

Abstract
Objective(s):
Abnormal lung cell death including autophagy and apoptosis is the central feature in acute lung injury (ALI). To identify the cellular mechanisms and the chronology by which different types of lung cell death are activated during lipopolysaccharide (LPS)-induced ALI, we decided to evaluate autophagy (by LC3-II and autophagosome) and apoptosis (by caspase-3) at different time points after LPS treatment in a rat model of LPS-induced ALI.
Materials and Methods:
Sprague-Dawley rats were randomly divided into two groups: control group and LPS group. ALI was induced by LPS intraperitoneal injection (3 mg/kg). The lung tissues were collected to measure lung injury score by histopathological evaluation, the protein expression of LC3-II and caspase-3 by Western blot, and microstructural changes by electron microscopy analysis.
Results:
During ALI, lung cell death exhibited modifications in the death process at different stages of ALI. At early stages (1 hr and 2 hr) of ALI, the mode of lung cell death started with autophagy in LPS group and reached a peak at 2 hr. As ALI process progressed, apoptosis was gradually increased in the lung tissues and reached its maximal level at later stages (6 hr), while autophagy was time-dependently decreased.
Conclusion:
These findings suggest that activated autophagy and apoptosis might play distinct roles at different stages of LPS-induced ALI. This information may enhance the understanding of lung pathophysiology at the cellular level during ALI and pulmonary infection, and thus help optimize the timing of innovating therapeutic approaches in future experiments with this model.
Keywords: Acute lung injury, Apoptosis, Autophagy, Lipopolysaccharide
Introduction
Acute lung injury (ALI) represents a leading cause of death in critically ill patients with a high mortality rate (35–45%) (1). Despite recent advances in ALI therapy, the efficacy of therapies has been modest. This slow progress may partially result from our poor understanding of the cellular and molecular basis of ALI. Additional experiments are needed to investigate the cellular and molecular basis of ALI.
Recently, dysregulation of lung cell death has been reported during ALI (2). The modes of cell death in the lungs at least include (2): (i) type I programmed cell death (apoptosis), characterized by the formation of apoptotic bodies, cell shrinkage, DNA fragmentation, and chromatin condensation. (ii) type II programmed cell death (autophagy), characterized by the formation of autophagosomes, degradation of cytoplasmic contents, and little chromatin condensation. (iii) necrosis, characterized by cellular swelling and membrane lysis. Among these, apoptosis was believed to be involved in lung injury because inhibition of apoptosis-associated signaling components such as the Fas/FasL system and caspase reportedly reduced the extent of lung injury in experimental animals (3, 4). However, the possible role of autophagy in ALI remains to be debated. On one hand, increased autophagy reportedly plays an important role in ischemia-reperfusion or ventilator-induced lung injury (5, 6). On the other hand, autophagy-associated LC3-II has been found to interact with the Fas pathway to prevent epithelial cell apoptosis under hyperoxia (7). Further studies are needed to confirm the role of autophagy and apoptosis in experimental ALI models. Those studies examining whether different types of cell death are dynamically altered in the lung during ALI should provide important information regarding the functional role of different modes of lung cell death.
Therefore, this study aimed to investigate the time-dependent changes of autophagy and apoptosis in the lungs of the rats injected with lipopolysaccharide (LPS) and their normal counterparts injected with normal saline. By comparing the lung injury, apoptosis, and autophagy in these animals, the specific role of autophagy and apoptosis in LPS-induced lung injury can be clarified.
Materials and Methods
Reagents and animals
LPS was obtained from Sigma-Aldrich; primary antibodies against LC-3 and Caspase-3 were obtained from Abcam Corporation.
Ninety-six healthy male Sprague-Dawley (SD) rats (180–200 g) were obtained from the Laboratory Animal Research Center of Tongji Medical College, Huazhong University of Science and Technology (Wuhan, China). Animals were maintained under specific pathogen-free conditions (temperature: 22 ± 2°C, relative humidity: 40%–80%), and allowed free access to standard laboratory food and water.
LPS-induced ALI model
Ninety-six healthy male SD rats were randomly divided into two groups: normal control group and LPS group. The LPS group was intraperitoneally injected with LPS (3 mg/kg) dissolved in 1 ml of sterile saline. The normal control group was injected with the same volume of saline. Lung tissues were collected prior to and 1, 2, 4, 6, and 8 hr after LPS (LPS-0 hr, LPS-1 hr, LPS-2 hr, LPS-4 hr, LPS-6 hr, LPS-8 hr) and normal saline injection (Ctrl-0 hr, Ctrl-1 hr, Ctrl-2 hr, Ctrl-4 hr, Ctrl-6 hr, and Ctrl-8 hr). Eight animals were used at each time point. All animal procedures were performed in accordance with National Institutes of Health Guide for the Care and Use of Laboratory Animals (8th Edition, revised 2011) and approved by the Animal Care and Use Committee of Hubei University of Science and Technology, China.
Measurement of Lung injury Score
For histopathological analysis, lung tissues were fixed in 4% formalin and embedded in paraffin. Subsequently, sections (5 μm thick) were prepared and stained with hematoxylin and eosin for pathological observation.
Lung injury score of each slide was assessed by two independent pathologists in a blinded manner as previously described (8). The score represents the average of these pathologists. Each section was scored according to the following four items: (i) alveolar septal congestion, (ii) alveolar hemorrhage, (iii) intra-alveolar cell infiltrates, and (iv) intra-alveolar fibrin deposition. Each item was scored from 0 to 3 according to injury field. Within each field, points were assigned according to the following criteria: alveolar septal congestion, 0: all septae are thin and delicate, 1: congested alveolar septae occur in <1/3 of the field, 2: congested alveolar septae occur in 1/3–2/3 of the field, 3: congested alveolar septae occur in >2/3 of the field; alveolar hemorrhage, 0: no hemorrhage, 1: at least 5 erythrocytes per alveolus in 1–5 alveoli, 2: at least 5 erythrocytes in 5–10 alveoli, 3: at least 5 erythrocytes in >10 alveoli; intra-alveolar fibrin deposition, 0: no intra-alveolar fibrin, 1: fibrin deposition in <1/3 of the field, 2: fibrin deposition in 1/3–2/3 of the field, 3: fibrin deposition in >2/3 of the field; intra-alveolar cell infiltrates, 0: 0–5 intra-alveolar cells per field, 1: 5–10 intra-alveolar cells per field, 2: 10–20 intra-alveolar cells per field, 3: >20 intra–alveolar cells per field. All of the points for each item were weighted according to their relative importance. The total injury score was calculated according to the following formula: total lung injury score = [(points of alveolar septal congestion/number of fields) + (points of alveolar hemorrhage/number of fields) + 2 × (points of intra-alveolar cell infiltrates/number of fields)+3 × (points of intra-alveolar fibrin deposition/number of fields)]/total number of alveoli counted.
Transmission electron microscopy
Lung tissues (1 mm3) were fixed in 2.5% glutaraldehyde, followed by 1% osmium tetroxide and embedded in epoxy resin 618. Subsequently, these samples were cut into ultrathin sections and examined using a Hitachi H-300 transmission electron microscope (FEI Tecnai G2 12-type, Holland, The Netherlands).
Western blot analysis
Lung tissues were homogenized with a lysis buffer containing: 50 mmol/l Tris, pH 7.5, 150 mmol/l NaCl, 1% Triton X-100, 1% sodium deoxycholate, 1.0 mmol/lphenylmethanesulfonyl fluoride, 50 mmol/l sodium fluoride, 1.0 mmol/l sodium orthovanadate, 50 μg/ml pepstatin, and 50 μg/ml leupeptin. The homogenate was incubated at 4 °C for 30 min and then centrifuged for 10 min at 4 °C. The supernatant was harvested and stored at −80 °C for further analysis. Total protein content in the supernatant was measured by the BCA protein assay. Subsequently, an equal amount of proteins was separated on 15% SDS-PAGE gels and then transferred to a polyvinylidene difluoride membrane (Millipore, Bedford, MA, USA). After being blocked with 5% fat-free milk, the membranes were incubated with primary antibodies against caspase-3, cleaved caspase-3, LC3-II, or β-actin overnight at 4°C. After washing three times with TBS-T, the membranes were incubated with horseradish peroxidase (HRP) conjugated secondary antibody (Santa Cruz, USA) for 1 hr at room temperature. Protein bands were visualized using the ECL system and analyzed with a gel imaging system (Kodak system EDAS120, Tokyo, Japan).
Statistical analysis
All data are presented as mean±SEM. For determination of time-dependent changes in LPS-induced lung changes, the differences in all measured parameters between different time points in control and LPS groups were analyzed and comparisons were made at each time point between control and LPS groups. Statistical analysis was performed using one-way ANOVA followed by a Student-Neuman-Keuls t-test. P-values<0.05 were considered significant.
Results
Time-dependent changes in LPS-induced lung injury
First, we characterized the time-dependent profiles of LPS-induced lung injury. As shown in Figure 1, a time-dependent increase of lung injury score was observed in the rats from 1-hr, 2-hr, 4-hr, 6-hr, and 8-hr LPS groups. At the 2-hr, 4-hr, 6-hr, and 8-hr time points, SD rats treated with LPS showed significantly higher lung injury scores than animals treated with saline (P<0.01) (Figure 1B). The lung injury was characterized by alveolar septal widening, increased neutrophil infiltration, microvascular expansion, and alveolar hemorrhage (Figure 1A).
Figure 1.
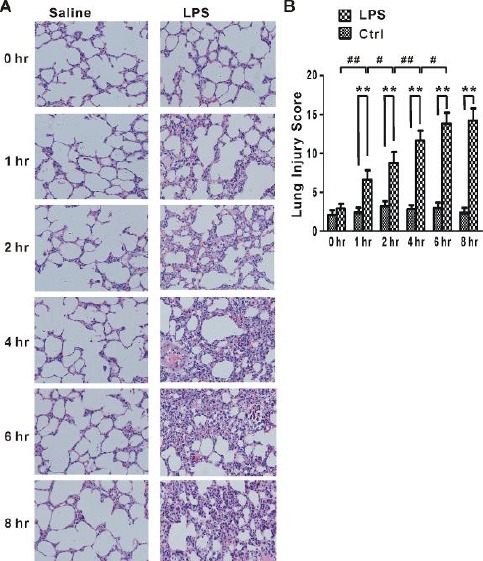
Histopathological changes and lung injury score
(A) Histological structures of control and LPS groups at different time points after LPS exposure determined by H&E staining. The results showed alveolar septal widening, inflammatory infiltrates and areas of hemorrhage. (original magnifications, ×200; HE) (B) Lung injury score was determined and graded according to Matute-Bello Scoring as described in Materials and Methods. One-way ANOVA followed by a Student-Neuman-Keuls t-test was used to analyze significant differences in lung injury score at each time point between control (Ctrl, n=8 for each time point) and LPS groups (LPS, n=8 for each time point) and between different time points in control (Ctrl, n=8 for each time point) and LPS (LPS, n=8 for each time point) groups. **P < 0.01 indicates significant differences in lung injury score at each time point between control and LPS groups. #P<0.05 and ##P<0.01 indicate significant differences in lung injury score between different time points in LPS groups
Time-dependent changes of autophagy in LPS-induced lung injury
Next, we determined whether autophagy was time-dependently altered during LPS-induced lung injury. Because the production of LC3-II is regarded as a critical step in autophagy events (9), this study determined the protein level of LC3-II by Western blot. As shown in Figure 2A, LC3-II level was significantly increased in LPS-1 hr, LPS-2 hr, LPS-4 hr, and LPS-6 hr groups when compared with normal control groups (Ctrl-1 hr, Ctrl-2 hr, Ctrl-4 hr, and Ctrl-6 hr). The up-regulation of LC3-II reached a maximum at 2 hr after LPS administration. Subsequently, the up-regulation of LC3-II gradually decreased over 8 hr. These data suggested that a time-dependent change of autophagy occurred in LPS-induced lung injury.
Figure 2.
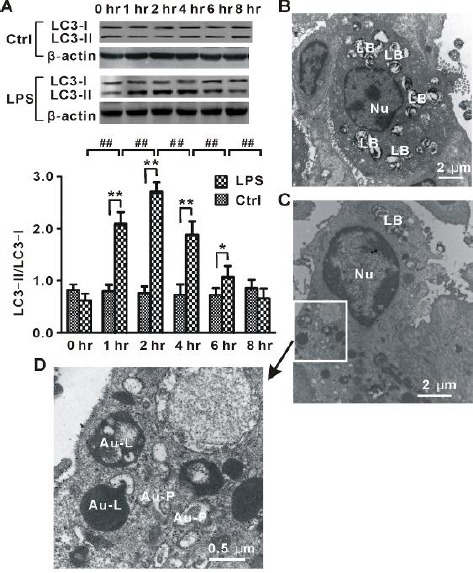
Autophagy in LPS-induced lung injury
(A) Time-dependent changes in the protein level of LC3-II in lung tissues from rats (LPS, n=8 for each time point) injected with LPS and normal control animals (Ctrl, n=8 for each time point) treated with saline. (B) Representative electron microscopy images for normal alveolar type II cell. (C) Representative electron microscopy images for autophagic alveolar type II cells in LPS-2 hr group rats. (D) High-resolution figure of part of autophagic alveolar type II cell in (C) showed increased formation of autophagosomes. LB, lamellar bodies; Nu, nuclear; Au-P, autophagosome; Au-L, autolysosome. One-way ANOVA followed by a Student-Neuman-Keuls t-test was used to analyze significant differences in the protein level of LC3-II at each time point between control and LPS groups and between different time points in control and LPS groups. *P<0.05 and **P<0.01 indicates significant differences in the protein level of LC3-II at each time point between control and LPS groups. ##P < 0.01 indicates significant differences in the protein level of LC3-II between different time points in LPS groups
Meanwhile, our electron microscopy analysis showed that the lungs from rats treated with LPS exhibited increased characteristic autophagosomes and autolysosomes in alveolar type II cells (Figures 2C and 2D); in contrast, few or no autophagosomes were observed in normal control groups (Figure 2B). Thus, autophagy was induced during LPS-induced lung injury.
Time-dependent changes of apoptosis in LPS-induced lung injury
To determine whether apoptosis is time-dependently altered during LPS-induced lung injury, protein expression of cleaved (active form) caspase-3, a critical executioner of apoptosis, was measured by Western blot. As shown in Figure 3A, the cleaved caspase-3 level was significantly increased in LPS-2 hr, LPS-4 hr, LPS-6 hr, and LPS-8 hr groups when compared with normal control groups (Ctrl-2 hr, Ctrl-4 hr, Ctrl-6 hr, and Ctrl-8 hr). The elevated caspase-3 level reached a maximum at 6 hr after LPS administration. At 8 hr after LPS administration, the up-regulation of cleaved caspase-3 was decreased. These data suggested that apoptosis was time-dependently altered in LPS-induced lung injury.
Figure 3.
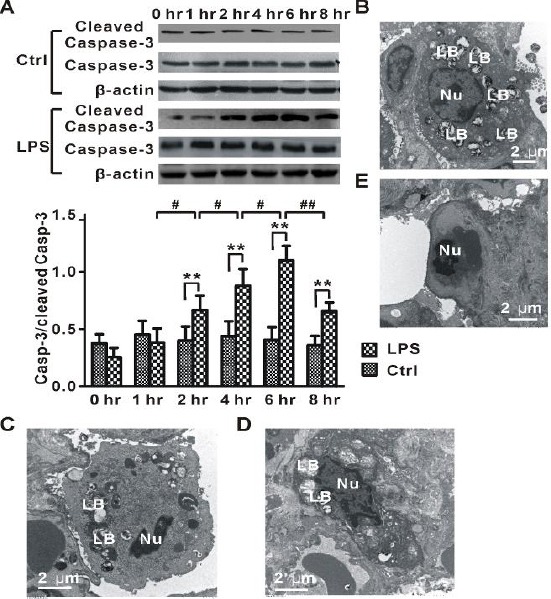
Apoptosis in LPS-induced lung injury
(A) Time-dependent changes in the protein level of cleaved caspase-3 in lung tissues from rats (LPS, n=8 for each time point) injected with LPS and normal control animals (Ctrl, n=8 for each time point) treated with saline. (B) Representative electron microscopy image for normal alveolar type II cell in Ctrl-6 hr group. (C) Representative electron microscopy image for apoptotic alveolar type II cell with chromatin condensed in the LPS-6 hr group. (D) Typical electron microscopy image for apoptotic alveolar type II cell with disruption of lamellar bodies, chromatin condensed at the periphery of the nucleus, and vacuolization of the nuclear membrane in the LPS-6 hr group. (E) Representative electron microscopy image for apoptotic endothelial cell with chromatin condensed in the LPS-6 hr group. LB, lamellar bodies; Nu, nuclear. One-way ANOVA followed by a Student-Neuman-Keuls t-test was used to analyze significant differences in the protein level of cleaved caspase-3 at each time point between control and LPS groups and between different time points in control and LPS groups. **P<0.01 indicates significant differences in the protein level of cleaved caspase-3 at each time point between control and LPS groups. #P<0.05 and ##P<0.01 indicate significant differences in the protein level of cleaved caspase-3 between different time points in LPS groups
Additionally, our electron microscopy analysis showed that the lungs from rats treated with LPS exhibited increased apoptosis in alveolar type II cells (Figures 3C and 3D) and endothelial cells (Figure 3E). These apoptotic cells showed features characteristic of apoptosis, including increased electron density of the cytoplasm, chromatin condensation, and vacuolization of the nuclear membrane.
Discussion
To clarify the possible role of autophagy and apoptosis in LPS-induced lung injury, the present study investigated the time-dependent changes of autophagy and apoptosis in the lungs of the rats treated with LPS and the normal control injected with normal saline. Our data showed that both autophagy and apoptosis in the lungs of the rats were time-dependently altered during LPS-induced ALI, but they reached a peak at different time points, suggesting that the two types of lung cell death might play different roles in LPS-induced ALI.
Autophagy and LPS-induced ALI
Autophagy is a fundamental homeostatic process and plays a key role in the regulation of intracellular protein and organelle and metabolic homeostasis. However, under pathological conditions, autophagy dysfunction has been implicated in several human diseases, including cancers (10), cardiac diseases (11), and lung diseases (12). Accumulating data suggest that autophagy is involved in the development of different kinds of lung injury, including cigarette smoke exposure (13) or ventilator-induced lung injury (5). In contrast, autophagy may play a protective role in hyperoxia-induced lung injury (7). Thus, the role of autophagy in lung injury appears to be context-dependent. However, the dynamic changes and functional significance of autophagy in LPS-induced ALI remain unknown.
In the present study, we observed the time course of autophagy in a rat model of LPS-induced ALI. This study showed that autophagy was time-dependently triggered by LPS administration, evidenced by increased expression of LC3-II and characteristic autophagosomes in alveolar type II cells. The up-regulation of LC3-II reached a peak at 2 hr after LPS administration, which preceded the extent of lung injury. Lung injury score in LPS treated groups was observed with a maximum at 6 hr. Therefore, our data suggest that at early stages of LPS-induced ALI, activated autophagy might act as an adaptive response to pathological stimuli, but not an injurious process in lung injury. In support of our hypothesis, recent evidence reveals that autophagy may play a protective role against lung injury/inflammation in sepsis (14, 15).
Apoptosis and LPS-induced ALI
Accumulating evidence reveals that apoptosis plays a key role in different kinds of lung injury, including LPS-induced ALI (3, 16–18). LPS has been widely used as a tool to study the mechanisms of ALI in animals (18, 19). LPS reportedly induced increased apoptosis of alveolar epithelial cells (20), pulmonary artery endothelial cells (21), and bronchial epithelial cells (22) of experimental animals. Several apoptotic signaling pathways including the Fas/FasL system (23) and the caspase family (18) have been implicated in LPS-induced ALI. Inhibition of these apoptotic signaling pathways can improve LPS-induced ALI. In the present study, our Western blot and transmission electron microscopy analysis showed that LPS administration activated apoptosis of lung cells. During LPS-induced ALI, the time course of apoptotic marker caspase-3 expression was similar to that of lung injury score. Thus, the results of our study and others suggest that apoptosis is involved in LPS-induced ALI.
Relationship between autophagy and Apoptosis in LPS-induced ALI
The relationship between autophagy and apoptosis may be very complicated. Both autophagy and apoptosis of lung cells can be triggered by extracellular stimuli such as hypoxia, oxide stress, and ischemia-reperfusion injury (24-26). Some intracellular signaling pathways including mitogen-activated protein kinases (24, 27) and NF-κB (27, 28) are involved in both types of cell death. A recent study by Tanaka et al (7) has revealed a crosstalk between autophagy- and apoptosis-associated intracellular signaling pathways in lung cells, suggesting that autophagy might act as a survival mechanism to antagonize Fas-mediated apoptosis. Another study by Lee et al (29) showed that activation of autophagy rescues amiodarone-induced apoptosis of lung epithelial cells and pulmonary toxicity in rats. In contrast, inhibition of autophagy by autophagic inhibitors enhances apoptosis in lung cancer cells (30). In this study, autophagy reached a peak at an earlier stage (2 hr), while apoptosis attained the maximum at a later stage (6 hr). Thus, our data and these above findings support a hypothesis that an imbalance between autophagy and apoptosis in lung cells may contribute to the pathogenesis of LPS-induced ALI. Further studies are needed to investigate the possible role of the imbalance between autophagy and apoptosis in ALI.
Conclusion
Collectively, our results showed different time courses of autophagy and apoptosis in the lungs of the rats during LPS-induce ALI, suggesting that the two types of lung cell death might play distinct roles at different stages of LPS-induced ALI. This information may enhance the understanding of lung pathophysiological alterations at the cellular levels during ALI and pulmonary infection, and thus help to optimize the timing of innovative therapeutic approaches in future experiments with this model. In addition, further studies using inhibitors or gene knockout models will be required to delineate the role of autophagy and apoptosis in experimental ALI models.
Conflict of interest
No conflicts of interest are declared by the authors.
Acknowledgment
This work was supported by the Natural Science Foundation of Hubei Province (no. 2014CFC1081) and Science Fund of Hubei University of Science and Technology (no. ZX1301 and 14zx06).
References
- 1.Phua J, Badia JR, Adhikari NK, Friedrich JO, Fowler RA, Singh JM, et al. Has mortality from acute respiratory distress syndrome decreased over time? A systematic review. Am J Respir Crit Care Med. 2009;179:220–227. doi: 10.1164/rccm.200805-722OC. [DOI] [PubMed] [Google Scholar]
- 2.Tang PS, Mura M, Seth R, Liu M. Acute lung injury and cell death: how many ways can cells die? Am J Physiol. 2008;294:L632–L641. doi: 10.1152/ajplung.00262.2007. [DOI] [PubMed] [Google Scholar]
- 3.Neff TA, Guo R-F, Neff SB, Sarma JV, Speyer CL, Gao H, et al. Relationship of Acute Lung Inflammatory Injury to Fas/FasL System. Am J Pathol. 2005;166:685–694. doi: 10.1016/S0002-9440(10)62290-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quadri SM, Segall L, De Perrot M, Han B, Edwards V, Jones N, et al. Caspase Inhibition Improves Ischemia-Reperfusion Injury After Lung Transplantation. Am J Transplant. 2005;5:292–299. doi: 10.1111/j.1600-6143.2004.00701.x. [DOI] [PubMed] [Google Scholar]
- 5.Gao M, Liu D, Du Y, Sun R, and Zhao L. Autophagy facilitates ventilator-induced lung injury partly through activation of NF-kappaB pathway. Med Sci Monit. 2013;19:1173–1175. doi: 10.12659/MSM.889746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang J, Wang J-S, Zheng Z-K, Tang J, Fan K, Guo H, et al. Participation of autophagy in lung ischemia-reperfusion injury in vivo. J Surg Res. 2013;182:e79–e87. doi: 10.1016/j.jss.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 7.Tanaka A, Jin Y, Lee S-J, Zhang M, Kim HP, Stolz DB, et al. Hyperoxia-Induced LC3B Interacts with the Fas Apoptotic Pathway in Epithelial Cell Death. Am J Respir Cell Mol Biol. 2012;46:507–514. doi: 10.1165/rcmb.2009-0415OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matute-Bello G, Winn RK, Jonas M, Chi EY, Martin TR, Liles WC. Fas (CD95) induces alveolar epithelial cell apoptosis in vivo:implications for acute pulmonary inflammation. Am J Pathol. 2001;158:153–161. doi: 10.1016/S0002-9440(10)63953-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gustafsson AB, Gottlieb RA. Autophagy in Ischemic Heart Disease. Circ Res. 2009;104:150–158. doi: 10.1161/CIRCRESAHA.108.187427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakahira K, Choi AMK. Autophagy: a potential therapeutic target in lung diseases. Am J Physiol. 2013;305:L93–L107. doi: 10.1152/ajplung.00072.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryter SW, Lee SJ, Choi AM. Autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. Expert Rev Respir Med. 2010;4:573–584. doi: 10.1586/ers.10.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lo S, Yuan SS, Hsu C, Cheng YJ, Chang YF, Hsueh HW, et al. Lc3 over-expression improves survival and attenuates lung injury through increasing autophago-somal clearance in septic mice. Ann Surg. 2013;257:352–363. doi: 10.1097/SLA.0b013e318269d0e2. [DOI] [PubMed] [Google Scholar]
- 15.Yen YT, Yang HR, Lo HC, Hsieh YC, Tsai SC, Hong CW, et al. Enhancing autophagy with activated protein C and rapamycin protects against sepsis-induced acute lung injury. Surgery. 2013;153:689–698. doi: 10.1016/j.surg.2012.11.021. [DOI] [PubMed] [Google Scholar]
- 16.Perl M, Chung C-S, Lomas-Neira J, Rachel T-M, Biffl WL, Cioffi WG, et al. Silencing of Fas, but Not Caspase-8, in Lung Epithelial Cells Ameliorates Pulmonary Apoptosis, Inflammation, and Neutrophil Influx after Hemorrhagic Shock and Sepsis. Am J Pathol. 2005;167:1545–1559. doi: 10.1016/S0002-9440(10)61240-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Messer MP, Kellermann P, Weber SJ, Hohmann C, Denk S, Klohs B, et al. Silencing of fas, fas-associated via death domain, or caspase 3 differentially affects lung inflammation, apoptosis, and development of trauma-induced septic acute lung injury. Shock. 2013;39:19–27. doi: 10.1097/SHK.0b013e318277d856. [DOI] [PubMed] [Google Scholar]
- 18.Kawasaki M, Kuwano K, Hagimoto N, Matsuba T, Kunitake R, Tanaka T, et al. Protection from Lethal Apoptosis in Lipopolysaccharide-Induced Acute Lung Injury in Mice by a Caspase Inhibitor. Am J Pathol. 2000;157:597–603. doi: 10.1016/S0002-9440(10)64570-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie K, Yu Y, Huang Y, Zheng L, Li J, Chen H, et al. Molecular Hydrogen Ameliorates Lipopolysaccharide-Induced Acute Lung Injury in Mice Through Reducing Inflammation and Apoptosis. Shock. 2012;37:548–555. doi: 10.1097/SHK.0b013e31824ddc81. [DOI] [PubMed] [Google Scholar]
- 20.Ma X, Xu D, Ai Y, Ming G, Zhao S. Fas inhibition attenuates lipopolysaccharide-induced apoptosis and cytokine release of rat type II alveolar epithelial cells. Mol Biol Rep. 2010;37:3051–3056. doi: 10.1007/s11033-009-9876-9. [DOI] [PubMed] [Google Scholar]
- 21.Thambiayya K, Wasserloos K, Kagan VE, Stoyanovsky D, Pitt BR. A critical role for increased labile zinc in reducing sensitivity of cultured sheep pulmonary artery endothelial cells to LPS-induced apoptosis. Am J Physiol. 2012;302:L1287–L1295. doi: 10.1152/ajplung.00385.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vernooy JHJ, Dentener MA, van Suylen RJ, Buurman WA, Wouters EFM. Intratracheal Instillation of Lipopolysaccharide in Mice Induces Apoptosis in Bronchial Epithelial Cells. Am J Respir Cell Mol Biol. 2001;24:569–576. doi: 10.1165/ajrcmb.24.5.4156. [DOI] [PubMed] [Google Scholar]
- 23.Kitamura Y, Hashimoto S, Mizuta N, Kobayashi A, Kooguchi K, Fujiwara I, et al. Fas/FasL-dependent Apoptosis of Alveolar Cells after Lipopolysaccharide-induced Lung Injury in Mice. Am J Respir Crit Care Med. 2001;163:762–769. doi: 10.1164/ajrccm.163.3.2003065. [DOI] [PubMed] [Google Scholar]
- 24.Lim SK, Jeong YW, Kim DI, Park MJ, Choi JH, Kim SU, et al. Activation of PRMT1 and PRMT5 mediates hypoxia- and ischemia-induced apoptosis in human lung epithelial cells and the lung of miniature pigs: The role of p38 and JNK mitogen-activated protein kinases. Biochem Biophys Res Commun. 2013;440:707–713. doi: 10.1016/j.bbrc.2013.09.136. [DOI] [PubMed] [Google Scholar]
- 25.Kuwano K. Epithelial cell apoptosis and lung remodeling. Cell Mol Immunol. 2007;4:419–429. [PubMed] [Google Scholar]
- 26.Ryter SW, Choi AM. Autophagy in the lung. Proc Am Thorac Soc. 2010;7:13–21. doi: 10.1513/pats.200909-101JS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pan H, Zhang Y, Luo Z, Li P, Liu L, Wang C, et al. Autophagy mediates avian influenza H5N1 pseudotyped particle-induced lung inflammation through NF-kappaB and p38 MAPK signaling pathways. Am J Physiol. 2014;306:L183–195. doi: 10.1152/ajplung.00147.2013. [DOI] [PubMed] [Google Scholar]
- 28.Li L, Wu W, Huang W, Hu G, Yuan W, Li W. NF-kappaB RNAi decreases the Bax/Bcl-2 ratio and inhibits TNF-alpha-induced apoptosis in human alveolar epithelial cells. Inflamm Res. 2013;62:387–397. doi: 10.1007/s00011-013-0590-7. [DOI] [PubMed] [Google Scholar]
- 29.Lee KY, Oh S, Choi YJ, Oh SH, Yang YS, Yang MJ, et al. Activation of autophagy rescues amiodarone-induced apoptosis of lung epithelial cells and pulmonary toxicity in rats. Toxicol Sci. 2013;136:193–204. doi: 10.1093/toxsci/kft168. [DOI] [PubMed] [Google Scholar]
- 30.Wu G, Li H, Ji Z, Jiang X, Lei Y, and Sun M. Inhibition of autophagy by autophagic inhibitors enhances apoptosis induced by bortezomib in non-small cell lung cancer cells. Biotechnol Lett. 2014;36:1171–1178. doi: 10.1007/s10529-014-1470-0. [DOI] [PubMed] [Google Scholar]