

Abstract
Objective(s):
Investigation of acute effect on cellular bioenergetics provides the opportunity to characterize the possible adverse effects of drugs more comprehensively. This study aimed to investigate the changes in biochemical and biophysical properties of heart mitochondria induced by captopril and nifedipine antihypertensive treatment.
Materials and Methods:
Male, 12-week-old Wistar rats in two experimental models (in vivo and in vitro) were used. In four groups, the effects of escalating doses of captopril, nifedipine and combination of captopril + nifedipine added to the incubation medium (in vitro) or administered per os to rat (in vivo) on mitochondrial ATP synthase activity and membrane fluidity were monitored.
Results:
In the in vitro model we observed a significant inhibitory effect of treatment on the ATP synthase activity (P<0.05) with nonsignificant differences in membrane fluidity. Decrease in the value of maximum reaction rate Vmax (P<0.05) without any change in the value of Michaelis-Menten constant Km, indicative of a noncompetitive inhibition, was presented. At the in vivo level, we did not demonstrate any significant changes in the ATP synthase activity and the membrane fluidity in rats receiving captopril, nifedipine, and combined therapy.
Conclusion:
In vitro kinetics study revealed that antihypertensive drugs (captopril and nifedipine) directly interact with mitochondrial ATP synthase. In vivo experiment did not prove any acute effect on myocardial bioenergetics and suggest that drugs do not enter cardiomyocyte and have no direct effect on mitochondria.
Keywords: Captopril, Heart, Membrane fluidity, Mitochondria, Mitochondrial proton-translocating ATPases, Nifedipine
Introduction
Pharmacological treatment of hypertension is one of the most common therapies due to its high prevalence in developed countries (1–3). Several basic groups of antihypertensive agents including angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, calcium channel blockers, diuretics, and beta-blockers are standardly used to treat high blood pressure (4–6). Furthermore, the combination of antihypertensive agents has been recently found to be effective in the treatment of hypertension (7). Unfortunately, in addition to the favorable properties of antihypertensive therapy, its adverse effects have also been observed (8–10). However, in the field of hypertension, especially concerning the heart, there is not enough information about the possible side effects of antihypertensive drugs on cellular bioenergetics in the myocardium. Moreover, the available studies provide diverse results (11-13). As for captopril and nifedipine, the study of Ziegelhöffer et al (14) deals with the changes in mitochondrial functional properties induced by this antihypertensive treatment in spontaneously hypertensive rats. Authors demonstrated the presence of the adaptation process to hypertension associated with endogenous protective mechanism observed in the case of hypoxia or acute diabetes mellitus (15-17). However, the study does not reveal whether described alterations are consequences of the negative adverse effect caused by captopril treatment.
For this purpose, the aim of this study was to inves-tigate the impact of antihypertensive pharmacotherapy on biochemical and biophysical characteristics determining mitochondrial utility with regard to their ability to synthesize and supply sufficient quantity of ATP to cardiomyocytes of normotensive rats. Using an experimental model in vivo, we assessed the possible side effects of antihypertensive treatment with captopril, nifedipine, and their combination on the subcellular level in terms of their effect on production of cellular energy. Realization of this experiment in vitro has allowed us to follow the mechanism of action of the antihypertensive agent on the membrane and the membrane components of heart mitochondria possibly after the drug enters the cardiomyocyte. The functional state of mitochondrial membrane was evaluated by the parameters of ATP synthase activity, activity of mitochondrial proton-translocating ATPase, measured in the presence of Mg2+ ions as the activity of DNP-stimulated Mg2+-ATPase, and the mitochondrial membrane fluidity, determined by using a hydrophobic fluorescence probe (DPH) (1,6-diphenyl-1,3,5-hexatriene). The degree of motional restriction of this probe (i.e. to the degree of their fluorescence anisotropy) is directly proportional to the apparent membrane microviscosity.
Materials and Methods
Animals
Experiments were performed on 10–12 weeks old male Wistar rats (240 ± 20 g) housed in cages in groups (max. 5 animals per cage) under conditions of natural light control (12 hr light, 12 hr darkness) at 22°C. The animals were fed a standard pellet diet with access to water ad libitum. According to experimental design of the in vivo model, animals received the drugs per os by gavage 2 times per day for 3 days; the daily doses used were: captopril (80 mg/kg; Sigma-Aldrich Chemie GmbH, Germany), nifedipine (2.5, 5, and 10 mg/kg; Sigma-Aldrich Chemie GmbH, Germany), and combination of captopril and nifedipine (20 and 2.5; 40 and 5; 80 and 10 mg/kg) (18, 19). The maximal used doses of captopril and nifedipine were selected on the basis of 2 studies, with the shorter duration of use to find the acute effects of drugs (20, 21). After each experiment, the animals were anesthetized with the solution of thiopental and heparin administered intraperitoneally at a dose of 50–60 mg/kg (500 IU i.p.; Léčiva, Czech Republic), and animals were subsequently killed. Hearts were excised from animals and immersed in an ice cold solution containing 180 mmol/l KCl, and 4 mmol/l ethyl-enediaminetetraacetic acid, pH 7.4 (16).
All animal experiments were performed in accor-dance with the rules issued by the State Veterinary Administration of the Slovak Republic, legislation No 289/2003 and with the regulations of the Animal Research and Care Committee of Institute for Heart Research.
Animal monitoring
Systolic blood pressure was measured by tail-cuff plethysmography using the Statham Pressure Transducer P23XL (Hugo Sachs, Germany) at the third day of drug administration.
Experimental design
In the in vivo experimental model animals were divided into four groups (n=7 for each group): control group not subject to drug administration, group administered with a single dose of captopril, group administered with nifedipine at three different doses, and group administered with the combination of captopril and nifedipine at three different doses. Animals included in experimental model in vitro, where the drugs were added into the incubation medium after isolation of mitochondrial membranes, were divided into the following groups (n=7 for each group): control group, group with captopril in five different concentrations, group with nifedipine in five different concentrations, and the group with the combined substances.
Isolation of mitochondrial membrane
Expired hearts damped with small volume of isolation solution were cut into small pieces and suffused with 20 ml of ice-cold isolation solution (180 mmol/l KCL, 4 mmol/l ethylenediamine-tetraacetic acid, and 1% bovine serum albumin, pH 7.4) with the added protease Sigma P 6141 (2.5 mg/g of tissue). Prepared samples were homogenized using a Teflon-glass homogenizer placed on ice and subsequently processed by differential centri-fugation. After 10 min centrifu-gation at 1000 g, the sediment with isolation solution was homogenized and centrifuged at 1000 g for 10 min. The supernatant was centrifuged again at 5000 g for 15 min. Sediment was resuspended in 20 ml of isolation solution without albumin and centrifuged again (5000 g, 15 min). The resultant sediment was the final mitochondrial fraction from which the amount of protein was determined using the Lowry colorimetric method (22). Samples were diluted which resulted in mitochondrial protein concen-tration of 1 μg/μl. The whole process of heart mitochondria isolation was performed at 4 °C.
Measurements of mitochondrial ATP synthase enzyme activity
ATP synthase enzyme activity was determined as 2,4-dinitrophenol (DNP)-stimulated activity of Mg2+-ATPase corresponding to the hydrolytic activity of ATP synthase in the presence of Mg2+ ions. The effect of antihypertensives on the enzyme activity was assessed in the same way in both, the in vivo model when drugs were administered by gavage per os, and the in vitro model as kinetic measurements, when the drugs were added directly into the test-tubes. Procedure was as follows: 650 μl of incubation medium consisting of 100 μl MgCl2 (40 mmol/l), 200 μl imidazole buffer (250 mmol/l, pH 7.4) in the presence and absence of 100 μl dinitrophenol (0.1 mmol/l) replenished with 250 μl or 350 μl distilled water was added to 50 μl of mitochondrial fraction. DNP-stimulated Mg2+-ATPase activity was obtained by adding dinitrophenol, which made all mitochondrial membranes leaky and thus permeable to Mg2+ ions. In the in vitro experimental model, we afterward added antihypertensive drugs - captopril and nifedipine (200 μl) in various concentrations in the ranges of 0.45 to 0.01 and 0.015 to 0.003 mmol/l. In the case of all in vitro experiments, nifedipine was dissolved in a 10% solution of dimethyl sulfoxide (DMSO) due to its poor solubility. The samples in the incubation medium were pre-incubated for 10 min at 37 °C and then the reaction was initiated by addition of 40 mmol/l ATP. After 20 min of incubation, the reaction was stopped by adding 1 ml of ice-cold 12% trichloroacetic acid. DNP-stimulated Mg2+-ATPase activity was determined spectrophotometrically at 700 nm as the amount of inorganic phosphate Pi liberated by ATP splitting in the presence of mitochondrial fraction and Mg2+ per unit of time (μmol. Pi/g/hr) (23).
Measurements of enzyme kinetics
The method of enzyme kinetics, consisting of measuring the dependence of ATP synthase activity (determined as DNP-stimulated Mg2+-ATPase activity) on the increasing concentration of ATP allows to determine the basic kinetic parameters Vmax-maximal reaction rate and Km-Michaelis-Menten constant and thus to determine one of the basic types of enzyme inhibition: competitive, noncompetitive, uncompetitive, and allosteric. The measurement procedure was identical to the above-mentioned determination of the mitochondrial ATP synthase activity with the difference in initiating the reaction by ATP with concentration range (0 - 40) mmol/l in the presence of inhibitors - drugs in the following concentrations: 0.3 mmol/l captopril, 0.01 mmol/l nifedipine, combination of 0.3 mmol/l captopril, and 0.01 mmol/l nifedipine. For measurement, the modified method of the study by Horvat et al and Vrbjar et al was used (24, 25).
Measurements of mitochondrial membrane fluidity
Membrane fluidity characterizing the physical state of the lipid bilayer of mitochondrial membrane was determined by fluorescence spectroscopy method as the anisotropy of the fluorescence probe 1,6-diphenyl-1,3,5-hexatriene, which is sensitive to the movement of acyl chains of the membrane phospholipids. A high degree of fluorescence anisotropy of DPH indicates a high degree of structural order of the phospholipids, which means lower membrane fluidity. Isolated mitochondria were incubated for 20 min in the incubation medium with the final volume of 2 ml containing 180 mmol/l of KCl, 4 mmol/l of ethylenediaminetetraacetic acid, and 200 μl of drug (captopril/nifedipine) in the concentration range from 0.45 to 0.01 mmol/l for captopril, from 0.015 to 0.003 mmol/l for nifedipine, or from 0.45 and 0.015 mmol/l to 0.015 and 0.03 mmol/l for combination of captopril and nifedipine in in vitro measurements. Subsequently, the sample with the volume of 1750 μl was suspended to the standardized concentration and labelled with 250 μl of DPH. The time dependence of fluorescence anisotropy of DPH was measured during 1800 sec (26).
Estimation of mitochondrial purity
Purity of mitochondrial preparation was tested by estimation of the markers - ATPase activities, a characteristic for sarcolemma (the Na+/K+-ATPase) and another for sarcoplasmic reticulum ATPase (the Mg2+/Ca2+-ATPase) in the absence and presence of their specific inhibitors using the technic described in detail by Ferko et al (16) and Malekova et al (27).
Statistical analysis
Data were expressed as means±standard deviations for normally distributed variables, otherwise median and quartile ranges from the lower quartile Q1 to the upper quartile Q3 were used. Differences between the groups were compared and tested at significance level α = 0.05 by non-parametric statistical tests: Mann-Whitney test for comparison of two groups and Kruskal-Wallis test to compare more than two groups. In the case of the significance of the global test, a post hoc Conover-Inman test for pairwise comparisons (pairs) was conducted.
Results
Estimation of mitochondrial purity
The degree of contamination of mitochondrial preparation in the membranes of the sarcolemma and sarcoplasmic reticulum was estimated by the presence of their marker enzymes and amounted to 0.84% and 1.59%, respectively (data not shown).
In vivo effect of antihypertensive drugs on blood pressure
No significant change of systolic blood pressure was observed on the third day of administration of captopril, nifedipine, and their combination in normotensive rats (data not shown).
Effect of dimethyl sulfoxide on functional and biophysical properties of mitochondrial membrane in vitro
The possible influence of DMSO on the fluidity and ATP synthase activity of mitochondrial membrane was experimentally excluded (Table 1).
Table 1.
Effect of 10% solution of dimethyl sulfoxide (DMSO) on DNP-stimulated Mg2+-ATPase activity and fluorescence anisotropy of DPH in vitro. The results are reported as the arithmetic mean±standard deviation, n = 5
| Concentration of DMSO [%] | 10% | Control |
|---|---|---|
| ATP synthase activity [μmol.Pi-1.hr-1] | 35.79 ± 1.17 | 34.42 ± 0.93 |
| Membrane fluidity [arb.unit] | 0.196 ± 0.019 | 0.194 ± 0.018 |
In vitro modulation of mitochondrial membranes
In vitro effect of antihypertensive drugs captopril, nifedipine, and their combination on the mitochondrial enzyme ATP synthase activity and the effect of the drugs depending on the concentration are presented in Figure 1. Results showed that both drugs, as well as their combination, had an inhibitory effect on the hydrolytic activity of ATP synthase. In the case of modulation by ACE inhibitor, captopril, we observed the largest decrease in the mitochondrial ATP synthase activity in the highest concentration and its activity was decreased almost to zero (0.912 μmol. Pi/g/hr). This decrease by 97.41% was statistically significant (P = 0.002) (Figure 1A). In the case of modulation by the blocker of calcium channels, nifedipine, the level of inhibition in its highest concentration (0.015 mmol/l) was manifested by a decrease in the activity by 33% in comparison with the control group (P = 0.003) (Figure 1B). The combination of captopril and nifedipine in the concentrations of 0.45 and 0.015 mmol/l respectively, caused the maximal level of inhibition with a decrease by 98.11% (P <0.0001) (Figure 1C).
Figure 1.
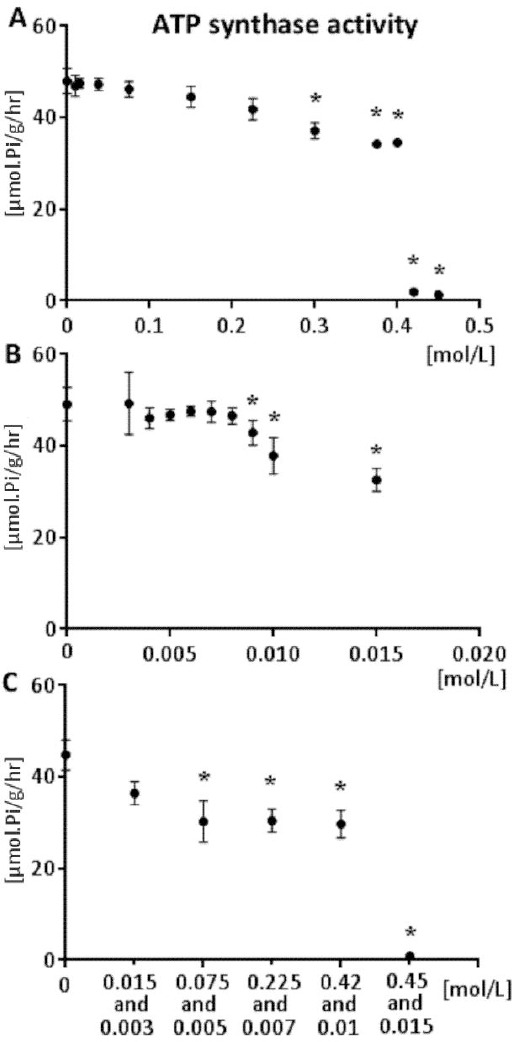
DNP-stimulated Mg2+-ATPase activity (ATP synthase activity) dependence on the concentration of captopril (A) nifedipine (B) and combination of captopril and nifedipine (C) in vitro. 0 represents the control group. The results are reported as the arithmetic mean ± standard deviation, n = 7, *- P<0.05 vs. control group
The measurement of enzyme activation with increasing concentration of ATP showed a statistically significant decrease in Vmax value (P = 0.02) in all groups affected by the presence of drugs (captopril 0.3 mmol/l, nifedipine 0.01 mmol/l, combination of captopril 0.42 mmol/l, and nifedipine 0.01 mmol/l). Simultaneously in the presence of drugs as inhibitors, we did not find any statistically significant change in the value of Michaelis constant Km in comparison with the control group (P = 0.88). Results from the fluorescence anisotropy measurements did not exhibit statistically significant differences in any of the experimental groups when compared to controls (P = 0.55; P = 0.75; P = 0.8) (Figure 2). We did not find a linear relationship between mitochondrial ATP synthase activity and membrane fluidity (correlation coefficient Kendall tau b = 0.2, P = 0.81).
Figure 2.
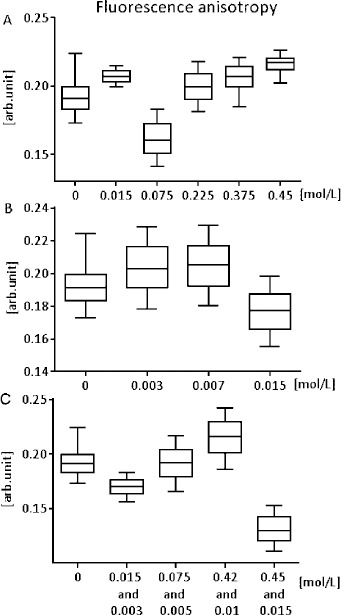
Fluorescence anisotropy of DPH (reciprocal of the membrane fluidity) dependence on the concentration of captopril (A), nifedipine (B), and combination of captopril and nifedipine (C) in vitro. Reported concentrations correspond to the concentrations added to the incubation medium. 0 represents the control group. The minimum, lower quartile, median, upper quartile, and maximum of the measured value are presented; n = 7
In vivo modulation of mitochondrial membrane
In in vivo conditions, ACE inhibitor had been administered for three days in a daily dosage of 80 mg/kg, which caused a statistically non-significant decrease in ATP synthase activity by 1.67% (P = 0.67) (Figure 3A). On the contrary, nifedipine with 5 mg/kg daily-dosage manifested non-significant activating effect with maximal average increase of ATP synthase activity by 21.31% (P = 0.21) (Figure 3B). Increasing doses of drugs administered in combination caused a gradual increase in mitochondrial enzyme activity, while in the group with 80 mg/kg and 10 mg/kg daily dosage of captopril and nifedipine, the level of activation amounted to 14% compared to the control group. Although, this ascending trend was not significant (P = 0.16) (Figure 3C).
Figure 3.
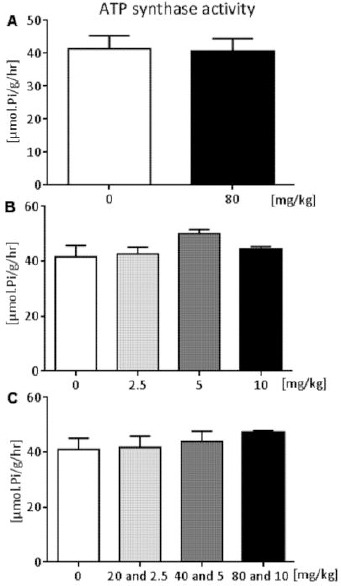
Dose-dependent effect of captopril (A), nifedipine (B), and combination of both (C) on DNP-stimulated Mg2+-ATPase activity (ATP synthase activity) of measurement in vivo. 0 represents the control group. The results are reported as the arithmetic mean ± standard deviation; n = 7
We found antihypertensive therapy not significantly effective on lipid acyl chain arrangement within mitochondrial membranes. In all groups, the therapy had minimal fluidizing effect. In the group administered with captopril at a daily dosage of 80 mg/kg, we recorded a decline in the fluorescence anisotropy of DPH (reciprocal of the membrane fluidity) by 2.5% compared to the controls (P = 0.8) (Figure 4A). In the group treated with nifedipine at a daily dosage of 5 and 2.5 mg/kg, we observed a decrease in fluorescence anisotropy of DPH by 3.25% (P = 0.78) (Figure 4B). Finally, in the group treated with the combination of captopril and nifedipine, the reduction in the fluorescence anisotropy was about 3% (P = 0.8) (Figure 4C).
Figure 4.
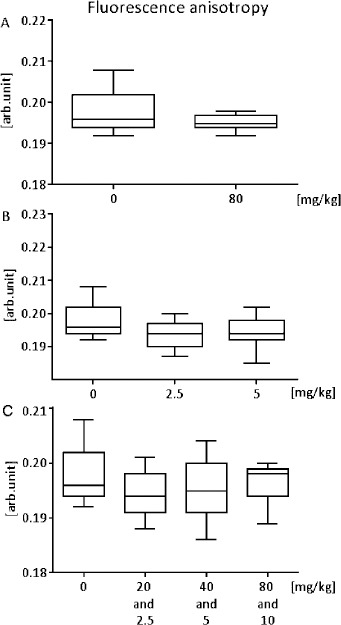
Dose-dependent effect of captopril (A), nifedipine (B), and combination of both (C) on the fluorescence anisotropy of DPH (reciprocal of the membrane fluidity); measurement in vivo. 0 represents the control group. The minimum, lower quartile, median, upper quartile, and maximum of the measured value are presented; n = 7
Discussion
Due to its high prevalence, hypertension remains the biggest risk factor for cardiovascular diseases, despite extensive knowledge about its pathogenesis and important progress in the field of pharmacological therapy (11,28–30), as well as increased knowledge about the alterations in the subcellular level concerning the heart in hypertension (1). However, alterations induced by the treatment of hypertension in cellular bioenergetics in the myocardium, which has recently been investigated, have not been fully explored yet. Animal studies about the mechanism of pharmacological action in terms of cellular bioenergetics provide an opportunity to characterize possible adverse effects of drugs more comprehensively.
Captopril, an angiotensin-converting enzyme inhibitor, and nifedipine, a dihydropyridine calcium channel blocker are widely used in the experiments dealing with the treatment of cardiovascular diseases like hypertension. Several studies have documented favorable and cardioprotective effects of captopril and nifedipine treatment (31–35). In contrast, other studies did not report any protective effect of captopril on mitochondrial function in spontaneously hypertensive rat hearts (14, 36, 37).
Mitochondria with their ability to control ATP production play a central role in the energetic metabolism of all tissues including myocardium, especially. However, the optimal state of mitochondria for its functional processes is indispensable (38). Notably, mitochondrial transport system and receptors necessitate for their proper function, certain mobility of molecules of the lipid bilayer membrane. Changes in the membrane properties incurred due to possible interaction of the membrane with the drugs may negatively affect mitochondrial function (39–41), for example by incorporation of the drug to the membrane. This can be manifested by the change in the structural ordering of membrane phospholipids, which can be reflected in the change of fluorescence of DPH – a marker of membrane fluidity. The optimal state of fluidity is crucial for mitochondria: it is not appropriate for the membrane to be too rigid, or too fluid (42, 43). High degree of membrane fluidity can adversely influence the permeability of the membrane, reduce the ability of the affected mitochondria to generate enough ATP (44), and can eventually end in apoptosis (45). On the contrary, the positive remodeling of mitochondrial membrane was demonstrated to be manifested in metabolic diseases such as acute diabetes mellitus (16, 46, 47). Compensatory conditions generated by the remodeling process of the mitochondrial membrane enabled myocardium to cope with the increased energy demands in the pathological state.
There is some evidence that hypertension similarly to acute diabetes mellitus (16, 17, 47) induced many adaptative changes in the properties and function of the heart mitochondria (36). In addition, the experimental studies of Ziegelhöffer et al and Mujkošová et al investigated the effect of antihypertensive treatment by captopril and its combination with nifedipine on the hypertension-induced alterations (14, 36). To exclude the changes linked with adaptation to hypertension, the experimental model of normotensive Wistar rats was used. The in vitro study of the mitochondrial membrane allowed us to assess whether the drugs acted by direct interaction with the enzyme ATP synthase or indirectly through the interaction and subsequent modulation of the mitochondrial membrane. In vivo experiments were established to assess the acute side effects of antihypertensive treatment on the functional properties of heart mitochondria.
The activity of ATP synthase determined as DNP-stimulated Mg2+-ATPase activity was modulated by the increased concentration of ACE inhibitor, captopril, in in vitro condition. Captopril as a modulator had an inhibitory effect. The observed statistically non-significant differences in the increased fluorescence anisotropy DPH in the presence of captopril suggest unimpaired physical condition of the mitochondrial membrane by the action of the drug. In the case of the blocker of calcium channels, nifedipine, the same inhibitory effect of ATP synthase activity was observed, as it was observed in the modulation by the ACE inhibitor, captopril. Due to the poor solubility of nifedipine, it was necessary to dissolve the drug in a solution of DMSO. Applied 10% concentration of DMSO has proven to be the lowest possible concentration with respect to precipitation with the lower concentrations of DMSO in the incubation medium. This concentration was still acceptable because DMSO used for dissolution of nifedipine did not interfere with the determination of ATP synthase activity and did not affect the fluidity of mitochondrial membrane (Table 1). The combination of both drugs inhibited the hydrolytic ATP synthase activity, with the decline in the activity for higher concentrations of 0.45 mmol/l (captopril) and 0.015 mmol/l (nifedipine) representing their toxic effect. Observation of any significant fluidizing effect of monotherapy and combination therapy suggested that drugs did not interact with membrane components and did not incorporate into the membrane as described previously in the study of Jaremko et al (48) and Pereira et al (49).
The non-significant differences in the membrane fluidity between the control and the group with the addition of antihypertensive drugs indicate that drugs directly interacted with the ATP synthase enzyme and not through interaction with the mitochondrial membrane. The kinetics measurement consisting of the activation of ATP synthase activity with the increasing concentrations of ATP revealed a decrease in Vmax values in all groups with the presence of inhibitor, compared to the control group. Unchanged Km values in the groups with inhibitors proved that the inhibitor did not cause any alterations in the affinity of the ATP-binding site of the enzyme to a substrate. The observed changes in kinetics parameters determined a noncompetitive type of drug inhibition. This type of inhibition leads to a direct interaction between the drug and enzyme at a place other than the active site. The observation of any significant change in membrane fluidity confirmed the fact that antihypertensive drugs do not incorporate into mitochondrial membrane and do not alter the biophysical properties of the mitochondrial membrane. On this basis, we can infer that antihypertensive drugs bind to one of the subunits of F1 part of the ATP synthase. This might change the conformation of the enzyme, thus, irreversibly inactivate the active site.
Assuming the immediate effect of the antihypertensive agents we designed the in vivo model with the aim to approach clinical conditions. Our special emphasis was given to the short drug action effect, because in the long term action of drugs, the alterations associated with the systemic response of organism possibly masking direct drug effect, are also involved. Moreover, captopril differs from other inhibitors of angiotensin-converting enzyme by its short elimination half-life (18). The equally short elimination half-life of nifedipine was proven (19). With respect to the short acting of both antihypertensive drugs and to study mainly acute drug effects, we have set the drug administration for in vivo testing to two times per day for three days. Results showed that captopril caused only a minimal, nonsignificant increase in mitochondrial membrane fluidity (decrease in fluorescence anisotropy) and a decrease in mitochondrial ATP synthase enzyme activity. Nevertheless, nifedipine caused a non-significant increase in mitochondrial membrane fluidity (decrease in fluorescence anisotropy) but an expressive increase of ATP synthase activity in the dosage of 5 mg/kg. We did not observe such an expressive increase in the enzyme activity in a higher concentration of antihypertensive agents. In the case of combined drug administration, nifedipine reduced the inhibitory effect of captopril observed in monotherapy. This resulted in an increase of the ATP synthase activity. We observed a decreasing trend in the average values of anisotropy, i.e. an increase in fluidity.
Conclusion
In summary, we performed both in vitro and in vivo experiments to assess the possible adverse effects of antihypertensive drugs captopril and nifedipine from the view of myocardial bioener-getics. Results of ATP synthase activity and mitochondrial membrane fluidity from in vivo measurements suggested that antihypertensive therapy represented by captopril and nifedipine have no harmful adverse effect on the functional properties of the heart mitochondrial membrane. Although, the in vitro kinetics study, allowing to get more detailed information about the mechanisms of action and not including all regulatory and metabolic processes as in vivo study, showed direct drug interaction with the mitochondrial ATP synthase enzyme is responsible for its noncompetitive inhibition. Based on the different results from in vivo and in vitro experiments, we propose that the drugs in vivo did not permeate through the cardiomyocyte membrane and had no direct effect on mitochondrial bioenergetics.
Conflict of Interest
Authors declare that there is no conflict of interest associated with this study.
Acknowledgment
The results in this paper were derived from a student thesis. Authors feel indebted to Csilla Viczenczova and Tamara Beňová from Department of Histochemistry and Electron microscopy, Institute for Heart Research, Slovak Academy of Sciences, Bratislava, Slovakia, for their valuable help in performing the measurements of blood pressure. The excellent help of Mrs Zlatica Hradecká and Mrs Miroslava Zádorová from Institute for Heart Research, Centre of Excellence SAS NOREG, Slovak Academy of Sciences, Bratislava, Slovakia, is gratefully acknowledged. Supported by Grants: APVV-01-02-11, and VEGA SR: 2/0133/15 and 2/0201/15.
References
- 1.Kuneš J, Kadlecová M, Vaněčková I, Zicha J. Critical developmental periods in the pathogenesis of hypertension. Physiol Res. 2012;61(Suppl 1):S9–17. doi: 10.33549/physiolres.932364. [DOI] [PubMed] [Google Scholar]
- 2.Paulis L, Steckelings UM, Unger T. Key advances in antihypertensive treatment. Nat Rev Cardiol. 2012;9:276–285. doi: 10.1038/nrcardio.2012.33. [DOI] [PubMed] [Google Scholar]
- 3.Pintérová M, Kuneš J, Zicha J. Altered neural and vascular mechanisms in hypertension. Physiol Res. 2011;60:381–402. doi: 10.33549/physiolres.932189. [DOI] [PubMed] [Google Scholar]
- 4.Elliott WJ, Ram CV. Calcium channel blockers. J Clin Hypertens (Greenwich) 2011;13(9):687–9. doi: 10.1111/j.1751-7176.2011.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Law M, Wald N, Morris J. Lowering blood pressure to prevent myocardial infarction and stroke: a new preventive strategy. Health Technol Assess. 2003;7:1–94. doi: 10.3310/hta7310. [DOI] [PubMed] [Google Scholar]
- 6.Wang JG, Pimenta E, Chwallek F. Comparative review of the blood pressure-lowering and cardiovascular benefits of telmisartan and perindopril. Vasc Health Risk Manag. 2014;10:189–200. doi: 10.2147/VHRM.S59429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taddei S. Combination therapy in hypertension: what are the best options according to clinical pharmacology principles and controlled clinical trial evidence? Am J Cardiovasc Drugs. 2015;15:185–194. doi: 10.1007/s40256-015-0116-5. [DOI] [PubMed] [Google Scholar]
- 8.Conlin PR, Williams GH. Use of calcium channel blockers in hypertension. Adv Intern Med. 1998;43:533–62. [PubMed] [Google Scholar]
- 9.White P. Calcium channel blockers. AACN Clin Issues Crit Care Nurs. 1992;3:437–446. doi: 10.4037/15597768-1992-2015. [DOI] [PubMed] [Google Scholar]
- 10.van Geijn HP, Lenglet JE, Bolte AC. Nifedipine trials: effectiveness and safety aspects. BJOG. 2005;112(Suppl 1):79–83. doi: 10.1111/j.1471-0528.2005.00591.x. [DOI] [PubMed] [Google Scholar]
- 11.Das AM, Harris DA. Mitochondrial ATP synthase regulation in heart: defects in hypertension are restored after treatment with captopril. Cardioscience. 1992;3:227–232. [PubMed] [Google Scholar]
- 12.Postnov YV, Orlov SN, Budnikov YY, Doroschuk AD, Postnov AY. Mitochondrial energy conversion distubance with decrease in ATP production as source of systemic arterial hypertension. Pathophysiology. 2007;14:195–204. doi: 10.1016/j.pathophys.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 13.Roman MJ, Saba PS, Pini R, Spitzer M, Pickering TG, Rosen S, Alderman MH, Devereux RB. Parallel cardiac and vascular adaptation in hypertension. Circulation. 1992;86:1909–1918. doi: 10.1161/01.cir.86.6.1909. [DOI] [PubMed] [Google Scholar]
- 14.Ziegelhöffer A, Mujkošová J, Ferko M, Vrbjar N, Ravingerová T, Uličná O, Waczulíková I, Ziegelhöffer B. Dual influence of spontaneous hypertension on membrane properties and ATP production in heart and kidney mitochondria in rat: effect of captopril and nifedipine, adaptation and dysadaptation. Can J Physiol Pharmacol. 2012;90:1311–1323. doi: 10.1139/y2012-107. [DOI] [PubMed] [Google Scholar]
- 15.Ziegelhöffer A, Ravingerová T, Waczulíková I, Cársky J, Neckár J, Ziegelhöffer-Mihalovicová B, Styk J. Energy transfer in acute diabetic rat hearts: adaptation to increased energy demands due to augmented calcium transients. Ann N Y Acad Sci. 2002;967:463–468. [PubMed] [Google Scholar]
- 16.Ferko M, Gvozdjaková A, Kucharská J, Mujkosová J, Waczulíková I, Styk J, Ravingerová T, Ziegelhöffer-Mihalovicová B, Ziegelhöffer A. Functional remodeling of heart mitochondria in acute diabetes: interrelationships between damage, endogenous protection and adaptation. Gen Physiol Biophys. 2006;25:397–413. [PubMed] [Google Scholar]
- 17.Ravingerová T, Adameová A, Matejíková J, Kelly T, Nemčeková M, Kucharská J, Pecháňová O, Lazou A. Subcellular mechanisms of adaptation in the diabetic myocardium: Relevance to ischemic preconditioning in the nondiseased heart. Exp Clin Cardiol Winter. 2010;15:68–76. [PMC free article] [PubMed] [Google Scholar]
- 18.Duchin KL, McKinstry DN, Cohen AI, Migdalof BH. Pharmacokinetics of captopril in healthy subjects and in patients with cardiovascular diseases. Clin Pharmacokinet. 1988;14:241–59. doi: 10.2165/00003088-198814040-00002. [DOI] [PubMed] [Google Scholar]
- 19.Snider ME, Nuzum DS, Veverka A. Long-acting nifedipine in the management of the hypertensive patient. Vascular Health and Risk Management. 2008;4:1249–1257. doi: 10.2147/vhrm.s3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kubo T, Fujie K, Yamashita M, Misu Y. Antihypertensive effects of nifedipine on conscious normotensive and hypertensive rats. J Pharmacobiodyn. 1981;4:294–300. doi: 10.1248/bpb1978.4.294. [DOI] [PubMed] [Google Scholar]
- 21.Miguel-Carrasco JL, Zambrano S, Blanca AJ, Mate A, Vázquez CM. Captopril reduces cardiac inflammatory markers in spontaneously hypertensive rats by inactivation of NF-kB. Journal of Inflammation (London, England) 2010;7:21. doi: 10.1186/1476-9255-7-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 23.Taussky HH, Shorr E. A microcolorimetric method for the determination of inorganic phosphorus. J Biol Chem. 1953;202:675–685. [PubMed] [Google Scholar]
- 24.Horvat A, Momić T, Petrović S, Nikezić G, Demajo M. Selective inhibition of brain Na,K-ATPase by drugs. Physiol Res. 2006;55:325–338. doi: 10.33549/physiolres.930696. [DOI] [PubMed] [Google Scholar]
- 25.Vrbjar N, Dzurba A, Ziegelhöffer A. Enzyme kinetics and the activation energy of (Na,K)-ATPase in ischaemic hearts: influence of the duration of ischaemia. Gen hysiol Biophys. 1994;13:405–411. [PubMed] [Google Scholar]
- 26.Shinitzky M. Membrane fluidity in malignancy. Adversative and recuperative. Biochim Biophys Acta. 1984;738:251–261. doi: 10.1016/0304-419x(83)90007-0. [DOI] [PubMed] [Google Scholar]
- 27.Malekova L, Kominkova V, Ferko M, Stefanik P, Krizanova O, Ziegelhöffer A, Szewczyk A, Ondrias K. Bongkrekic acid and atractyloside inhibits chloride channels from mitochondrial membranes of rat heart. Biochim Biophys Acta. 2007;1767:31–44. doi: 10.1016/j.bbabio.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 28.Millgård J, Hägg A, Sarabi M, Lind L. Captopril, but not nifedipine, improves endothelium-dependent vasodilation in hypertensive patients. J Hum Hypertens. 1998;12:511–516. doi: 10.1038/sj.jhh.1000665. [DOI] [PubMed] [Google Scholar]
- 29.Weir MR. Targeting mechanisms of hypertensive vascular disease with dual calcium channel and renin-angiotensin system blockade. J Hum Hypertens. 2007;21:770–779. doi: 10.1038/sj.jhh.1002254. [DOI] [PubMed] [Google Scholar]
- 30.Meyers RS, Siu A. 2011. Pharmacotherapy review of chronic pediatric hypertension. Clin Ther. 2011;33:1331–1356. doi: 10.1016/j.clinthera.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 31.Ago T, Yang Y, Zhai P, Sadoshima J. Nifedipine inhibits cardiac hypertrophy and left ventricular dysfunction in response to pressure overload. J Cardiovasc Transl Res. 2010;3:304–313. doi: 10.1007/s12265-010-9182-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen JB, Rao BF, Chang J, Liao XG, Cao YD. Effects of captopril on myocardial energy metabolism in mice with viral myocarditis] Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 2003;15:485–488. [PubMed] [Google Scholar]
- 33.Vavrínková H, Tutterová M, Stopka P, Divisová J, Kazdová L, Drahota Z. The effect of captopril on nitric oxide formation and on generation of radical forms of mitochondrial respiratory chain compounds in ischemic rat heart. Physiol Res. 2001;50:481–489. [PubMed] [Google Scholar]
- 34.Yanagishita T, Tomita M, Itoh S, Mukae S, Arata H, Ishioka H, Geshi E, Konno N, Katagiri T. Protective effect of captopril on ischemic myocardium. Jpn Circ J. 1997;61:161–169. doi: 10.1253/jcj.61.161. [DOI] [PubMed] [Google Scholar]
- 35.Mrak RE, Carry MM, Murphy ML, Peng CF, Straub KD. Reperfusion injury in ischemic myocardium: effects of nifedipine and verapamil. Am J Cardiovasc Pathol. 1990;3:61–68. [PubMed] [Google Scholar]
- 36.Mujkosová J, Ulicná O, Waczulíková I, Vlkovicová J, Vancová O, Ferko M, Polák S, Ziegelhöffer A. Mitochondrial function in heart and kidney of spontaneously hypertensive rats: influence of captopril treatment. Gen Physiol Biophys. 2010;29:203–207. [PubMed] [Google Scholar]
- 37.Rossi MA, Ramos SG, Prado CM. Chronic inhibition of nitric oxide synthase induces hypertension and cardiomyocyte mitochondrial and myocardial collagen remodelling in the absence of hypertrophy. J Hypertens. 2003;21:993–1001. doi: 10.1097/00004872-200305000-00025. [DOI] [PubMed] [Google Scholar]
- 38.Shi C, Wu F, Xu J. Incorporation of β-sitosterol into mitochondrial membrane enhances mitochondrial function by promoting inner mitochondrial membrane fluidity. J Bioenerg Biomembr. 2013;45:301–305. doi: 10.1007/s10863-012-9495-3. [DOI] [PubMed] [Google Scholar]
- 39.Schneider JM, Younes A. Binding of bepridil to isolated rat heart mitochondria. Basic Res Cardiol. 1989;84:623–630. doi: 10.1007/BF01906947. [DOI] [PubMed] [Google Scholar]
- 40.Almotrefi AA, Dzimiri N. Effects of beta-adrenoceptor blockers on mitochondrial ATPase activity in guinea pig heart preparations. Eur J Pharmacol. 1992;215:231–236. doi: 10.1016/0014-2999(92)90032-y. [DOI] [PubMed] [Google Scholar]
- 41.Bernardo TC, Cunha-Oliveira T, Serafim TL, Holy J, Krasutsky D, Kolomitsyna O, Krasutsky P, Moreno AM, Oliveira PJ. Dimethylaminopyridine derivatives of lupane triterpenoids cause mitochondrial disruption and induce the permeabilitym transition. Bioorg Med Chem. 2013;21:7239–7249. doi: 10.1016/j.bmc.2013.09.066. [DOI] [PubMed] [Google Scholar]
- 42.Chvanov M. Metabolic control of elastic properties of the inner mitochondrial membrane. J Phys Chem B. 2006;110:22903–22909. doi: 10.1021/jp0638181. [DOI] [PubMed] [Google Scholar]
- 43.Unsay JD, Cosentino K, Subburaj Y, García-Sáez AJ. Cardiolipin effects on membrane structure and dynamics. Langmuir. 2013;29:15878–15887. doi: 10.1021/la402669z. [DOI] [PubMed] [Google Scholar]
- 44.Eckmann J, Eckert SH, Leuner K, Muller WE, Eckert GP. Mitochondria: mitochondrial membranes in brain ageing and neurodegeneration. Int J Biochem Cell Biol. 2013;45:76–80. doi: 10.1016/j.biocel.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 45.Richter C, Schweizer M, Cossarizza A, Franceschi C. Control of apoptosis by the cellular ATP level. FEBS Lett. 1996;378:107–110. doi: 10.1016/0014-5793(95)01431-4. [DOI] [PubMed] [Google Scholar]
- 46.Ziegelhöffer A, Waczulíková I, Ravingerová T, Ziegelhöffer-Mihalovičová B, Neckář J, Styk J. Augmented Energy Transfer in Rat Heart Mitochondria: Compensatory Response to Abnormal Household of Energy in Acute Diabetes, Atherosclerosis, Hypertension and Diabetes, Progress in Experimental Cardiology. 2003;8:439–453. [Google Scholar]
- 47.Waczulikova I, Habodaszova D, Cagalinec M, Ferko M, Ulicna O, Mateasik A, Sikurova L, Ziegelhöffer A. Mitochondrial membrane fluidity, potential, and calcium transients in the myocardium from acute diabetic rats. Can J Physiol Pharmacol. 2007;85:372–381. doi: 10.1139/y07-035. [DOI] [PubMed] [Google Scholar]
- 48.Jaremko L, Jaremko M, Giller K, Becker S, Zweckstetter M. Structure of the mitochondrial translocator protein in complex with a diagnostic ligand. Science. 2014;343:1363–1366. doi: 10.1126/science.1248725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pereira LC, Miranda LF, de Souza AO, Dorta DJ. BDE-154 induces mitochondrial permeability transition and impairs mitochondrial bioenergetics. J Toxicol Environ Health A. 2014;77:24–36. doi: 10.1080/15287394.2014.861337. [DOI] [PubMed] [Google Scholar]