaptic depotentiation in mice lacking cytoplasmic polyadenylation element binding protein 3 (CPEB3; Huang et al., 2014). Coupled with the finding that CPEB3 KO mice exhibit deficits in the extinction of contextual fear memory (Chao et al., 2013), this further supports the notion that CaMKII inhibition could enhance extinction learning, and highlights a potential involvement of an LTD-like synaptic mechanism in this process. Additionally, there are a few reports that CaMKII also regulates memory reconsolidation. For example, reconsolidation of an amphetamine-conditioned place preference was disrupted by infusion of KN-93 into the hippocampus before re-exposure to the conditioned context (Sakurai et al., 2007). Likewise, CaMKII activity in the hippocampus is important for the maintenance of a spatial memory after retrieval (Da Silva et al., 2013). On the other hand, the reconsolidation of a cocaine-associated contextual memory was not affected by intra-BLA CaMKII inhibition (Arguello et al., 2014). Although inconsistent with our results, the procedural differences between the two studies, including manipulating contextual rather than discrete cue memories, likely explains the differential findings.
Role of CaMKII signaling in regulating drug-associated behaviors
CaMKIIα phosphorylation events have recently been linked to drug-related learning. (Easton et al., 2013, 2014; Salling et al., 2016). For example, mice without the capacity for CaMKIIα autophosphorylation (T286A mice) exhibited a delay in the establishment of a preference for cocaine, (Easton et al., 2014), and increased phosphorylation of CaMKIIα at T286 was observed in the amygdala after rats lever press for alcohol (Salling et al., 2016). In addition, reduction of CaMKIIα function in the nucleus accumbens shell prevented amphetamine-associated plasticity and reduced self-administration (Loweth et al., 2013). Together, these studies are consistent with our conclusions that CaMKIIα activity is a critical component of drug-related memory processes. Our proteomics results further suggest a very precise and novel, memory-specific regulation of CaMKIIα at S331. Phosphorylation of S331 has not been previously reported to change in a biological context, but a recent study has found S331 to be a putative autophosphorylation site that exists in vivo (Baucum et al., 2015). Interestingly, S331 is located within the linker region of CaMKIIα, and the linker region has the potential to regulate the function of CaMKII via multiple mechanisms in addition to kinase activity (Chao et al., 2011; Stratton et al., 2013). The present study is the first to report that S331 phosphorylation may inhibit kinase activity, suggesting that CaMKIIα may have the ability to autoinhibit, potentially providing a negative feedback mechanism and/or a mechanism for mediating LTD. Future studies will further explore how and when this novel phosphorylation site on CaMKIIα is regulated, if memory manipulation-specific phosphorylation events occur in distinct subpopulations of BLA neurons, and how CaMKIIα S331 phosphorylation affects synaptic plasticity.
Summary
Together, the present study identified several signaling cascades regulated by the extinction and reconsolidation of a memory associated with self-administered cocaine. In particular, we identified a novel mechanism by which CaMKIIα can regulate both memory processes. Furthermore, our results suggest that inhibiting CaMKII or related signaling cascades could be a novel approach for combined behavioral and pharmacological therapy in the treatment of addictive disorders.
Footnotes
This work was supported by USPHS Grants K01DA031745 (M.M.T.), DA018343 (A.C.N.), DA015222 (J.R.T.), T32007433 (M.T.R.), T32031111 (M.T.R.), and the Pennsylvania Department of Health. We thank Vidhya Nagarajan, Laura Rupprecht, and Megan Bertholomey for technical assistance, and Mary LoPresti and Jean Kanyo for assistance with the proteomics sample preparation and data collections, respectively.
The authors declare no competing financial interests.
References
- Agren T, Engman J, Frick A, Björkstrand J, Larsson EM, Furmark T, Fredrikson M. Disruption of reconsolidation erases a fear memory trace in the human amygdala. Science. 2012;337:1550–1552. doi: 10.1126/science.1223006. [DOI] [PubMed] [Google Scholar]
- Arguello AA, Hodges MA, Wells AM, Lara H, 3rd, Xie X, Fuchs RA. Involvement of amygdalar protein kinase A, but not calcium/calmodulin-dependent protein kinase II, in the reconsolidation of cocaine-related contextual memories in rats. Psychopharmacology. 2014;231:55–65. doi: 10.1007/s00213-013-3203-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- Baucum AJ, 2nd, Shonesy BC, Rose KL, Colbran RJ. Quantitative proteomics analysis of CaMKII phosphorylation and the CaMKII interactome in the mouse forebrain. ACS Chem Neurosci. 2015;6:615–631. doi: 10.1021/cn500337u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer KU, LeBel E, McDonald GL, O'Leary H, Schulman H, De Koninck P. Transition from reversible to persistent binding of CaMKII to postsynaptic sites and NR2B. J Neurosci. 2006;26:1164–1174. doi: 10.1523/JNEUROSCI.3116-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossert JM, Marchant NJ, Calu DJ, Shaham Y. The reinstatement model of drug relapse: recent neurobiological findings, emerging research topics, and translational research. Psychopharmacology. 2013;229:453–476. doi: 10.1007/s00213-013-3120-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley CT, Sekiya F, Kim YJ, Rhee SG, Caldwell KK. Identification of phospholipase C-gamma1 as a mitogen-activated protein kinase substrate. J Biol Chem. 2004;279:41807–41814. doi: 10.1074/jbc.M407851200. [DOI] [PubMed] [Google Scholar]
- Carroll KM, Onken LS. Behavioral therapies for drug abuse. Am J Psychiatry. 2005;162:1452–1460. doi: 10.1176/appi.ajp.162.8.1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cestari V, Costanzi M, Castellano C, Rossi-Arnaud C. A role for ERK2 in reconsolidation of fear memories in mice. Neurobiol Learn Mem. 2006;86:133–143. doi: 10.1016/j.nlm.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Chalifoux JR, Carter AG. GABAB receptor modulation of voltage-sensitive calcium channels in spines and dendrites. J Neurosci. 2011;31:4221–4232. doi: 10.1523/JNEUROSCI.4561-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CY, Picotti P, Hüttenhain R, Heinzelmann-Schwarz V, Jovanovic M, Aebersold R, Vitek O. Protein significance analysis in selected reaction monitoring (SRM) measurements. Mol Cell Proteomics. 2012;11 doi: 10.1074/mcp.M111.014662. M111 014662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao HW, Tsai LY, Lu YL, Lin PY, Huang WH, Chou HJ, Lu WH, Lin HC, Lee PT, Huang YS. Deletion of CPEB3 enhances hippocampus-dependent memory via increasing expressions of PSD95 and NMDA receptors. J Neurosci. 2013;33:17008–17022. doi: 10.1523/JNEUROSCI.3043-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao LH, Stratton MM, Lee IH, Rosenberg OS, Levitz J, Mandell DJ, Kortemme T, Groves JT, Schulman H, Kuriyan J. A mechanism for tunable autoinhibition in the structure of a human Ca2+/calmodulin-dependent kinase II holoenzyme. Cell. 2011;146:732–745. doi: 10.1016/j.cell.2011.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap SJ, Bayer KU. CaMKII regulation in information processing and storage. Trends Neurosci. 2012;35:607–618. doi: 10.1016/j.tins.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap SJ, Freund RK, O'Leary H, Sanderson JL, Roche KW, Dell'Acqua ML, Bayer KU. Autonomous CaMKII mediates both LTP and LTD using a mechanism for differential substrate site selection. Cell Rep. 2014;6:431–437. doi: 10.1016/j.celrep.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Silva WC, Cardoso G, Bonini JS, Benetti F, Izquierdo I. Memory reconsolidation and its maintenance depend on L-voltage-dependent calcium channels and CaMKII functions regulating protein turnover in the hippocampus. Proc Natl Acad Sci U S A. 2013;110:6566–6570. doi: 10.1073/pnas.1302356110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvarci S, Nader K, LeDoux JE. Activation of extracellular signal-regulated kinase-mitogen-activated protein kinase cascade in the amygdala is required for memory reconsolidation of auditory fear conditioning. Eur J Neurosci. 2005;21:283–289. doi: 10.1111/j.1460-9568.2004.03824.x. [DOI] [PubMed] [Google Scholar]
- Easton AC, Lucchesi W, Mizuno K, Fernandes C, Schumann G, Giese KP, Müller CP. alphaCaMKII autophosphorylation controls the establishment of alcohol-induced conditioned place preference in mice. Behav Brain Res. 2013;252:72–76. doi: 10.1016/j.bbr.2013.05.045. [DOI] [PubMed] [Google Scholar]
- Easton AC, Lourdusamy A, Havranek M, Mizuno K, Solati J, Golub Y, Clarke TK, Vallada H, Laranjeira R, Desrivières S, Moll GH, Mössner R, Kornhuber J, Schumann G, Giese KP, Fernandes C, Quednow BB, Müller CP. αCaMKII controls the establishment of cocaine's reinforcing effects in mice and humans. Transl Psychiatry. 2014;4:e457. doi: 10.1038/tp.2014.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Gaamouch F, Buisson A, Moustié O, Lemieux M, Labrecque S, Bontempi B, De Koninck P, Nicole O. Interaction between alphaCaMKII and GluN2B controls ERK-dependent plasticity. J Neurosci. 2012;32:10767–10779. doi: 10.1523/JNEUROSCI.5622-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs RA, Branham RK, See RE. Different neural substrates mediate cocaine seeking after abstinence versus extinction training: a critical role for the dorsolateral caudate-putamen. J Neurosci. 2006;26:3584–3588. doi: 10.1523/JNEUROSCI.5146-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs RA, Bell GH, Ramirez DR, Eaddy JL, Su ZI. Basolateral amygdala involvement in memory reconsolidation processes that facilitate drug context-induced cocaine seeking. Eur J Neurosci. 2009;30:889–900. doi: 10.1111/j.1460-9568.2009.06888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamache K, Pitman RK, Nader K. Preclinical evaluation of reconsolidation blockade by clonidine as a potential novel treatment for posttraumatic stress disorder. Neuropsychopharmacology. 2012;37:2789–2796. doi: 10.1038/npp.2012.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirosawa M, Hoshida M, Ishikawa M, Toya T. MASCOT: multiple alignment system for protein sequences based on three-way dynamic programming. Comput Appl Biosci. 1993;9:161–167. doi: 10.1093/bioinformatics/9.2.161. [DOI] [PubMed] [Google Scholar]
- Hofmann SG, Sawyer AT, Asnaani A. d-Cycloserine as an augmentation strategy for cognitive behavioral therapy for anxiety disorders: an update. Curr Pharm Des. 2012;18:5659–5662. doi: 10.2174/138161212803530916. [DOI] [PubMed] [Google Scholar]
- Holmes A, Quirk GJ. Pharmacological facilitation of fear extinction and the search for adjunct treatments for anxiety disorders: the case of yohimbine. Trends Pharmacol Sci. 2010;31:2–7. doi: 10.1016/j.tips.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WH, Chao HW, Tsai LY, Chung MH, Huang YS. Elevated activation of CaMKIIα in the CPEB3-knock-out hippocampus impairs a specific form of NMDAR-dependent synaptic depotentiation. Front Cell Neurosci. 2014;8:367. doi: 10.3389/fncel.2014.00367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins MA, Christel CJ, Jiao Y, Abiria S, Kim KY, Usachev YM, Obermair GJ, Colbran RJ, Lee A. Ca2+-dependent facilitation of Cav1.3 Ca2+ channels by densin and Ca2+/calmodulin-dependent protein kinase II. J Neurosci. 2010;30:5125–5135. doi: 10.1523/JNEUROSCI.4367-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW. The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci. 2009;10:561–572. doi: 10.1038/nrn2515. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Volkow ND. The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry. 2005;162:1403–1413. doi: 10.1176/appi.ajp.162.8.1403. [DOI] [PubMed] [Google Scholar]
- Kantak KM, Nic Dhonnchadha BÁ. Pharmacological enhancement of drug cue extinction learning: translational challenges. Ann N Y Acad Sci. 2011;1216:122–137. doi: 10.1111/j.1749-6632.2010.05899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Kenneth Lloyd G, Mason BJ. Development of pharmacotherapies for drug addiction: a Rosetta stone approach. Nat Rev Drug Discov. 2009;8:500–515. doi: 10.1038/nrd2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- Lee JL, Gardner RJ, Butler VJ, Everitt BJ. d-Cycloserine potentiates the reconsolidation of cocaine-associated memories. Learn Mem. 2009;16:82–85. doi: 10.1101/lm.1186609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux M, Labrecque S, Tardif C, Labrie-Dion É, Lebel É, De Koninck P. Translocation of CaMKII to dendritic microtubules supports the plasticity of local synapses. J Cell Biol. 2012;198:1055–1073. doi: 10.1083/jcb.201202058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Yasuda R, Raghavachari S. Mechanisms of CaMKII action in long-term potentiation. Nat Rev Neurosci. 2012;13:169–182. doi: 10.1038/nrn3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loweth JA, Li D, Cortright JJ, Wilke G, Jeyifous O, Neve RL, Bayer KU, Vezina P. Persistent reversal of enhanced amphetamine intake by transient CaMKII inhibition. J Neurosci. 2013;33:1411–1416. doi: 10.1523/JNEUROSCI.4386-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Isozaki K, Roche KW, Nicoll RA. Synaptic targeting of AMPA receptors is regulated by a CaMKII site in the first intracellular loop of GluA1. Proc Natl Acad Sci U S A. 2010;107:22266–22271. doi: 10.1073/pnas.1016289107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon AM, Tesmer JJ. Structural insights into phospholipase C-beta function. Mol Pharmacol. 2013;84:488–500. doi: 10.1124/mol.113.087403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlo E, Milton AL, Goozée ZY, Theobald DE, Everitt BJ. Reconsolidation and extinction are dissociable and mutually exclusive processes: behavioral and molecular evidence. J Neurosci. 2014;34:2422–2431. doi: 10.1523/JNEUROSCI.4001-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton AL, Everitt BJ. The psychological and neurochemical mechanisms of drug memory reconsolidation: implications for the treatment of addiction. Eur J Neurosci. 2010;31:2308–2319. doi: 10.1111/j.1460-9568.2010.07249.x. [DOI] [PubMed] [Google Scholar]
- Monfils MH, Cowansage KK, Klann E, LeDoux JE. Extinction-reconsolidation boundaries: key to persistent attenuation of fear memories. Science. 2009;324:951–955. doi: 10.1126/science.1167975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nic Dhonnchadha BA, Szalay JJ, Achat-Mendes C, Platt DM, Otto MW, Spealman RD, Kantak KM. d-Cycloserine deters reacquisition of cocaine self-administration by augmenting extinction learning. Neuropsychopharmacology. 2010;35:357–367. doi: 10.1038/npp.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nic Dhonnchadha BÁ, Lin A, Leite-Morris KA, Kaplan GB, Man HY, Kantak KM. Alterations in expression and phosphorylation of GluA1 receptors following cocaine-cue extinction learning. Behav Brain Res. 2013;238:119–123. doi: 10.1016/j.bbr.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 6. Amsterdam; Boston: Academic/Elsevier; 2007. [Google Scholar]
- Pi HJ, Otmakhov N, Lemelin D, De Koninck P, Lisman J. Autonomous CaMKII can promote either long-term potentiation or long-term depression, depending on the state of T305/T306 phosphorylation. J Neurosci. 2010;30:8704–8709. doi: 10.1523/JNEUROSCI.0133-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price KL, Saladin ME, Baker NL, Tolliver BK, DeSantis SM, McRae-Clark AL, Brady KT. Extinction of drug cue reactivity in methamphetamine-dependent individuals. Behav Res Ther. 2010;48:860–865. doi: 10.1016/j.brat.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price KL, Baker NL, McRae-Clark AL, Saladin ME, Desantis SM, Santa Ana EJ, Brady KT. A randomized, placebo-controlled laboratory study of the effects of d-cycloserine on craving in cocaine-dependent individuals. Psychopharmacology. 2013;226:739–746. doi: 10.1007/s00213-011-2592-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raveendran R, Devi Suma Priya S, Mayadevi M, Steephan M, Santhoshkumar TR, Cheriyan J, Sanalkumar R, Pradeep KK, James J, Omkumar RV. Phosphorylation status of the NR2B subunit of NMDA receptor regulates its interaction with calcium/calmodulin-dependent protein kinase II. J Neurochem. 2009;110:92–105. doi: 10.1111/j.1471-4159.2009.06108.x. [DOI] [PubMed] [Google Scholar]
- Ressler KJ, Rothbaum BO, Tannenbaum L, Anderson P, Graap K, Zimand E, Hodges L, Davis M. Cognitive enhancers as adjuncts to psychotherapy: use of d-cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry. 2004;61:1136–1144. doi: 10.1001/archpsyc.61.11.1136. [DOI] [PubMed] [Google Scholar]
- Rodrigues SM, Farb CR, Bauer EP, LeDoux JE, Schafe GE. Pavlovian fear conditioning regulates Thr286 autophosphorylation of Ca2+/calmodulin-dependent protein kinase II at lateral amygdala synapses. J Neurosci. 2004;24:3281–3288. doi: 10.1523/JNEUROSCI.5303-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai S, Yu L, Tan SE. Roles of hippocampal N-methyl-d-aspartate receptors and calcium/calmodulin-dependent protein kinase II in amphetamine-produced conditioned place preference in rats. Behav Pharmacol. 2007;18:497–506. doi: 10.1097/FBP.0b013e3282ee7b62. [DOI] [PubMed] [Google Scholar]
- Salling MC, Faccidomo SP, Li C, Psilos K, Galunas C, Spanos M, Agoglia AE, Kash TL, Hodge CW. Moderate alcohol drinking and the amygdala proteome: identification and validation of calcium/calmodulin dependent kinase II and AMPA receptor activity as novel molecular mechanisms of the positive reinforcing effects of alcohol. Biol Psychiatry. 2016;79:430–442. doi: 10.1016/j.biopsych.2014.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez H, Quinn JJ, Torregrossa MM, Taylor JR. Reconsolidation of a cocaine-associated stimulus requires amygdalar protein kinase A. J Neurosci. 2010;30:4401–4407. doi: 10.1523/JNEUROSCI.3149-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanhueza M, Lisman J. The CaMKII/NMDAR complex as a molecular memory. Mol Brain. 2013;6:10. doi: 10.1186/1756-6606-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanhueza M, Fernandez-Villalobos G, Stein IS, Kasumova G, Zhang P, Bayer KU, Otmakhov N, Hell JW, Lisman J. Role of the CaMKII/NMDA receptor complex in the maintenance of synaptic strength. J Neurosci. 2011;31:9170–9178. doi: 10.1523/JNEUROSCI.1250-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, LeDoux JE. Memory consolidation of auditory pavlovian fear conditioning requires protein synthesis and protein kinase A in the amygdala. J Neurosci. 2000;20:RC96. doi: 10.1523/JNEUROSCI.20-18-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See RE. Neural substrates of cocaine-cue associations that trigger relapse. Eur J Pharmacol. 2005;526:140–146. doi: 10.1016/j.ejphar.2005.09.034. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Stevens CF, Tonegawa S, Wang Y. Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992a;257:201–206. doi: 10.1126/science.1378648. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Paylor R, Wehner JM, Tonegawa S. Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992b;257:206–211. doi: 10.1126/science.1321493. [DOI] [PubMed] [Google Scholar]
- Sorg BA. Reconsolidation of drug memories. Neurosci Biobehav Rev. 2012;36:1400–1417. doi: 10.1016/j.neubiorev.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton MM, Chao LH, Schulman H, Kuriyan J. Structural studies on the regulation of Ca2+/calmodulin dependent protein kinase II. Curr Opin Struct Biol. 2013;23:292–301. doi: 10.1016/j.sbi.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JR, Olausson P, Quinn JJ, Torregrossa MM. Targeting extinction and reconsolidation mechanisms to combat the impact of drug cues on addiction. Neuropharmacology. 2009;56:186–195. doi: 10.1016/j.neuropharm.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torregrossa MM, Kalivas PW. Neurotensin in the ventral pallidum increases extracellular gamma-aminobutyric acid and differentially affects cue- and cocaine-primed reinstatement. J Pharmacol Exp Ther. 2008;325:556–566. doi: 10.1124/jpet.107.130310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torregrossa MM, Taylor JR. Learning to forget: manipulating extinction and reconsolidation processes to treat addiction. Psychopharmacology. 2013;226:659–672. doi: 10.1007/s00213-012-2750-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torregrossa MM, Sanchez H, Taylor JR. d-Cycloserine reduces the context specificity of pavlovian extinction of cocaine cues through actions in the nucleus accumbens. J Neurosci. 2010;30:10526–10533. doi: 10.1523/JNEUROSCI.2523-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torregrossa MM, Corlett PR, Taylor JR. Aberrant learning and memory in addiction. Neurobiol Learn Mem. 2011;96:609–623. doi: 10.1016/j.nlm.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torregrossa MM, Gordon J, Taylor JR. Double dissociation between the anterior cingulate cortex and nucleus accumbens core in encoding the context versus the content of pavlovian cocaine cue extinction. J Neurosci. 2013;33:8370–8377. doi: 10.1523/JNEUROSCI.0489-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronson NC, Taylor JR. Molecular mechanisms of memory reconsolidation. Nat Rev Neurosci. 2007;8:262–275. doi: 10.1038/nrn2090. [DOI] [PubMed] [Google Scholar]
- Tronson NC, Wiseman SL, Neve RL, Nestler EJ, Olausson P, Taylor JR. Distinctive roles for amygdalar CREB in reconsolidation and extinction of fear memory. Learn Mem. 2012;19:178–181. doi: 10.1101/lm.025783.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui J, Inagaki M, Schulman H. Calcium/calmodulin-dependent protein kinase II (CaMKII) localization acts in concert with substrate targeting to create spatial restriction for phosphorylation. J Biol Chem. 2005;280:9210–9216. doi: 10.1074/jbc.M407653200. [DOI] [PubMed] [Google Scholar]
- Unrod M, Drobes DJ, Stasiewicz PR, Ditre JW, Heckman B, Miller RR, Sutton SK, Brandon TH. Decline in cue-provoked craving during cue exposure therapy for smoking cessation. Nicotine Tob Res. 2014;16:306–315. doi: 10.1093/ntr/ntt145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vurbic D, Gold B, Bouton ME. Effects of d-cycloserine on the extinction of appetitive operant learning. Behav Neurosci. 2011;125:551–559. doi: 10.1037/a0024403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan X, Torregrossa MM, Sanchez H, Nairn AC, Taylor JR. Activation of exchange protein activated by cAMP in the rat basolateral amygdala impairs reconsolidation of a memory associated with self-administered cocaine. PLoS One. 2014;9:e107359. doi: 10.1371/journal.pone.0107359. [DOI] [PMC free article] [PubMed] [Google Scholar]