

Abstract
As neural structures grow in size and increase metabolic demand, the CNS vasculature undergoes extensive growth, remodeling, and maturation. Signals from neural tissue act on endothelial cells to stimulate blood vessel ingression, vessel patterning, and acquisition of mature brain vascular traits, most notably the blood–brain barrier. Using mouse genetic and in vitro approaches, we identified retinoic acid (RA) as an important regulator of brain vascular development via non-cell-autonomous and cell-autonomous regulation of endothelial WNT signaling. Our analysis of globally RA-deficient embryos (Rdh10 mutants) points to an important, non-cell-autonomous function for RA in the development of the vasculature in the neocortex. We demonstrate that Rdh10 mutants have severe defects in cerebrovascular development and that this phenotype correlates with near absence of endothelial WNT signaling, specifically in the cerebrovasculature, and substantially elevated expression of WNT inhibitors in the neocortex. We show that RA can suppress the expression of WNT inhibitors in neocortical progenitors. Analysis of vasculature in non-neocortical brain regions suggested that RA may have a separate, cell-autonomous function in brain endothelial cells to inhibit WNT signaling. Using both gain and loss of RA signaling approaches, we show that RA signaling in brain endothelial cells can inhibit WNT-β-catenin transcriptional activity and that this is required to moderate the expression of WNT target Sox17. From this, a model emerges in which RA acts upstream of the WNT pathway via non-cell-autonomous and cell-autonomous mechanisms to ensure the formation of an adequate and stable brain vascular plexus.
SIGNIFICANCE STATEMENT Work presented here provides novel insight into important yet little understood aspects of brain vascular development, implicating for the first time a factor upstream of endothelial WNT signaling. We show that RA is permissive for cerebrovascular growth via suppression of WNT inhibitor expression in the neocortex. RA also functions cell-autonomously in brain endothelial cells to modulate WNT signaling and its downstream target, Sox17. The significance of this is although endothelial WNT signaling is required for neurovascular development, too much endothelial WNT signaling, as well as overexpression of its target Sox17, are detrimental. Therefore, RA may act as a “brake” on endothelial WNT signaling and Sox17 to ensure normal brain vascular development.
Keywords: brain vascular development, cerebrovasculature, endothelial cell, retinoic acid, VEGF, WNT
Introduction
Expansion and maturation of the vasculature is essential to support brain growth and to establish a vascular plexus that can sustain brain function. Mouse CNS vascular development begins at approximately embryonic day 9 (E9), when vessels from the perineural vascular plexus (PNVP) that surround the CNS ingress, starting at the spinal cord and soon moving into more rostral brain structures (Nakao et al., 1988). Angiogenic growth occurs in response to vascular endothelial growth factor-A (VEGFA) (Breier et al., 1992; Haigh et al., 2003; Raab et al., 2004; James et al., 2009) and WNT ligands (Stenman et al., 2008; Daneman et al., 2009) secreted by neural progenitors in the ventricular zone (VZ) and, later, WNT ligands from postmitotic neurons. Parallel with vascular growth, CNS endothelial cells (ECs) acquire blood–brain barrier (BBB) properties, including the expression of tight junctional proteins and transporters such as glucose transporter-1 (GLUT-1) that ensure influx and efflux of substances across the BBB (Bauer et al., 1993; Daneman et al., 2010). CNS vascular development is complex, in part because vascular growth and maturation occur against the backdrop of a rapidly changing neural environment that produces most neuro-angiogenic ligands. How CNS ECs successfully integrate diverse angiogenic and maturation cues from the neural environment to create a stable vasculature is not well understood.
Retinoic acid (RA) is a lipid soluble hormone produced by cell types within and around the CNS and it has diverse developmental roles (Napoli, 1999; Toresson et al., 1999; Li et al., 2000; Maden, 2001; Schneider et al., 2001; Smith et al., 2001; Zhang et al., 2003; Siegenthaler et al., 2009). RA signaling is mediated by RA receptors (RARs) that act as receptors and transcription factors to control gene transcription (Al Tanoury et al., 2013). RA is required for vasculogenesis in the early embryo (Lai et al., 2003; Bohnsack et al., 2004) and there is some evidence that it may have a role in angiogenesis and vessel maturation in the CNS. RA is implicated in BBB development through the regulation of BBB protein expression, specifically VE-cadherin (Mizee et al., 2013; Lippmann et al., 2014). Mice that lack both retinoid receptors RARα and RARγ have significant defects in CNS development and visible brain hemorrhaging, notably in the cerebral hemispheres (Lohnes et al., 1994). RAR receptors are expressed in fetal human and mouse brain ECs (Mizee et al., 2013), suggesting that ECs in the developing CNS are RA responsive. Collectively, these data indicate RA may have a significant role in controlling brain vascular development.
Using global RA-deficient mouse mutants (Rdh10 mutants) and EC-specific disruption of RA signaling (PdgfbiCre;dnRAR403-flox), we show RA has separate, non-cell-autonomous and cell-autonomous roles with regard to endothelial WNT signaling. Rdh10 mutant embryos have impaired neocortical development (Siegenthaler et al., 2009) and we describe herein vascular growth defects specific to the neocortex. Reduced cerebrovascular growth in Rdh10 mutants is accompanied by disruption in VEGF-A and WNT. However, elevated Vegfa expression is not limited to the neocortex and may reflect widespread brain hypoxia. In contrast, endothelial WNT signaling is specifically diminished in the Rdh10 mutant cerebrovasculature. This is accompanied by significantly elevated levels of WNT inhibitors in the Rdh10 mutant neocortex, but no other brain regions. Combined with our data showing that RA suppresses gene expression of WNT inhibitors in cultured neocortical progenitors, our analysis of cerebrovascular defects in Rdh10 mutants points to RA functioning non-cell-autonomously in the neocortex to create a permissive environment for endothelial WNT signaling. Vascular development is relatively normal in other regions of Rdh10 mutant brains and, strikingly, endothelial WNT signaling is increased. This finding suggested that RA may act cell-autonomously in brain ECs to inhibit WNT signaling. In support of this, we find PdgfbiCre;dnRAR403-flox mutants have increased endothelial WNT signaling and expression of the WNT transcriptional targets LEF-1 and Sox17. Collectively, this work shows that RA regulates brain vascular development by acting upstream of WNT signaling through different non-cell-autonomous and cell-autonomous mechanisms.
Materials and Methods
Animals.
Mice used for experiments were housed in specific-pathogen-free facilities approved by the Association for Assessment and Accreditation of Laboratory Animal Care and were handled in accordance with protocols approved by the University of California–San Francisco (UCSF) Committee on Animal Research and the University of California Anschutz Medical Campus Institutional Animal Care and Use Committee. The following mouse lines were used in this study: PdgfbiCre (Claxton et al., 2008), Ctnnb1-flox (Brault et al., 2001), Bat-gal-lacZ (Maretto et al., 2003), Ephrin-B2-H2B-GFP (Davy et al., 2006), and dnRAR403-flox (Rosselot et al., 2010). The Rdh10 ENU point mutation mutant allele has been described previously (Ashique et al., 2012) and were obtained from Andy Peterson at Genentech. Tamoxifen (Sigma-Aldrich) was dissolved in corn oil (Sigma-Aldrich; 20 mg/ml) and 100 μl was injected intraperitoneally into pregnant females at E9 and E10 to generate PdgfbiCre;dnRAR403-flox mutant animals. For the generation of PdgfbiCre;Ctnnb1-fl/fl mutants, tamoxifen was administered to pregnant females on E11 and E12. The RA-enriched diet (final concentration 0.175 mg/g food) consisted of all-trans-RA (atRA; Sigma-Aldrich) dissolved in corn oil and mixed with Bioserv Nutra-Gel Diet, Grain-Based Formula, Cherry Flavor. atRA diet was prepared fresh daily and provided ad libitum from the afternoon of E10 through the day of collection (E14.5 or E16.5).
Immunohistochemistry.
Fetuses (E12.5–E18.5) were collected and whole heads or brains were fixed overnight in 4% paraformaldehyde. All tissues were cryoprotected with 20% sucrose in PBS and subsequently frozen in optimal cutting temperature medium. Tissue was cryosectioned in 12 μm increments. Immunohistochemistry was performed on tissue sections as described previously (Zarbalis et al., 2007; Siegenthaler et al., 2009) using the following antibodies: rabbit anti-β-galactosidase (anti-β-gal, 1:500; Cappel), rabbit anti-GLUT-1 (1:500; Lab Vision-Thermo Scientific), goat anti-Sox17 (1:100; R&D Systems), chicken anti-GFP (1:500; Invitrogen), mouse anti-BrdU (1:50; BD Biosciences), mouse anti-CoupTFII (1:100; R&D Systems), rabbit anti-Claudin-3 (1:200; Invitrogen), rabbit anti-LEF-1 (1:100; Cell Signaling Technology), rabbit anti-Pax6 (1:200; BioLegend), chicken anti-Tbr2 (1:100; Millipore), and rat anti-Ctip2 (1:1000; Abcam). After incubation with primary antibody(s), sections were incubated with appropriate Alexa Fluor-conjugated secondary antibodies (Invitrogen), Alexa Fluor 633-conjugated isolectin-B4 (Invitrogen), and DAPI (Invitrogen). For LEF-1, immunostaining was performed using the Tyramide System Amplification Kit (Invitrogen) per manufacturer's instructions. Immunofluorescent (IF) images were captured using a Retiga CCD-cooled camera and associated QCapture Pro software (QImaging), a Nikon i80 research microscope with Cool-Snap CCD-cooled camera, or Zeiss 780 LSM confocal microscope.
Cell proliferation and trans-well migration assay with bEnd.3 cell line.
The mouse brain endothelioma cell line (bEnd.3) was from ATCC (catalog #CRL-2299). All experiments were performed on cells from passages 2–4 and cells were grown in Dulbecco's minimal essential media with 4.5 g/L glucose, 1.5 g/L sodium bicarbonate, 4 mm l-glutamine (Invitrogen), 10% fetal bovine serum (FBS) (Invitrogen), and penicillin (0.0637 g/L)–streptomycin (0.1 g/L) (UCSF Cell Culture Facility or Invitrogen). On day 1 of the cell proliferation assays, 7 × 104 cells were plated in each well of an 8-well glass chambered slide (Nunc) and allowed to adhere for ∼5 h, after time the medium was changed to DMEM with 1% FBS. On day 2, atRA (50 nm; Sigma-Aldrich) and/or WNT3a (0.05, 0.1 or 0.3 μg/ml; R&D Systems) was added to the medium. On day 5, 1 mm BrdU (Roche) was added to the medium in each well and, 2 h later, cells were fixed for 15 min with 4% paraformaldehyde. Cells were immunostained to detect BrdU incorporation (mouse anti-BrdU 1:50; BD Bioscience) and stained with DAPI to visualize all cell nuclei. For analysis of cell proliferation, four 10 × images were obtained for each treatment condition (two wells per treatment in each replicate) and the percentage of BrdU+ cells was determined for each image (no. of BrdU+ cells/no. of DAPI+ cells). The value for each replicate is an average from the four images. For the Transwell migration assay, 8 × 104 cells in 100 μl of medium was pipetted into the top chamber of a Millicell cell culture insert with a 8 μm filter pore size (Millipore catalog #PI8P01250). The culture well immediately below the insert contained 500 μl of medium with RA (50 nm) and/or WNT3a (0.1 or 0.3 μg/ml) and WNT7a (5 μg/ml). The cells were allowed to migrate through the pores for 20 h, cells were fixed for 15 min with 4% paraformaldehyde, and a cotton swab was used to remove the cells still within the top chamber. The filter was cut away from the insert, stained with DAPI to visualize the cell nuclei, and filters were mounted onto slides for imaging. For analysis of cell migration, four 10 × images were obtained for each treatment condition (two Transwell filters per treatment in each replicate) and the number of DAPI+ nuclei were assessed in a counting area within each 10 × image field. For WNT7a-RA experiments, the entire 10 × field was counted. For both the cell proliferation and Transwell migration assays, a minimum of three independent replicates (n ≥ 3) were performed for each treatment condition.
Quantitative analysis of fetal neurovasculature.
Vessel density and β-gal+ endothelial cell analysis was performed on E12.5 and E14.5 control (Rdh10+/+ or Rdh10+/−) and Rdh10-mutant animals (thalamus, midbrain, and hindbrain), E14.5 and E16.5 Bat-gal-LacZ/+ animals (forebrain), and E18.5 PdgfbiCre;dnRAR403-fl control and mutant animals (forebrain) on a minimum of 3 separate brains per genotype/treatment/embryonic day point (n ≥ 3). To determine mean vessel density, the sum length of Ib4+ cerebral vessels was determined from a single, 20 × field and divided by the area of the tissue analyzed. All density measurements were performed using ImageJ software on a minimum of five 20 × fields per brain. For quantification of β-gal+ ECs in fetuses expressing the Bat-gal-lacZ/+ allele, the number of β-gal+/Ib4 ECs was counted in a single, 20 × image and divided by the sum length of Ib4+ blood vessels within the image. This was performed on a minimum of five 20 × fields per brain. To quantify cell proliferation in the Rdh10 E14.5 control and mutant PNVP and in the neocortical plexus, pregnant dams were injected with BrdU (50 mg/kg body weight; Roche) and embryos were collected 2 h later. After processing for GLUT-1/BrdU/Ib4/DAPI IF, the total number of BrdU+/GLUT-1+ ECs was divided by the total number of GLUT-1+ ECs in a 20 × field. Analysis was performed separately for the PNVP and vessels with the neocortical plexus. All cell proliferation analysis was performed using ImageJ software on a minimum of five 20 × fields per brain. Cell proliferation analysis was performed on a minimum of 3 separate brains per genotype (n ≥ 3).
Luciferase assays.
HEK293 cells were grown in 1:1 DMEM:F12 supplemented with 10% FBS and penicillin–streptomycin. Twenty-four hours before transfection cells, were plated in antibiotic ad libitum medium at a density of 4 × 105 per well of poly-l-lysine-treated 12-well plates. Cells were transfected using Lipofectamine 2000 (Invitrogen) with 500 ng of the following expression plasmids: RARα.pCMV-Sport6 (Open Biosystems), RXRβ.pCMV-Sport6 (Open Biosystems), or dnRARα.pCIG (subcloned with dnRARa403 (Addgene plasmid no. 15153) and pCIG (Megason and McMahon, 2002) and 100 ng of the reporter plasmids M50-TOP-Flash or M51-FOP-Flash (Addgene). pCIG was added to normalize total DNA concentration. Four hours after transfection, cells were treated with recombinant mouse WNT3a (0.1 μg/ml; R&D Systems), RA (1 μm; Sigma Aldrich), or vector control. Luciferase levels were measured 18 h after transfection using the Dual Luciferase Assay Kit according to the manufacturer's instructions (Promega). Luciferase assays were performed in triplicate and normalized to total protein concentration. All assays were repeated in three independent experiments and the results of one such experiment are shown in Figure 5.
Figure 5.

Endothelial WNT signaling is increased in fetal brain vasculature of PdgfbiCre;dnRAR403-flox mutants. A, B, Open arrows indicate β-gal+ (green), Ib4+ (red) ECs in the striatum of E18.5 PdgfbiCre/+;Bat-gal-LacZ/+ and PdgfbiCre;dnRAR403-fl/fl;Bat-gal-lacZ/+. C, Graph depicting quantification of β-gal+ ECs per vessel length in E18.5 control (PdgfbiCre/+;Bat-gal-LacZ/+ or dnRAR403-flox;Bat-gal-LacZ/+) and mutant (PdgfbiCre;dnRAR403-fl/+;Bat-gal-lacZ/+, PdgfbiCre;dnRAR403-fl/fl;Bat-gal-lacZ/+) cortical, striatal, and thalamic vasculature. *Significance from control; # significance from PdgfbiCre;dnRAR403-fl/+. D, E, LEF-1 (green), Ib4+ (red) ECs in the neocortex of PdgfbiCre/+ and PdgfbiCre;dnRAR403-fl/fl. F, LEF-1 (54 kDa), and β-actin (52 kDa) immunoblots on protein lysate from E18.5 control (□: PdgfbiCre/+ or dnRAR403-flox) or mutant (■: PdgfbiCre;dnRAR403-fl/fl) neocortices. G, LEF-1 (green) and Ib4 (red) immunofluorescence in the head area of E18.5 PdgfbiCre/+ and PdgfbiCre;dnRAR403-fl/fl animals. Arrows indicate Ib4+/LEF-1− vessels. Scale bars, 100 μm.
Microvessel isolation, multigene transcriptional profiling.
Isolation of RNA from microvessels from E18.5 control (PdgfbiCre/+;Ctnnb1-fl/+) and mutant (PdgfbiCre/+;Ctnnb1-fl/fl) brains was performed as described previously (Siegenthaler et al., 2013). Multigene transcriptional profiling, a form of quantitative RT-PCR, was used to determine the number of mRNA copies per cell normalized to 18S rRNA abundance (106 18S-rRNA copies/cell; Shih and Smith, 2005). For each sample, mRNA copy numbers for Sox17, Lef1, and Axin2 were normalized to CD144 copy number to correct for variability in microvessel isolation between brains. Analysis was performed on microvessels isolated from 3 control and 3 mutant E18.5 brains (n = 3). For RT-PCR of RAR gene expression, RNA was isolated from E18.5 wild-type microvessels and postnatal day 7 meninges and cDNA was generated from 100 ng of RNA using the SuperScript VILO cDNA Synthesis Kit (Invitrogen). Primer sequences are as follows: Lef1 forward: AGGGCGACTTAGCCGACAT, Lef1 reverse: GGGCTTGTCTGACCACCTCAT; Axin2 forward: GTGCCGACCTCAAGTGCAA, Axin2 reverse: GGTGGCCCGAAGAGTTTTG; Sox17 forward: GGCCGATGAACGCCTTTAT, Sox17 reverse: AGCTCTGCGTTGTGCAGATCT; Rara forward: AGCTCTGCGTTGTGCAGATCT, Rara reverse: AGAGTGTCCAAGCCCTCAGA; Rarb forward: TTCAAAGCAGGAATGCACAG, Rarb reverse: GGCAAAGGTGAACACAAGGT; Rarg forward: CACAGCCTGCCAGTCTACAA, Rarg reverse: CTGGCAGAGTGAGGGAAAAG; Rxra forward: CTGCCGCTCGACTTCTCTAC, Rxra reverse: ATATTTCCTGAGGGATGGGC; Rxrb forward: TGGGGGTGAGAAAAGAGATG, Rxrb reverse: GAGCGACACTGTGGAGTTGA; Rxrg forward: AATGCTCTTGGCTCTCCGTA, Rxrg reverse: TGAAGAAGCCTTTGCAACCT.
Tissue and neocortical progenitor cell (NPC) culture/isolation, qPCR.
Meninges were removed from E14 wild-type (n = 5) and RDH10 mutant brains (n = 4). RNA was isolated separately from the neocortices and the non-neocortical brain regions using the RNeasy Mini Kit (Qiagen). E14 cortical progenitor cells (R&D systems) were seeded onto 15 μg/ml poly-l-ornithine (Sigma-Aldrich)- and 1 μg/ml laminin (Sigma-Aldrich)-coated 6 well plates as a monolayer culture. Cell culture medium was composed of DMEM/F-12 with Glutamax (Life Technologies), 1 × N2 supplement composed of insulin, human transferrin, putrescine, selenite, and progesterone (Life Technologies) and glucose (Sigma-Aldrich). The culture medium was supplemented with 10 ng/ml human basic fibroblast growth factor (R&D Systems) and 10 ng/ml human epidermal growth factor (R&D Systems) every day until cell lysate collection to maintain cortical progenitors cells in an undifferentiated state. After 24 h of exposure to the treatment conditions, total cellular RNA was isolated from vehicle-treated, 1 μm atRA-treated, and 1 μm atRA +1 μm pan-RAR antagonist-treated (Santa Cruz Biotechnology) cells using the RNeasy Mini Kit. Experiments using cortical progenitor cells were performed 3 separate times (n = 3). To synthesize cDNA, specifications were followed using the iScript cDNA synthesis kit with 1 μg (brain samples) or 500 ng (cultured cells) of RNA from each sample. To assess Vegfa, Ldha, Pdk, Cox4-2, Slc2a1, WNT7a, WNT7b, Sfrp1, Sfrp2, Sfrp4, Sfrp5, and Dkk1 transcript levels, qRT-PCR was performed according to the SYBR Green (Bio-Rad) protocol using the Bio-Rad CFX96 Real Time PCR Detection System. For an internal control, Actb transcript levels were also assessed. To identify differences in expression between control and mutant genotypes, delta-delta CT analysis was applied. Primer sequences were as follows: Vegfa forward: CAGGCTGCTGTAACGATGAA, Vegfa reverse: TTTGACCCTTTCCCTTTCCT; Ldha forward: AGCAGGTGGTTGAGAGTGCT, Ldha reverse: GGCCTCTTCCTCAGAAGTCA; Pdk1 forward: CCCCGATTCAGGTTCACG, Pdk1 reverse: CCCGGTCACTCATCTTCACA; Cox4-2 forward: GGTTGTCACCCTGACGGAAG, Cox4-2 reverse: GAGGGGAGGGGATGATTGTC; Slc2a1 forward: TCAGGCGGAAGCTAGGAAC, Slc2a1 reverse: GGAGGGAAACATGCAGTCATC; WNT7a forward: GCAATAAGACAGCCCCTCAG, WNT7a reverse: ATCCTGCCTGTGATCTGACC; WNT7b forward: CAGCCAATCTTCCATTCCAT, WNT7b reverse: CCTGACCTCTCCTGAACCTG; Sfrp1 forward: GAGTTTTGTTGCGGACCTGT, Sfrp1 reverse: GCCAGGGACAAAGCTAATGA; Sfrp2 forward: GCTTGTGGGTCCCAGACTTA, Sfrp2 reverse: GCATCATGCAATGAGGAATG; Sfrp4 forward: GACCCTGGCAACATACCTGA, Sfrp4 reverse: CATCTTGATGGGGCAGGATA; Sfrp5 forward: TGGAGCCCAGAAGAAGAAGA, Sfrp5 reverse: TTCTTGTCCCAGCGGTAGAC; Dkk1 forward: GCCTCCGATCATCAGACTGT, Dkk1 reverse: GCTGGCTTGATGGTGATCTT; Actb forward: CTAGGCACCAGGGTGTGAT, Actb reverse: TGCCAGATCTTCTCCATGTC.
Immunoblots.
Cortices (E18.5) from PdgfbiCre;dnRAR403-fl from 4 separate animals per genotype (n = 4) were collected and lysed in RIPA buffer (Sigma-Aldrich) containing a protease inhibitor cocktail tablet (Roche). Protein concentration was determined using a BCA kit (Pierce). Lysates were combined with 4 × sample buffer (300 mm Tris, 5% SDS, 50% glycerol, 0.025% bromophenol blue, 250 mm β-mercaptoethanol) and 70 μg (E18.5) or 15 μg (E16.5) of protein per sample was run on Protean Tris-HCI 4–20% gradient gel (Bio-Rad) and then transferred onto PVDF membranes (Bio-Rad) or nitrocellulose membranes (Bio-Rad) using the Trans-Blot Turbo System (Bio-Rad). Immunoblots were blocked with 5% non-fat dehydrated milk (NFDM) in Tris-buffered saline (TBS) with 0.1% Tween (TBS-T) for 1.5 h and then incubated overnight at 4°C in 2.5% NFDM in TBS-T-containing primary antibodies for rabbit anti-Sox17 (1:500; Abcam) or rabbit anti-LEF-1 (1:500; Cell Signaling Technology). After primary incubation, blots were washed and then incubated in the 2.5% NFDM containing the appropriate horseradish peroxidase-linked secondary antibody (1:5000; Santa Cruz Biotechnology) for 45 min at room temperature. Clarity ECL substrate (Bio-Rad) and the ChemiDoc MP system (Bio-Rad) were used to visualize immunotagged protein bands. Blots were stripped with stripping buffer (Restore Plus; ThermoScientific) and reprobed with a mouse anti-β-actin (1:2000; Cell Signaling Technology) antibody as a loading control. Densitometry of bands was performed using ImageLab software (Bio-Rad); density values were corrected for loading variations within each blot using the amount of β-actin expression.
Statistics.
To detect statistically significant differences in mean values between a control and mutant gentoype at one developmental time point (vessel density, β-gal+ ECs per vessel length, cell proliferation density, qPCR analysis), Student's t tests were used. Analysis that compared more than two groups (e.g., control and two mutant gentoypes, multiple developmental time points, multiple cell culture treatment conditions, etc.), a one-way ANOVA with Tukey's post hoc analysis was used to detect statistically significant differences between genotypes or treatment conditions using pairwise analysis. The SEM is reported on all graphs.
Results
Cerebrovascular development is impaired in Rdh10 mutant embryos
Mouse mutants with an ENU-induced point mutation in the RA-biosynthetic enzyme Rdh10 have reduced levels of RA and display developmental defects consistent with RA deficiency (Ashique et al., 2012). Rdh10 mutants survive until E14.5, thus permitting analysis of RA-related neurovascular defects. E14.5 Rdh10 mutants display severe defects in eye and craniofacial development, as well as significant expansion of the dorsal telencephalon (Fig. 1A). The latter phenotype is caused by expansion of neocortical progenitors at the expense of neuron generation, resulting in an elongated, “ballooned” neocortex (Siegenthaler et al., 2009). In sections at the level of the forebrain, notably fewer (Fig. 1A, arrow) or, in some areas, no, isolectin-B4+ (Ib4+) blood vessels (Fig. 1A, open arrow) were present in the long, thin neocortex in the Rdh10 mutant brain. Avascular neocortical regions were not observed consistently though were usually seen in regions where the neocortex was very thin. Higher-magnification images of the neocortex revealed fewer, though larger diameter vessels in the notably thinned Rdh10 mutant neocortex (Fig. 1B, arrow). Numerous large-diameter vessels were seen in the PNVP vasculature adjacent to the Rdh10 mutant neocortex (Fig. 1B, open arrows). In contrast to the neocortical vasculature, Ib4+ vessels in the thalamus of Rdh10 mutants were not overtly different from control (Fig. 1B), indicating that severe vascular defects may be limited to the neocortex.
Figure 1.

Neocortical vascular development in E14.5 Rdh10 mutant embryos. A, Ib4-labeled blood vessels in E14.5 wild-type and Rdh10 mutant forebrain. Open arrow indicates avascular area of the neocortex; arrow indicates reduced vascular plexus in expanded neocortex. B, High-magnification images of E14.5 vascular plexus in the neocortex and thalamus of wild-type and Rdh10 mutants. Open arrows and arrows indicate enlarged, dysplastic vessels in PNVP and within the neocortex, respectively. C, Representative images of GLUT-1/BrdU labeling in the two vascular plexus in the neocortex (NC): the superficial PNVP, and plexus within the neocortex. Open arrows indicate BrdU+/Glut+ cells in both panels. D, Graphs depicting quantification of EC proliferation index in the NC PNVP and NC plexus in E14.5 wild-type and Rdh10 mutants. Asterisks indicate significance from wild-type value. E, Low-magnification images of E12.5 and E14.5 wild-type and Rdh10 mutant forebrains. F, High-magnification images of neocortical PNVP and internal vascular plexus at E12.5 and E14.5 in wild-type and Rdh10 mutants. G, Graph depicting vascular density in the two genotypes in the neocortex and thalamus at E12.5 and E14.5. *Significance from E12.5 value of the same genotype; #significance from E14.5 wild-type value. Scale bars: A, E, 500 μm; B, C, 100 μm. Ncx, Neocortex.
Blood vessels in the developing cortex appeared reduced in number, whereas vessels in the PNVP appeared more numerous. Decreased EC proliferation within the neocortex and increased EC proliferation within the PVNP could account for these differences. We examined this possibility by quantifying the percentage of GLUT-1+ ECs in the neocortical plexus and PNVP that incorporate the thymidine analog BrdU (EC proliferation index). Significantly more GLUT-1+/BrdU+ ECs were observed in Rdh10 mutant PNVP overlying the neocortex (Fig. 1C,D), whereas EC proliferation was significantly reduced in the vascular plexus within the Rdh10 neocortex (Fig. 1C,D). Rdh10 mutants expression of GLUT-1, a glucose transporter enriched in CNS ECs with expression that is induced early in the CNS vasculature by WNT signaling (Daneman et al., 2010), appeared decreased in neocortical blood vessels and elevated in the neuroepithelial cells of the VZ compared with control (Fig. 1C).
We next compared E14.5 cerebrovascular density with that at E12.5, an earlier time point when neocortical defects in Rdh10 mutant are not as severe. At E12.5, the thickness of the neocortical wall was comparable in Rdh10 mutants to littermate control tissue (Fig. 1E,F, left panels) and the vascular density in the neocortex was not significantly different between control and Rdh10 mutant embryos (Fig. 1G). However, vessels in the Rdh10 mutant embryos appeared enlarged at this age (Fig. 1F, open arrows), indicating that vascular defects are potentially present at this time point. In control mice, both the neocortical wall and vasculature show significant growth between E12.5 and E14.5. However, from E12.5 to E14.5 in Rdh10 mutants, there was substantial lateral expansion but very little radial expansion of the neocortex and blood vessel growth was significantly impaired (Fig. 1E–G). We next quantified vascular density in the striatum and thalamus of control and Rdh10 mutants at both E12.5 and E14.5 and found no differences in vascular growth between Rdh10 mutant and control samples (Fig. 1G). This analysis demonstrates that cerebrovascular defects may emerge early in Rdh10 mutants during neocortical development and worsen over time and that vascular growth defects in Rdh10 mutants are specific to the neocortical region.
Elevated Vegfa expression is associated with an upregulation of hypoxia-inducible genes in Rdh10 mutant neocortices and non-neocortical brain regions
Neuroepithelial-derived VEGFA is a major regulator of vascular growth in the CNS (Haigh et al., 2003; Raab et al., 2004; James et al., 2009). Reduced VEGF-A from neural progenitors in the neocortical VZ of Rdh10 mutants could contribute to aberrant vascular growth in the neocortex. To test this, we quantified Vegfa gene expression using RNA isolated from neocortex only or all other non-neocortical brain structures (striatum, thalamus, midbrain, hindbrain) at E13.5. Vegfa expression was substantially increased in both the Rdh10 mutant neocortical and non-neocortical samples compared with littermate controls (Fig. 2A). Vegfa expression is induced in response to hypoxia, so the increase in Vegfa expression that we observed in the Rdh10 mutants could be due to tissue hypoxia. We tested this possibility by analyzing the expression of known hypoxia-inducible genes Ldha, Pdk1, and Cox4i2 (Firth et al., 1994; Kim et al., 2006; Fukuda et al., 2007). All of these hypoxia-inducible genes were also upregulated (Fig. 2A), indicating that the elevated Vegfa expression in the neocortex is likely due to tissue hypoxia. Interestingly, increased expression of hypoxia genes were also observed in the non-neocortical regions of the Rdh10 mutants even though vascular development was not significantly affected in these regions (Fig. 2A). Expression of Slc2a1, which encodes the GLUT-1 protein, is also increased by hypoxia through a similar hypoxia-inducible factor-mediated mechanism (Chen et al., 2001). We found that GLUT-1 appeared to be upregulated in the neuroepithelium of Rdh10 mutant neocortices (Fig. 1C) and that Slc2a1 expression was upregulated in the neocortex but not in the non-neocortex of the Rdh10 mutants (Fig. 2B). Furthermore, quantification of GLUT-1 immunofluorescence intensity in neocortical VZ and in non-neocortical brain regions (striatum and thalamus) showed that VZ GLUT-1 expression was significantly increased in the Rdh10 mutant neocortex, but not in other brain regions (Fig. 2C). This is evident in low-magnification images of E14.5 control and Rdh10 mutant brains, in which GLUT-1 expression was limited to blood vessels in the control and in non-neocortical brain regions of Rdh10 mutants; however, regions of high neural GLUT-1 expression were observed specifically in the Rdh10 mutant neocortex (Fig. 2D, arrows, E). Collectively, these data indicates that Rdh10 mutants have tissue hypoxia throughout the embryonic brain, possibly due to systemic defects in embryonic development. However, focal upregulation of GLUT-1 in the neocortex suggests that hypoxia is more pronounced in the neocortex, likely due to impaired vascular growth specifically in this brain structure.
Figure 2.

Hypoxia-inducible targets VEGFA and GLUT-1 are elevated in Rdh10 mutant neocortices. A, qPCR for the hypoxia-inducible genes Vegfa, Ldha, Pdk, and Cox4i2 transcript expression in control and Rdh10 mutant neocortices and non-neocortical brain structures. B, qPCR for Slc2a1 (GLUT-1) transporter transcript expression in control and Rdh10 mutant neocortices and non-neocortical brain structures. C, Quantification of average intensity signal for GLUT-1 in the VZ of neocortical and striatum/thalamus brain regions of control (wild-type, Rdh10 heterozygous) and Rdh10 mutants. D, Low-magnification images of GLUT-1 labeling in E14.5 wild-type and Rdh10 mutant brains at the level of the cortex and striatum. Arrows indicate regions of high neuroepithelial GLUT-1 signal in the Rdh10 mutant neocortical VZ. E, High-magnification images of GLUT-1 labeling in the neocortical VZ and striatum of wild-type and Rdh10 mutants. *Significance from control (p < 0.05). Scale bars, 500 μm.
Endothelial WNT signaling is diminished in the Rdh10 mutant cerebrovasculature and correlates with elevated expression of WNT inhibitors in the neocortex
WNT signaling in CNS ECs, activated by the neural-derived WNT ligands WNT7a and WNT7b, is important for vascular growth, stabilization, and acquisition of BBB properties. The neocortical vascular growth defects and altered expression of GLUT-1 in the vasculature and neuroepithelium in Rdh10 mutants (Figs. 1, 2) is similar to mutant mice in which WNT7a and WNT7b are both deleted (Stenman et al., 2008) and when the WNT signaling component β-catenin is conditionally deleted from ECs (Daneman et al., 2009; Zhou et al., 2014). Therefore, we next looked at the integrity of the WNT pathway (e.g., endothelial WNT signaling, WNT ligands, and inhibitors) in Rdh10 mutant neocortices. We used the WNT signaling reporter mouse line Bat-gal-lacZ to assess endothelial WNT signaling in the Rdh10 mutant neocortical vasculature. β-gal+ ECs, as determined by colocalization with Ib4, were readily apparent in the control neocortical vasculature (Fig. 3A, arrows); however, β-gal+ ECs were nearly absent in the Rdh10 mutant neocortical vasculature and overlying PNVP (Fig. 3A, right). β-gal+ neural cells in the neocortex (Fig. 3A, open arrows) and in the overlying skin mesenchyme (Fig. 3A, double arrows) were present in Rdh10 mutants. We quantified the number of β-gal+ ECs per vessel length at E12.5 and E14.5 in the neocortices of control and Rdh10 mutant embryos. The density of β-gal+ ECs significantly increased across developmental time points in wild-type neocortices, but was significantly reduced at both time points in Rdh10 mutants (Fig. 3B).
Figure 3.

Diminished WNT signaling in Rdh10 mutant cerebrovasculature. A, β-gal (green) and Ib4 (red) coimmunolabeling in neocortical blood vessels at E14.5 of Bat-gal-LacZ/+ and Rdh10 mutant Bat-gal-LacZ/+ animals. Arrows indicate β-gal+ ECs, open arrows indicate β-gal+ neural cells, and double-head arrows point to β-gal+ cells in the skin. B, Quantification of number of β-gal+ ECs per vessel length in in the neocortex of control (wild-type and Rdh10 heterozygous) and Rdh10 mutant animals at E12.5 and E14.5. *Significance between control at E12.5 and E14.5; #significance from E12.5 wild-type; *#significance from E14.5 wild-type. C, D, Arrows indicate Ib4+ (red) vessels with Claudin-3 (green) signal in the neocortical region of a control, Bat-gal/+ brain. Open arrows in the control and mutant samples indicate Claudin-3 signal in the skin overlying the brain. Double arrows indicate Claudin-3-/Ib4+ vessels in the Rdh10 mutants. E, Arrows indicate LEF-1+ (green) ECs (Ib4 in red) in the neocortex of Bat-gal-LacZ/+ and Rdh10 mutant Bat-gal-LacZ/+ animals. F, qPCR for transcript expression of WNT ligands (Wnt7a, Wnt7b) and WNT inhibitors (Sfrp1, Sfrp2, Sfrp5, and Dkk1) in wild-type and Rdh10 mutant E13.5 neocortices and non-neocortical brain structures. *Significance between control and Rdh10 mutants. G, qPCR for transcription expression of the WNT inhibitors Sfrp5 and Dkk1 in cultured neocortical progenitors treated with RA or a pan RAR inhibitor; #significance from vehicle. Scale bars, 100 μm.
We assayed expression of two known targets of WNT-mediated gene transcription in the CNS vasculature, Claudin-3 (Liebner et al., 2008) and LEF-1 (Filali et al., 2002). Consistent with Bat-gal-LacZ expression analysis, Claudin-3 (Fig. 3C,D) and LEF-1 (Fig. 3E) expression were appreciably decreased in the neocortical vasculature of Rdh10 mutants. In conjunction with our quantitative analysis using the WNT signaling reporter, decreased expression of vascular LEF-1 and Claudin-3 in Rdh10 mutants demonstrates decreased endothelial WNT signaling within the neocortex of these mutants.
We next investigated whether the expression of WNT7a and WNT7b transcripts were reduced in neocortices of Rdh10 mutants; however, qPCR analysis showed no difference between wild-type and Rdh10 mutants at E13.5 (Fig. 3F). RA plays a crucial role in the development of the lung primordium by suppressing the expression of the WNT inhibitor Dkk1 (Chen et al., 2010). It is possible that RA inhibits the expression of Dkk1 in the neocortex to ensure that proper endothelial WNT signaling occurs. Expression of Dkk1, as well as certain WNT inhibitors called soluble frizzled receptor proteins (sFRPs) (Sfrp1, Sfrp2, and Sfrp5), were significantly upregulated in Rdh10 mutant neocortices (Fig. 3F). Elevated expression of WNT inhibitors was specific to the neocortex of Rdh10 mutants because no significant changes in WNT inhibitor expression were observed in non-neocortical regions (Fig. 3F).
Dkk1 and Sfrp5 were the most robustly upregulated of the WNT inhibitors assayed in the Rdh10 mutant neocortices and RA has been shown to suppress Dkk1 transcription directly in other developing organs (Chen et al., 2010). We used cultured NPCs derived from E14 mouse neocortex to test the idea that RA may be required to suppress the expression of Dkk1 and Sfrp5 in the developing neocortex. Treatment with RA significantly downregulated Dkk1 and Sfrp5 expression in NPCs (Fig. 3G). RA-mediated inhibition of Dkk1 and Sfrp5 expression was abrogated by the addition of a pan-RAR inhibitor, suggesting that RARs are required to mediate the effect of RA on Sfrp5 and Dkk1 expression (Fig. 3G). We tested whether RA modulated expression of Dkk1 and Sfrp5 in cultured cortical neurons; however, Dkk1 and Sfrp5 were undetectable in cultured neurons (data not shown). Collectively, these data show that severe cerebrovascular growth defects in Rdh10 mutants correlate with diminished endothelial WNT signaling, a pathway required for brain vascular development. Further, our data indicate that RA may function in the neocortex to suppress expression WNT inhibitors in neocortical progenitors, thus creating a permissive environment for WNT-mediated cerebrovascular growth.
RA functions cell-autonomously in brain ECs to modulate WNT signaling
Severe vascular growth defects and increased expression of WNT inhibitors was only observed in the Rdh10 mutant neocortex, indicating a specific non-cell-autonomous role for RA in this brain structure through regulating WNT inhibitor expression by neocortical progenitors. RARs are expressed by brain ECs, indicating that RA signaling is likely active in brain ECs and may have an important, cell-autonomous function in this cell type. Our first indication of this was an observation from our analysis of endothelial WNT signaling in non-neocortical brain regions of Rdh10 mutants using endothelial Bat-gal-lacZ expression as a readout of WNT activity. In the E14.5 thalamus, β-gal+ ECs were evident in the thalamic vasculature of both Bat-gal/+ and Rdh10; Bat-gal/+ mutant samples; however, the number and intensity of β-gal+ ECs was increased in the Rdh10 mutant (Fig. 4A, open arrows). Quantification of the number of β-gal+ ECs per vessel length in the striatal and thalamic vasculature at E14.5 revealed a significant increase in β-gal+ ECs in Rdh10 mutants (β-gal+/Ib4+ cells per 100 μm vessel length; wild-type: 1.8 ± 0.06 SEM vs Rdh10 mutant: 2.4 ± 0.17 SEM n ≥ 3, p = 0.03). These data show that endothelial WNT signaling is increased in non-neocortical regions of the Rdh10 mutant brain.
Figure 4.

Elevated WNT signaling in non-cortical Rdh10 mutant vasculature and neurovascular development in PdgfbiCre;dnRAR403-flox animals. A, β-gal (green) and Ib4 (red) coimmunolabeling in the thalamic vasculature of E14.5 Bat-gal-LacZ/+ and Rdh10 mutant Bat-gal-LacZ/+ animals. Open arrows indicate β-gal+ ECs. B, Top, Depiction of prenatal tamoxifen injection timing for PdgfbiCre;dnRAR403-flox animals. Bottom, GFP (green) immunostaining and Ib4 (red) labeling in E14.5 PdgfbiCreERT2-IRES-GFP (also known as PdgfbiCre) brain to illustrate specific expression of transgene in blood vessels. Arrows indicate GFP+/Ib4+ blood vessels, open arrows indicate GFP−/Ib4+ microglia. C, Whole fetus images of E18.5 control (dnrar403-fl/+) and mutant (PdgfbiCre;dnRAR403-fl/+ or fl/fl). D, Low-magnification image of whole brains from PdgfbiCre/+ animals with zero or two copies of the dnRAR403-flox allele. Arrows indicate hemorrhage within the cerebral hemispheres (CH). E, GLUT-1- (green), Ib4- (red), and DAPI-stained cortical sections of E18.5 PdgfbiCre/+ and PdgfbiCre;dnRAR403-fl/fl mutant. Open arrows indicate microhemorrhages. Inset shows GLUT-1+ red blood cells in the brain parenchyma, indicative of hemorrhage. Arrow in inset indicates activated Ib4+ microglia with amoeboid morphology. F, Ib4+ cerebrovasculature in E18.5 PdgfbiCre/+ and PdgfbiCre;dnRAR403-fl/fl mutant sections. Arrows indicate enlarged vessels in mutant sample. G, Neocortical progenitor markers Pax6, Tbr2, and deep-layer cortical neuronal marker Ctip2 in E16.5 PdgfbiCre/+ and PdgfbiCre;dnRAR403-fl/fl mutant sections. Scale bars, A, G, 100 μm; E, F, 200 μm.
RA signaling through its receptors has been shown to inhibit WNT signaling in a variety of cell types (Easwaran et al., 1999; Mulholland et al., 2005; Chanda et al., 2013), raising the possibility that RA may regulate WNT signaling directly in brain ECs. To begin to test this idea, we developed a mouse model in which RA signaling is specifically disrupted in brain ECs using an inducible EC-specific CreERT2 line (Pdgfbi-CreERT2, referred to here as PdgfbiCre; Claxton et al., 2008) and a conditional, dominant-negative version of RARα allele located in the ROSA26R locus (dnRAR403-flox) (Rosselot et al., 2010). DnRARα403 is a truncation mutant of the human RARα that can bind to endogenous RARs, but when expressed in a cell, disrupts endogenous RA signaling activity (Tsai et al., 1992; Damm et al., 1993). To investigate the effect of disrupted endothelial RA signaling on prenatal brain vascular development, pregnant females were injected with tamoxifen at E9 and E10 to induce Cre-mediated expression of dnRARα403 in ECs and fetuses were collected at E14.5, E16.5, and E18.5 (Fig. 4B). To confirm vascular-specific expression of the PdgfbiCre transgene in the brain, we took advantage of the IRES-EGFP present in the transgene and used a GFP antibody to detect transgene expression. At E14.5, GFP expression was observed in Ib4+ blood vessels in the brain and this was not Ib4+ microglia, which could be distinguished by their ramified cell morphology (Fig. 4B). Grossly, E18.5 fetuses expressing one or two copies of the dnRAR403-flox allele (PdgfbiCre;dnRAR403-fl/+ and PdgfbiCre;dnRAR403-fl/fl) had no obvious phenotype (Fig. 4C). In the brain, small hemorrhages were evident in E18.5 cerebral hemispheres in PdgfbiCre;dnRAR403-fl/fl animals (Fig. 4D). This was seen as extravasated GLUT-1+ red blood cells in sections (Fig. 4E, open arrows) next to amoeboid-shaped Ib4+ microglia (Fig. 4E, arrow in inset), indicative of activated microglia caused by microbleeds. Cerebrovascular density at E18.5 was not overtly affected when RA signaling was disrupted in ECs (Ib4+ vessel length/area of analysis: control PdgfbiCre/+ or dnRAR403-flox, 0.35 ± 0.007 vs PdgfbiCre;dnRAR403-fl/+, 0.36 ± 0.012 vs PdgfbiCre;dnRAR403-fl/fl, 0.37 ± 0.004 n = 3, p = 0.5). This is consistent with our analysis of non-neocortical vasculature in Rdh10 mutant embryos and brain vascular development in embryos exposed to RAR inhibitors (Mizee et al., 2013). However, enlarged vessels were evident in the mutant cerebrovasculature (Fig. 4F, arrows) and cerebrovascular vessel diameter was significantly increased in PdgfbiCre;dnRAR403-fl/fl mutants at E18.5 (control PdgfbiCre/+ or dnRAR403-flox, 5.8 ± 0.09 μm vs PdgfbiCre;dnRAR403-fl/fl, 7.0 ± 0.232 μm, n = 3, p = 0.035). These data show that disrupting RA signaling in brain ECs causes morphological changes in blood vessels and focal vascular instability (e.g., microbleeds), but does not appear to alter angiogenic growth.
It is possible that disrupting RA signaling in the vasculature could abrogate neurodevelopmental processes such as neural progenitor proliferation and differentiation. We examined this in the E16.5 neocortex of PdgfbiCre;dnRAR403-fl control and mutant animals by looking at expression of established progenitor cell (Pax6 and Tbr2) and postmitotic neuronal markers (Ctip2). Qualitatively, the Pax6+- and Tbr2+-expressing progenitor populations appeared similar in PdgfbiCre;dnRAR403-flox control and mutant mice, as did the positioning of Ctip2+ neurons in the lower part of the cortical plate (Fig. 4G). These data indicate that disruption of endothelial RA signaling and any subsequent effects on vascular development and stability (e.g., microbleeds) does not grossly affect corticogenesis.
To test directly whether RA signaling functions cell-autonomously in brain ECs to inhibit WNT transcriptional activity, we bred the WNT transcriptional reporter line Bat-gal-lacZ into the PdgfbiCre;dnRAR403-flox control and mutant background and analyzed EC β-gal expression in the forebrain regions (e.g., neocortex, striatum, and thalamus). β-gal+ ECs were more numerous in E18.5 PdgfbiCre;dnRAR403-fl/fl fetal brain compared with control (Fig. 5A,B, open arrows), indicating that endothelial WNT signaling is more active when endothelial RA signaling is disrupted. Quantification of β-gal+ ECs per vessel length showed a significant increase in PdgfbiCre;dnRAR403-fl/+ and even more so in PdgfbiCre;dnRAR403-fl/fl mutants (Fig. 5C). Expression of LEF-1, a direct transcriptional target of WNT signaling expressed by brain ECs, appeared elevated in PdgfbiCre;dnRAR403-fl/fl mutants compared with control (Fig. 5D,E) and quantification of LEF-1 protein expression in cortical lysate showed a significant increase in PdgfbiCre;dnRAR403-fl/fl mutant samples (LEF-1 band density relative to β-actin: PdgfbiCre/+ or dnRAR403-flox, 0.85 ± 0.09 vs PdgfbiCre;dnRAR403-fl/fl, 1.4 ± 0.2, p = 0.046, n = 4). We looked at the expression of LEF-1 in the head vasculature of control and PdgfbiCre;dnRAR403-fl/fl mutants to determine whether disrupted RA signaling in non-CNS vessels leads to ectopic WNT activity. LEF-1 was expressed strongly expressed in the skin, but was not detectable in Ib4+ blood vessels in either genotype (Fig. 5G, arrows). This indicates that the interaction between RA and WNT signaling in ECs is likely limited to the brain vasculature. Further, this shows that expression of the dnRAR403-flox allele alone does not activate endothelial WNT signaling. Collectively, our analysis of non-cortical vasculature in Rdh10 mutants and Pdgfbi-Cre;dnRAR403-flox mutants demonstrates that disruption of RA signaling in brain ECs causes increased WNT signaling and points to a novel, cell-autonomous function for RA as an inhibitor of endothelial WNT signaling in the developing brain.
RA exposure inhibits endothelial WNT signaling both in vivo and in cultured ECs
We next tested whether RA is sufficient to inhibit WNT activity in brain ECs by feeding pregnant Bat-gal-lacZ/+ mice an RA-enriched diet from E10 to E14.5 or E16.5 and then analyzing β-gal+ EC density in the neocortical vasculature (Fig. 6A). Exposure to RA did not significantly alter β-gal+ endothelial cell density at E14.5 (Fig. 6B). Between E14.5 and E16.5, there was a significant increase in the β-gal+ EC density in fetuses from control diet females, but this was not observed in RA-exposed animals, resulting in a significant difference between the control and RA diet at E16.5 (Fig. 6B). The RA-dependent reduction in WNT signaling did not affect neocortical vascular density at either age (Fig. 6C), indicating that the alterations in RA and WNT signaling caused by exogenous RA exposure did not affect neurovascular growth overtly.
Figure 6.

RA inhibits endothelial WNT signaling in vivo and in vitro. A, Depiction of RA treatment paradigm for pregnant Bat-gal-LacZ/+ animals. B, Graph depicting quantification of β-gal+ ECs per 100 μm vessel length in control and RA-exposed fetuses at E14.5 and E16.5. *Significant difference from E14.5 control diet; #significant difference from control diet at E16.5. C, Graph depicting quantification of vessel density in control and RA-treated animals at E14.5 and E16.5. D, Graph depicting quantification of transwell migration assay with bEnd.3 cell line after treatment with RA, WNT3a, or RA + WNT3a. *Significance from untreated cells (CTL). E, Quantification of cell proliferation of bEnd.3 cells after 3 d of treatment with RA, WNT3a, or both. *Significance from untreated cells (CTL); #significant difference from WNT3a treatment. F, RT-PCR for RARs and RXRs using E18.5 microvessel and postnatal day 7 meninges cDNA. The housekeeping gene GAPDH is used to show equal amount of RNA to generate the cDNA used in the RT-PCR. G, Transfection of a RARα construct decreases the response of cells to WNT stimulation. Two-way ANOVA revealed a significant difference due to construct (F(1, 16) = 1301, p < 0.001) and treatment (F(3, 16) = 518.1, p < 0.001), as well as a significant interaction between both factors (F(3, 16) = 200.1, p < 0.001). H, RXRβ does not alter the response of cells to WNT stimulation. Two-way ANOVA revealed a significant difference due to treatment (F(3, 16) = 90.17, p < 0.001), but no significant difference due to construct (F(1, 16) = 4.358, p > 0.05) and no interaction between the two factors (F(3, 16) = 1.188, p > 0.05). I, dnRARα increases the response of cells to WNT stimulation. Two-way ANOVA revealed a significant difference due to construct (F(1, 16) = 110.7, p < 0.001) and treatment (F(3, 16) = 110.7, p < 0.001), as well as a significant interaction between the two factors (F(3, 16) = 49.98, p < 0.001). For G–I, asterisks directly above the bar indicate significance from untreated pCIG control and hash marks indicate significance from WNT3a treatment alone; within-group differences are indicated by connected lines.
Our in vivo data point to an inhibitory effect of RA on WNT signaling, but it is not clear whether it can block WNT-mediated effects on brain EC behavior. We tested this in culture by determining whether RA inhibits the effect of WNT ligands on brain EC migration and proliferation. Treatment with the WNT ligand WNT7a promotes transwell migration of the mouse brain endothelioma cell line bEnd.3 (Daneman et al., 2009) and we observed the same effect with WNT3a (Fig. 6D) and WNT7a. RA in the nanomolar range had no effect on bEnd.3 cell transwell migration, but did block the promigratory effect of WNT3a (Fig. 6D) and WNT7a on migration (no. of cells per 10 field: control, 963 ± 112 SEM; RA (50 nm), 1070 ± 146 SEM; WNT7a (5 μg/ml), 1256 ± 37 SEM; RA + WNT7a, 945 ± 72 SEM; control vs WNT7a, p = 0.0062; WNT7a vs RA + WNT7a, p = 0.0027, n = 3). The same concentration of WNT3a inhibited bEnd.3 cell proliferation, an effect that was blocked when cells were cotreated with RA (Fig. 6E). These data further confirm that RA can regulate endothelial WNT signaling directly and can modulate WNT-mediated endothelial cell behavior.
We next sought to determine whether the effect of RA on WNT signaling was at the level of RARs. We tested RARα specifically because it was the most abundant RAR expressed by fetal brain microvessels, which contain ECs (Fig. 6F). To do this, we manipulated RA signaling in cultured cells expressing a WNT-β-catenin signaling reporter. HEK293 cells were transfected with TOP-Flash (containing seven copies of the TCF/LEF binding site upstream of a firefly luciferase gene) or FOP-Flash (containing seven mutated copies of the TCF/LEF binding site upstream of a firefly luciferase gene). Activation of WNT signaling induces accumulation and subsequent translocation of β-catenin to the nucleus, which interacts with TCF/LEF transcription factors, activating the TOP-Flash reporter construct, but not the FOP-Flash reporter construct. Cells were cotransfected with control (pCIG), RARα, or RXRβ expression vectors. Cells transfected with control vector and treated with WNT3a showed enhanced TOP-Flash activity over FOP-Flash activity (p < 0.001), whereas treatment with RA only had no significant effect on reporter activity with control vector (Fig. 6G). Cotreatment of WNT3a and RA to cells transfected with control vector led to reduced activation of the TOP-flash reporter compared with WNT3a alone (Fig. 6G). Cotransfection of RARα had a significant, inhibitory effect on WNT signaling and decreased TOP-Flash activation by 70.6% after WNT3a treatment (p < 0.001), by 81.1% after RA treatment (p < 0.001), and by 90.2% after cotreatment with WNT3a and RA (p < 0.001) compared with vector controls (Fig. 6G). Interestingly, cotransfection of another retinoid receptor, RXRβ, did not alter WNT signaling activation after WNT3a, RA, or combined WNT3a and RA treatment compared with similarly treated vector controls (Fig. 6H). These results show that RARα can regulate WNT transcriptional activity.
We next sought to determine whether disruption of RA signaling in cells altered their responsiveness to WNT ligands. To do this, cells were cotransfected with the same dominant-negative RARα construct (dnRARα403) used to construct the dnRAR403-flox allele used in our in vivo experiments (Damm et al., 1993; Sen et al., 2005). Expression of this truncated construct interferes with endogenous RA signaling because the transcriptional regulatory domain of the receptor is deleted (Damm et al., 1993; Sen et al., 2005; Rajaii et al., 2008). Expression of the dnRARα403 construct in cells without treatment of WNT3a or RA had no effect on TOP-Flash reporter activity (Fig. 6I), showing that the expression of the dominant-negative receptor does not activate WNT transcriptional activity directly. In cells expressing the dnRARα403 construct, WNT3a-mediated activation of the TOP-Flash reporter was substantially increased compared with the WNT3a-treated cells with control vector (Fig. 6I). This shows that expression of dnRARα403 disrupts the normal RAR-mediated inhibition of WNT signaling within cells, possibly by displacing endogenous receptors in retinoid receptor complexes. We observed an RA-dependent component because cotreatment with RA and WNT3a dampened the activation effect of dnRARα (Fig. 6I). Previous studies have shown that dnRARα403 can still bind RA ligand, although with less affinity than wild-type RARα (Damm et al., 1993). Together, these studies confirm a reciprocal relationship between WNT and RA signaling at the level of RARs.
Sox17 is a target of WNT signaling in fetal brain ECs and is upregulated after disruption of RA signaling
WNT signaling regulates neurovascular development in the CNS and our evidence points to RA signaling as a modulator of WNT signaling in brain ECs. Sox17 is a transcription factor that is required for vascular development and its expression is regulated by endothelial WNT signaling in the postnatal CNS vasculature (Ye et al., 2009; Corada et al., 2013). We tested whether the latter was also the case for the fetal brain vasculature using mice with EC conditional knock-down of the WNT signaling component β-catenin (PdgfbiCre;Ctnnb1-flox). At E14.5, Sox17 was expressed to varying degrees by ECs in the neocortex, whereas Sox17 expression was appreciably decreased in dysplastic blood vessels of PdgfbiCre;Ctnnb1-fl/fl mutants (Fig. 7A). Moreover, the expression of Sox17 and the WNT transcriptional targets Lef1 and Axin2 was significantly reduced in the fetal brain microvasculature isolated from E18.5 PdgfbiCre;Ctnnb1-fl/fl mutant brains (Fig. 7B). These data show that Sox17 is regulated by WNT-β-catenin signaling in the fetal brain vasculature.
Figure 7.

Elevated expression of Sox17 in PdgfbiCre;dnRAR403-fl/fl neurovasculature. A, Immunostaining for Sox17 (green) in Ib4+ (red) cerebral vessels in tissue from control and an EC-specific knock-down of WNT signaling component β-catenin at E14.5 (PdgfbiCre;Ctnnb1-fl/fl). B, Graph depicting Lef1, Axin2, and Sox17 transcript levels in microvessels isolated from E18.5 PdgfbiCre/+;Ctnnb1-fl/+ and PdgfbiCre/+;Ctnnb1-fl/fl brains. Asterisks indicate significance from PdgfbiCre;Ctnnb1-fl/+ value. C, Representative Sox17 (green) immunostaining in Ib4+ (red) cerebral vessels at E18.5 from PdgfbiCre/+ and PdgfbiCre;dnRAR403-fl/fl brains. Open arrows indicate weakly Sox17+ vessels; arrows indicate vessels with high Sox17 expression. D, Sox17 (44 kDa) and β-actin (52 kDa) immunoblots on protein lysate from E18.5 control (□: PdgfbiCre/+ or dnRAR403-flox) or mutant (■: PdgfbiCre;dnRAR403-fl/fl) neocortices. Scale bars, 100 μm.
We next investigated Sox17 in the context of disrupted RA signaling using PdgfbiCre;dnRAR403-fl/fl mutants that have elevated endothelial WNT transcriptional activity. High expression of Sox17 was observed in some vessels in the E18.5 control cortex (Fig. 7C, left, arrows), whereas other vessels had low Sox17 expression (Fig. 7C, left, open arrows). In contrast, Sox17 was strongly expressed by all blood vessels in the PdgfbiCre;dnRAR403-fl/fl fetal neocortex (Fig. 7C, right, arrows) and Sox17 protein expression, quantified via immunoblotting, was significantly elevated in fetal cortical lysate compared with control (Fig. 7D; Sox17 band density relative to β-actin: PdgfbiCre/+ or dnRAR403-flox, 1.3 ± 0.07 vs PdgfbiCre;dnRAR403-fl/fl, 1.8 ± 0.14, p = 0.019, n = 4). These data show that brain ECs with disrupted RA signaling and increased WNT signaling have increased Sox17 expression.
Sox17 is expressed by arterial ECs and is required for expression of artery-specific markers (Corada et al., 2013). In the fetal brain vasculature, we found that Sox17 was weakly expressed by venous blood vessels, identified by nuclear receptor Coup-TFII (Fig. 8A, open arrows). Sox17 was highly expressed by CoupTFII-negative vessels (Fig. 8A, arrow) and arterial vessels identified by Ephrin-B2-GFP in the EC nuclei (Fig. 8C, arrow). Expression of Sox17 was appreciable higher in Coup-TFII+ venous ECs in PdgfbiCre;dnRAR403-fl/fl fetal brains compared with control brain vasculature (Fig. 8B, open arrows). Coup-TFII was also expressed by perivascular mural cells (Fig. 8A, B, double arrow) and some neurons (Fig. 8B, triple arrow). High expression of Sox17 was limited to Ephrin B2-GFP+ vessels in control brain, whereas high Sox17 was observed in both Ephrin B2-GFP+ and Ephrin-B2-GFP− ECs in PdgfbiCre;dnRAR403-fl/fl fetal brain vasculature (Fig. 8C, D, arrows: Ephrin-B2-GFP+/Sox17+, open arrows: Ephrin-B2-GFP−/Sox17+). GFP signal was visible in EC membrane in PdgfbiCre;dnRAR403-fl/fl sections, but not control sections, due to IRES-GFP present in the PdgfbiCre allele (Fig. 8D, triple arrow). The increase in Sox17 in the vasculature, including venous blood vessels that normally have low levels of Sox17, in PdgfbiCre;dnRAR403-fl/fl fetal brains did not result in defects in arterial–venous specification. This is based on the observation that mutants retained expression of venous marker Coup-TFII and had both Ephrin-B2-GFP+ and Ephrin-B2-GFP− vessels (Fig. 8B,D). Collectively, our data suggest that RA signaling in endothelial cells may act as a balance to ensure normal WNT-driven brain vascular development and to moderate endothelial Sox17 expression levels.
Figure 8.
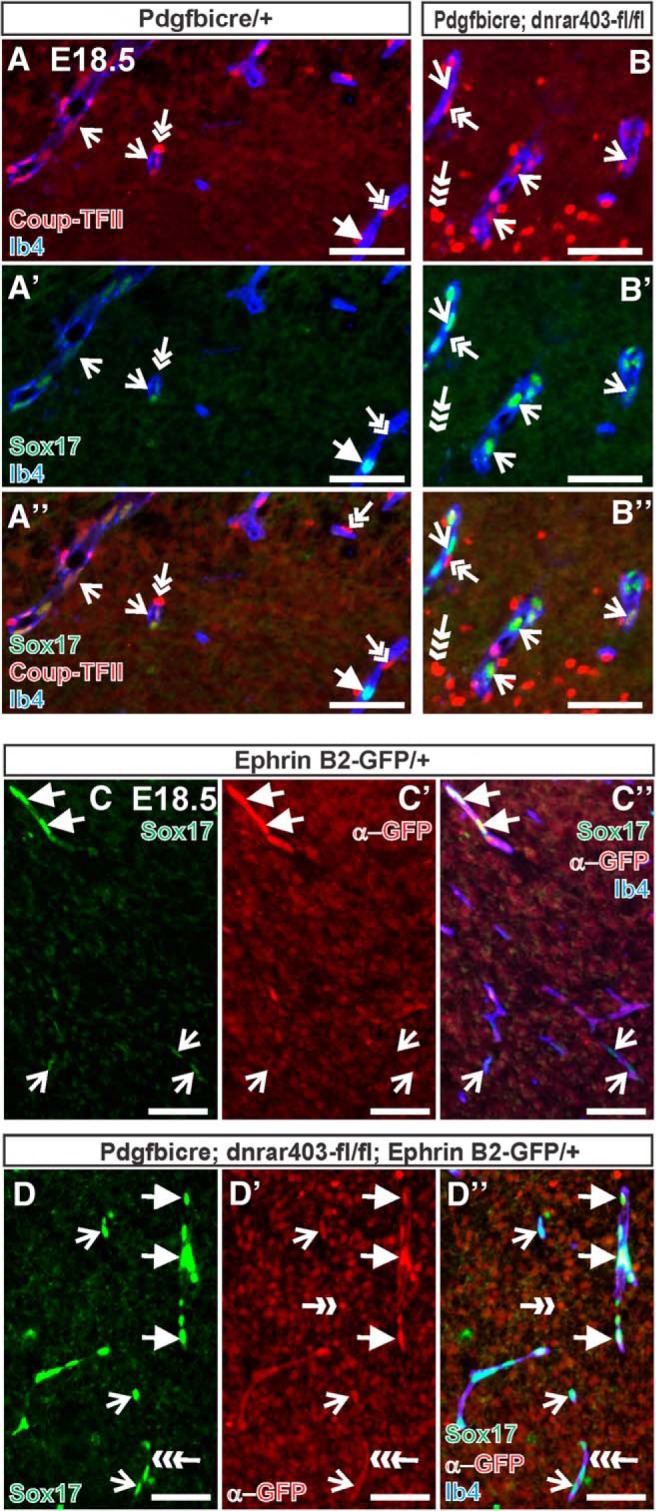
Elevated Sox17 expression in PdgfbiCre;dnRAR403-fl/fl venous and arterial vessels. A, B, Immunostaining for Sox17 (green) and Coup-TFII (red) on E18.5 PdgfbiCre/+ (A) and PdgfbiCre;dnRAR403-fl/fl (B) brains. Open arrows indicate Ib4+ (blue) vessels with Coup-TFII+ ECs (presumptive venous blood vessel). Arrow in A indicates blood vessel in control brain tissue that is Coup-TFII−/Sox17+ (presumptive arterial vessel). Double arrows indicate Coup-TFII+ mural cells; triple arrow indicates Coup-TFII+ neural cell. C, D, GFP (red) and Sox17 (green) immunostaining in control and PdgfbiCre;dnRAR403-fl/fl animals expressing Ephrin B2-GFP that labels arterial EC nuclei. Arrows indicate GFP+/Sox17+ arterial EC nuclei; open arrows indicate Sox17 expression in GFP- EC nuclei. C′′ and D′′ show overlay with Ib4 to label the vasculature (blue). Ephrin-B2-GFP is also expressed by some neurons (double-headed arrow). GFP IF is present in the endothelial cell membrane of images in D due to the IRES-GFP present in the PdgfbiCre allele construct (triple arrow). Scale bars, 100 μm.
Discussion
Here, we demonstrate that RA has separate functions during brain vascular development. In the developing neocortex, RA functions non-cell-autonomously to promote endothelial WNT signaling and cerebrovascular growth via a mechanism that involves suppressing expression of WNT inhibitors by neocortical progenitors and possibly neurons (Fig. 9A). RA also functions cell-autonomously in brain ECs to inhibit endothelial WNT signaling and prevent ectopic expression of WNT target genes such as Sox17 (Fig. 9B). Our results implicate for the first time a factor upstream of the WNT pathway in brain vascular development and reveal a multifaceted mechanism through which RA acts on both neural and vascular cells to target endothelial WNT signaling activity.
Figure 9.
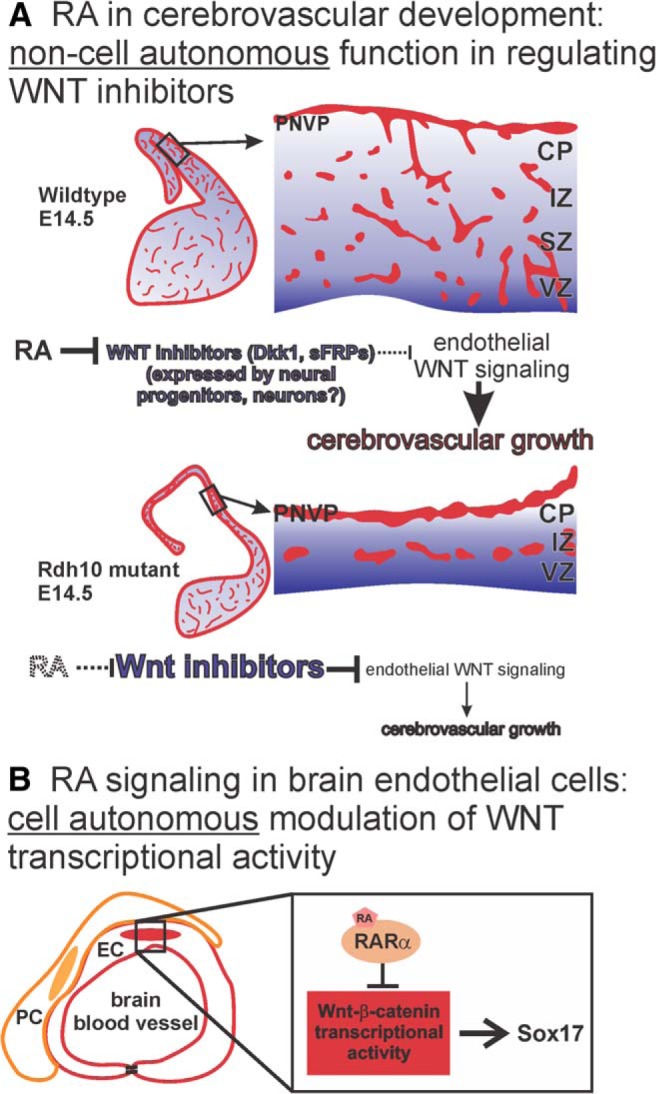
Model of RA functions during brain vascular development. A, RA in the developing neocortex normally functions to suppress expression of WNT inhibitors (Dkk1, sFRPs) to create a permissive environment for endothelial WNT signaling that drives cerebrovascular development. In RA-deficient Rdh10 mutants, ectopic expression of WNT inhibitors impedes endothelial WNT signaling, which disrupts growth of the cerebrovasculature. B, RA functions cell-autonomously in brain ECs, likely through its receptor RARα, to inhibit WNT-β-catenin transcriptional and limit expression of its target gene Sox17.
Rdh10 mutants globally lack RA and have significant developmental defects consistent with RA deficiency. Here, we show that, in addition to the defects in neocortical development, growth of the cerebrovasculature is severely impaired in Rdh10 mutants. Other brain regions have relatively normal vasculature, pointing to a unique role for RA in cerebrovascular development. We provide data that two major neuro-angiogenic pathways, VEGFA and WNT, are disrupted in Rdh10 mutant neocortices. With regard to VEGFA, Vegfa and several other hypoxia-inducible genes are upregulated in both the Rdh10 mutant neocortex and non-neocortical brain regions. These data indicate widespread hypoxia in the developing brain, possibly caused by other developmental defects in Rdh10 mutants. Tissue hypoxia appears to be more pronounced in the Rdh10 mutant neocortex, as evidenced by selective neural upregulation of GLUT-1 in this brain region, possibly due to severe cerebrovascular growth defects. Despite elevated Vegfa gene expression, we did not observe vascular overgrowth or impaired vascular integrity (e.g., hemorrhage) in the Rdh10 mutant brain, two features that have been reported in mutant mice with conditional upregulation of Vegfa in the neuroepithelium (Yang et al., 2013). Possibly, tissue hypoxia and Vegfa upregulation only begin to emerge at the end of Rdh10 mutant viability (E14.5), so VEGF-A protein levels are only elevated at late time points. At earlier developmental time points (E12.5), VEGF-A could be decreased in the Rdh10 mutant neocortex and possibly contribute to defects in cerebrovascular development, namely enlarged vasculature, seen at these time points. Our analysis does not differentiate between Vegfa transcripts expressed by different cell types present in the tissue samples. VEGF-A is expressed by neural progenitors, where it is required for vascular growth in the brain; however, VEGF-A expressed by ECs is reported to be required for neocortical and vascular development (Li et al., 2013). Increased VEGF-A from different cell sources in the neocortex could affect vascular and neocortical development differentially; however, more studies are needed to address this specifically.
Perhaps more compelling is our evidence demonstrating a near absence of endothelial WNT signaling concurrent with cerebrovascular defects in Rdh10 mutants. Endothelial WNT signaling, stimulated by WNT7a and WNT7b produced by progenitors and neurons in the developing brain, is required for brain vascular growth, stability, and BBB formation (Stenman et al., 2008; Daneman et al., 2009; Zhou et al., 2014). Therefore, reduced endothelial WNT signaling is likely a major factor contributing to defective cerebrovascular development in Rdh10 mutants. We provide evidence of a non-cell-autonomous function for RA as the underlying cause of reduced endothelial WNT signaling in Rdh10 mutants. We show that the WNT inhibitors Dkk1 and several sFRPs are specifically upregulated in the Rdh10 mutant neocortex, but no other brain regions. Dkk1 is a potent inhibitor of canonical WNT signaling through direct binding to WNT coreceptors low-density lipoprotein receptor-related 5 and 6 (LRP5/6), whereas sFRPs antagonize WNT signaling by interfering with the interaction between WNT ligands and receptors (Mao et al., 2001). Dkk1 and sFRP5 show the most substantial increase in gene expression in the Rdh10 neocortex and we provide cell culture data showing that RA, functioning through RARs, is sufficient to suppress Dkk1 and Sfrp5 gene expression in neocortical progenitors. This sets up a model in which RA deficiency in Rdh10 mutants leads to loss of RA-mediated suppression WNT inhibitors in neocortical progenitors and possibly postmitotic neurons, and the resulting ectopic expression of WNT inhibitors causes impairment of endothelial WNT signaling in the neocortex (Fig. 9A). Equally important to consider is that the cerebrovascular defects and diminished endothelial WNT signaling are occurring within a severely dysplastic neocortex caused by lack of RA. Reduced numbers of neocortical progenitors and neurons caused by aberrant proliferation and differentiation likely play some role in the altered expression of WNT pathway proteins. This is indicated by analysis showing that vascular growth defects are most pronounced at E14.5, a time point when the proliferative and postmitotic regions of the Rdh10 mutant neocortex are substantially thinner than control animals. An intriguing possibility is that persistent tissue hypoxia in the neocortex could be contributing to the aberrant progenitor proliferation and differentiation in the Rdh10 mutant cortex. In this way, the vascular defects could be a major contributor or at least exacerbate defects in corticogenesis. Recent work demonstrated that the neocortical progenitors switch from self-renewing divisions to neurogenerating divisions, which coincided with cerebrovascular growth and reduced levels of tissue hypoxia (Lange et al., 2016). Further studies are needed to understand how defective corticogenesis and impaired cerebrovascular development are connected in Rdh10 mutant animals.
In the non-neocortical brain regions of Rdh10 mutants, we found that endothelial WNT signaling was elevated. This was our first indication that RA may function cell-autonomously in brain ECs to inhibit WNT signaling. This observation was supported by increased endothelial WNT signaling in mutants with EC-specific disruption of RA signaling and data showing that exposure of embryos to excess RA diminishes brain endothelial WNT signaling. It is important to note that analysis of endothelial WNT signaling in PdgfbiCre;dnRAR403-flox mutants and RA-treated embryos encompassed neocortical and non-neocortical (striatum, thalamus) structures. This suggests that the cell-autonomous function for RA signaling in brain ECs throughout the brain is to inhibit endothelial WNT signaling. In the neocortex, however, our data demonstrate that RA has a separate, non-cell-autonomous function with regard to endothelial WNT signaling: controlling expression of WNT inhibitors to create a permissive environment for WNT-mediated cerebrovascular growth. Presumably, loss of RA in the neocortex of Rdh10 mutants lessens the inhibitory effect of RA signaling on endothelial WNT transcriptional activity. This is observed in other Rdh10 mutant brain regions. However, the substantial increase in WNT inhibitors resulting from the loss of RA acting on other cell types likely severely impairs activation of endothelial WNT pathways by WNT ligands. The significance of RA having non-cell-autonomous and cell-autonomous functions with regard to endothelial WNT signaling specifically in the neocortex is not clear, but will be addressed in future studies.
In the developing CNS, nascent vessels are surrounded by WNT ligands from neural sources. These signals ensure vessel integrity and help to initiate and maintain barrier properties in the neurovasculature, features that are required by all CNS ECs (Liebner et al., 2008; Stenman et al., 2008; Daneman et al., 2009; Zhou et al., 2014). Why, then, is RA acting as an inhibitor to this key pathway in brain ECs? Ectopic WNT signaling in the developing embryonic vasculature leads to widespread arterialization (Corada et al., 2010), so RA might act as an important “brake” on WNT signaling in the neurovasculature to prevent inappropriate acquisition of arterial traits. We do not, however, find evidence of arterialization of brain vessels in PdgfbiCre;dnRAR403-flox mutants. Possibly, fetal brain ECs do not respond to elevated WNT signaling in the same way as newly specified ECs. In support of this, when an inducible Cre line was used to express constitutively active β-catenin in ECs after E9.5, the widespread arterialization of the embryonic vasculature was not seen (Corada et al., 2010). We hypothesize that RA modulates WNT signaling through its receptor RARα to prevent overexpression of its target Sox17 (Fig. 9B). Forcing expression of Sox17 in ECs causes defects in brain and retinal vascular development, most notably increased vascular growth (Lee et al., 2014). We found dysplastic vessels and microbleeds in PdgfbiCre;dnRAR403-flox mutants with ectopic Sox17 expression. Forthcoming experiments will address whether the microbleeds and increased vascular diameter in PdgfbiCre;dnRAR403-flox mutants is caused by elevated Sox17 expression and will explore the transcriptional targets of Sox17 in brain ECs that mediate its function in the brain endothelium.
Our data showing repression of WNT signaling by RA in CNS ECs is consistent with the established literature on cross-talk between RA and WNT pathways both in development and disease. RA inhibits WNT signaling during hematopoietic stem cell development (Chanda et al., 2013) and in a variety of cancer cell lines with oncogenic β-catenin activity (Mulholland et al., 2005). Modulation of WNT signaling by RA signaling likely occurs at the level of RARα, which we show here is the main RAR expressed in fetal brain ECs. RARs can interact with components of the WNT transcriptional complex, which includes β-catenin, TCF members, and Lef1, and through these interactions modulate WNT-mediated transcription (Easwaran et al., 1999; Shah et al., 2003). Future work looking at the direct interactions between proteins in these two pathways will provide insight into how brain ECs integrate RA and WNT signaling appropriately during brain vascular development.
Footnotes
This work was supported by the National Institutes of Health (National Institute of Neurological Disorders and Stroke Grant K99-R00 NS070920 to J.A.S. and National Institute on Drug Abuse Grant R01 DA017627 to S.J.P.) and the American Health Association/American Academy of Neurology (Lawrence M. Brass, M.D. Stroke Research Postdoctoral Fellowship to J.A.S.). J.A.S. would like to recognize the invaluable guidance and support of Virginia Lancaster and Dr. Joseph Gerber throughout her life and career.
The authors declare no competing financial interests.
References
- Al Tanoury Z, Piskunov A, Rochette-Egly C. Vitamin A and retinoid signaling: genomic and nongenomic effects. J Lipid Res. 2013;54:1761–1775. doi: 10.1194/jlr.R030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashique AM, May SR, Kane MA, Folias AE, Phamluong K, Choe Y, Napoli JL, Peterson AS. Morphological defects in a novel Rdh10 mutant that has reduced retinoic acid biosynthesis and signaling. Genesis. 2012;50:415–423. doi: 10.1002/dvg.22002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer HC, Bauer H, Lametschwandtner A, Amberger A, Ruiz P, Steiner M. Neovascularization and the appearance of morphological characteristics of the blood-brain barrier in the embryonic mouse central nervous system. Brain Res Dev Brain Res. 1993;75:269–278. doi: 10.1016/0165-3806(93)90031-5. [DOI] [PubMed] [Google Scholar]
- Bohnsack BL, Lai L, Dolle P, Hirschi KK. Signaling hierarchy downstream of retinoic acid that independently regulates vascular remodeling and endothelial cell proliferation. Genes Dev. 2004;18:1345–1358. doi: 10.1101/gad.1184904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, Kemler R. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- Breier G, Albrecht U, Sterrer S, Risau W. Expression of vascular endothelial growth factor during embryonic angiogenesis and endothelial cell differentiation. Development. 1992;114:521–532. doi: 10.1242/dev.114.2.521. [DOI] [PubMed] [Google Scholar]
- Chanda B, Ditadi A, Iscove NN, Keller G. Retinoic acid signaling is essential for embryonic hematopoietic stem cell development. Cell. 2013;155:215–227. doi: 10.1016/j.cell.2013.08.055. [DOI] [PubMed] [Google Scholar]
- Chen C, Pore N, Behrooz A, Ismail-Beigi F, Maity A. Regulation of glut1 mRNA by hypoxia-inducible factor-1: interaction between H-ras and hypoxia. J Biol Chem. 2001;276:9519–9525. doi: 10.1074/jbc.M010144200. [DOI] [PubMed] [Google Scholar]
- Chen F, Cao Y, Qian J, Shao F, Niederreither K, Cardoso WV. A retinoic acid-dependent network in the foregut controls formation of the mouse lung primordium. J Clin Invest. 2010;120:2040–2048. doi: 10.1172/JCI40253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claxton S, Kostourou V, Jadeja S, Chambon P, Hodivala-Dilke K, Fruttiger M. Efficient, inducible Cre-recombinase activation in vascular endothelium. Genesis. 2008;46:74–80. doi: 10.1002/dvg.20367. [DOI] [PubMed] [Google Scholar]
- Corada M, Nyqvist D, Orsenigo F, Caprini A, Giampietro C, Taketo MM, Iruela-Arispe ML, Adams RH, Dejana E. The Wnt/beta-catenin pathway modulates vascular remodeling and specification by upregulating Dll4/Notch signaling. Dev Cell. 2010;18:938–949. doi: 10.1016/j.devcel.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corada M, Orsenigo F, Morini MF, Pitulescu ME, Bhat G, Nyqvist D, Breviario F, Conti V, Briot A, Iruela-Arispe ML, Adams RH, Dejana E. Sox17 is indispensable for acquisition and maintenance of arterial identity. Nat Commun. 2013;4:2609. doi: 10.1038/ncomms3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damm K, Heyman RA, Umesono K, Evans RM. Functional inhibition of retinoic acid response by dominant negative retinoic acid receptor mutants. Proc Natl Acad Sci U S A. 1993;90:2989–2993. doi: 10.1073/pnas.90.7.2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R, Agalliu D, Zhou L, Kuhnert F, Kuo CJ, Barres BA. Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc Natl Acad Sci U S A. 2009;106:641–646. doi: 10.1073/pnas.0805165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468:562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davy A, Bush JO, Soriano P. Inhibition of gap junction communication at ectopic Eph/ephrin boundaries underlies craniofrontonasal syndrome. PLoS Biol. 2006;4:e315. doi: 10.1371/journal.pbio.0040315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easwaran V, Pishvaian M, Salimuddin, Byers S. Cross-regulation of beta-catenin-LEF/TCF and retinoid signaling pathways. Curr Biol. 1999;9:1415–1418. doi: 10.1016/S0960-9822(00)80088-3. [DOI] [PubMed] [Google Scholar]
- Filali M, Cheng N, Abbott D, Leontiev V, Engelhardt JF. Wnt-3A/beta-catenin signaling induces transcription from the LEF-1 promoter. J Biol Chem. 2002;277:33398–33410. doi: 10.1074/jbc.M107977200. [DOI] [PubMed] [Google Scholar]
- Firth JD, Ebert BL, Pugh CW, Ratcliffe PJ. Oxygen-regulated control elements in the phosphoglycerate kinase 1 and lactate dehydrogenase A genes: similarities with the erythropoietin 3′ enhancer. Proc Natl Acad Sci U S A. 1994;91:6496–6500. doi: 10.1073/pnas.91.14.6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–122. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- Haigh J, Morelli PI, Gerhardt H, Haigh K, Tsien J, Damert A, Miquerol L, Muhlner U, Klein R, Ferrara N, Wagner EF, Betsholtz C, Nagy A. Cortical and retinal defects caused by dosage-dependent reductions in VEGF-A paracrine signaling. Dev Biol. 2003;262:225–241. doi: 10.1016/S0012-1606(03)00356-7. [DOI] [PubMed] [Google Scholar]
- James JM, Gewolb C, Bautch VL. Neurovascular development uses VEGF-A signaling to regulate blood vessel ingression into the neural tube. Development. 2009;136:833–841. doi: 10.1242/dev.028845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Lai L, Bohnsack BL, Niederreither K, Hirschi KK. Retinoic acid regulates endothelial cell proliferation during vasculogenesis. Development. 2003;130:6465–6474. doi: 10.1242/dev.00887. [DOI] [PubMed] [Google Scholar]
- Lange C, Turrero Garcia M, Decimo I, Bifari F, Eelen G, Quaegebeur A, Boon R, Zhao H, Boeckx B, Chang J, Wu C, Le Noble F, Lambrechts D, Dewerchin M, Kuo CJ, Huttner WB, Carmeliet P. Relief of hypoxia by angiogenesis promotes neural stem cell differentiation by targeting glycolysis. EMBO J. 2016 doi: 10.15252/embj.201592372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Lee S, Yang H, Song S, Kim K, Saunders TL, Yoon JK, Koh GY, Kim I. Notch pathway targets proangiogenic regulator sox17 to restrict angiogenesis. Circ Res. 2014;115:215–226. doi: 10.1161/CIRCRESAHA.115.303142. [DOI] [PubMed] [Google Scholar]
- Li H, Wagner E, McCaffery P, Smith D, Andreadis A, Dräger UC. A retinoic acid synthesizing enzyme in ventral retina and telencephalon of the embryonic mouse. Mech Dev. 2000;95:283–289. doi: 10.1016/S0925-4773(00)00352-X. [DOI] [PubMed] [Google Scholar]
- Li S, Haigh K, Haigh JJ, Vasudevan A. Endothelial VEGF sculpts cortical cytoarchitecture. J Neurosci. 2013;33:14809–14815. doi: 10.1523/JNEUROSCI.1368-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebner S, Corada M, Bangsow T, Babbage J, Taddei A, Czupalla CJ, Reis M, Felici A, Wolburg H, Fruttiger M, Taketo MM, von Melchner H, Plate KH, Gerhardt H, Dejana E. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J Cell Biol. 2008;183:409–417. doi: 10.1083/jcb.200806024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippmann ES, Al-Ahmad A, Azarin SM, Palecek SP, Shusta EV. A retinoic acid-enhanced, multicellular human blood-brain barrier model derived from stem cell sources. Sci Rep. 2014;4:4160. doi: 10.1038/srep04160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohnes D, Mark M, Mendelsohn C, Dollé P, Dierich A, Gorry P, Gansmuller A, Chambon P. Function of the retinoic acid receptors (RARs) during development (I). Craniofacial and skeletal abnormalities in RAR double mutants. Development. 1994;120:2723–2748. doi: 10.1242/dev.120.10.2723. [DOI] [PubMed] [Google Scholar]
- Maden M. Role and distribution of retinoic acid during CNS development. Int Rev Cytol. 2001;209:1–77. doi: 10.1016/S0074-7696(01)09010-6. [DOI] [PubMed] [Google Scholar]
- Mao B, Wu W, Li Y, Hoppe D, Stannek P, Glinka A, Niehrs C. LDL-receptor-related protein 6 is a receptor for Dickkopf proteins. Nature. 2001;411:321–325. doi: 10.1038/35077108. [DOI] [PubMed] [Google Scholar]
- Maretto S, Cordenonsi M, Dupont S, Braghetta P, Broccoli V, Hassan AB, Volpin D, Bressan GM, Piccolo S. Mapping Wnt/beta-catenin signaling during mouse development and in colorectal tumors. Proc Natl Acad Sci U S A. 2003;100:3299–3304. doi: 10.1073/pnas.0434590100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megason SG, McMahon AP. A mitogen gradient of dorsal midline Wnts organizes growth in the CNS. Development. 2002;129:2087–2098. doi: 10.1242/dev.129.9.2087. [DOI] [PubMed] [Google Scholar]
- Mizee MR, Wooldrik D, Lakeman KA, van het Hof B, Drexhage JA, Geerts D, Bugiani M, Aronica E, Mebius RE, Prat A, de Vries HE, Reijerkerk A. Retinoic acid induces blood-brain barrier development. J Neurosci. 2013;33:1660–1671. doi: 10.1523/JNEUROSCI.1338-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland DJ, Dedhar S, Coetzee GA, Nelson CC. Interaction of nuclear receptors with the Wnt/beta-catenin/Tcf signaling axis: Wnt you like to know? Endocr Rev. 2005;26:898–915. doi: 10.1210/er.2003-0034. [DOI] [PubMed] [Google Scholar]
- Nakao T, Ishizawa A, Ogawa R. Observations of vascularization in the spinal cord of mouse embryos, with special reference to development of boundary membranes and perivascular spaces. Anat Rec. 1988;221:663–677. doi: 10.1002/ar.1092210212. [DOI] [PubMed] [Google Scholar]
- Napoli JL. Retinoic acid: its biosynthesis and metabolism. Prog Nucleic Acid Res Mol Biol. 1999;63:139–188. doi: 10.1016/S0079-6603(08)60722-9. [DOI] [PubMed] [Google Scholar]
- Raab S, Beck H, Gaumann A, Yüce A, Gerber HP, Plate K, Hammes HP, Ferrara N, Breier G. Impaired brain angiogenesis and neuronal apoptosis induced by conditional homozygous inactivation of vascular endothelial growth factor. Thromb Haemost. 2004;91:595–605. doi: 10.1160/TH03-09-0582. [DOI] [PubMed] [Google Scholar]
- Rajaii F, Bitzer ZT, Xu Q, Sockanathan S. Expression of the dominant negative retinoid receptor, RAR403, alters telencephalic progenitor proliferation, survival, and cell fate specification. Dev Biol. 2008;316:371–382. doi: 10.1016/j.ydbio.2008.01.041. [DOI] [PubMed] [Google Scholar]
- Rosselot C, Spraggon L, Chia I, Batourina E, Riccio P, Lu B, Niederreither K, Dolle P, Duester G, Chambon P, Costantini F, Gilbert T, Molotkov A, Mendelsohn C. Non-cell-autonomous retinoid signaling is crucial for renal development. Development. 2010;137:283–292. doi: 10.1242/dev.040287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider RA, Hu D, Rubenstein JL, Maden M, Helms JA. Local retinoid signaling coordinates forebrain and facial morphogenesis by maintaining FGF8 and SHH. Development. 2001;128:2755–2767. doi: 10.1242/dev.128.14.2755. [DOI] [PubMed] [Google Scholar]
- Sen J, Harpavat S, Peters MA, Cepko CL. Retinoic acid regulates the expression of dorsoventral topographic guidance molecules in the chick retina. Development. 2005;132:5147–5159. doi: 10.1242/dev.02100. [DOI] [PubMed] [Google Scholar]
- Shah S, Hecht A, Pestell R, Byers SW. Trans-repression of beta-catenin activity by nuclear receptors. J Biol Chem. 2003;278:48137–48145. doi: 10.1074/jbc.M307154200. [DOI] [PubMed] [Google Scholar]
- Shih SC, Smith LE. Quantitative multi-gene transcriptional profiling using real-time PCR with a master template. Exp Mol Pathol. 2005;79:14–22. doi: 10.1016/j.yexmp.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Siegenthaler JA, Ashique AM, Zarbalis K, Patterson KP, Hecht JH, Kane MA, Folias AE, Choe Y, May SR, Kume T, Napoli JL, Peterson AS, Pleasure SJ. Retinoic acid from the meninges regulates cortical neuron generation. Cell. 2009;139:597–609. doi: 10.1016/j.cell.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegenthaler JA, Choe Y, Patterson KP, Hsieh I, Li D, Jaminet SC, Daneman R, Kume T, Huang EJ, Pleasure SJ. Foxc1 is required by pericytes during fetal brain angiogenesis. Biol Open. 2013;2:647–659. doi: 10.1242/bio.20135009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D, Wagner E, Koul O, McCaffery P, Dräger UC. Retinoic acid synthesis for the developing telencephalon. Cereb Cortex. 2001;11:894–905. doi: 10.1093/cercor/11.10.894. [DOI] [PubMed] [Google Scholar]
- Stenman JM, Rajagopal J, Carroll TJ, Ishibashi M, McMahon J, McMahon AP. Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science. 2008;322:1247–1250. doi: 10.1126/science.1164594. [DOI] [PubMed] [Google Scholar]
- Toresson H, Mata de Urquiza A, Fagerström C, Perlmann T, Campbell K. Retinoids are produced by glia in the lateral ganglionic eminence and regulate striatal neuron differentiation. Development. 1999;126:1317–1326. doi: 10.1242/dev.126.6.1317. [DOI] [PubMed] [Google Scholar]
- Tsai S, Bartelmez S, Heyman R, Damm K, Evans R, Collins SJ. A mutated retinoic acid receptor-alpha exhibiting dominant-negative activity alters the lineage development of a multipotent hematopoietic cell line. Genes Dev. 1992;6:2258–2269. doi: 10.1101/gad.6.12a.2258. [DOI] [PubMed] [Google Scholar]
- Yang D, Baumann JM, Sun YY, Tang M, Dunn RS, Akeson AL, Kernie SG, Kallapur S, Lindquist DM, Huang EJ, Potter SS, Liang HC, Kuan CY. Overexpression of vascular endothelial growth factor in the germinal matrix induces neurovascular proteases and intraventricular hemorrhage. Sci Transl Med. 2013;5:193ra190. doi: 10.1126/scitranslmed.3005794. [DOI] [PubMed] [Google Scholar]
- Ye X, Wang Y, Cahill H, Yu M, Badea TC, Smallwood PM, Peachey NS, Nathans J. Norrin, frizzled-4, and Lrp5 signaling in endothelial cells controls a genetic program for retinal vascularization. Cell. 2009;139:285–298. doi: 10.1016/j.cell.2009.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarbalis K, Siegenthaler JA, Choe Y, May SR, Peterson AS, Pleasure SJ. Cortical dysplasia and skull defects in mice with a Foxc1 allele reveal the role of meningeal differentiation in regulating cortical development. Proc Natl Acad Sci U S A. 2007;104:14002–14007. doi: 10.1073/pnas.0702618104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Smith D, Yamamoto M, Ma L, McCaffery P. The meninges is a source of retinoic acid for the late-developing hindbrain. J Neurosci. 2003;23:7610–7620. doi: 10.1523/JNEUROSCI.23-20-07610.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wang Y, Tischfield M, Williams J, Smallwood PM, Rattner A, Taketo MM, Nathans J. Canonical WNT signaling components in vascular development and barrier formation. J Clin Invest. 2014;124:3825–3846. doi: 10.1172/JCI76431. [DOI] [PMC free article] [PubMed] [Google Scholar]