

Abstract
Production of reactive nitrogen species (NO) is a key step in the immune response following infections. NO induces lesions to bacterial DNA, thus limiting bacterial growth within hosts. Using two pathogenic bacteria, Bacillus cereus and Shigella flexneri, we show that the DNA-repair protein Mfd (Mutation-Frequency-Decline) is required for bacterial resistance to the host-NO-response. In both species, a mutant deficient for mfd does not survive to NO, produced in vitro or by phagocytic cells. In vivo, the ∆mfd mutant is avirulent and unable to survive the NO-stress. Moreover, NO induces DNA-double-strand-breaks and point mutations in the Δmfd mutant. In overall, these observations demonstrate that NO damages bacterial DNA and that Mfd is required to maintain bacterial genomic integrity. This unexpected discovery reveals that Mfd, a typical housekeeping gene, turns out to be a true virulence factor allowing survival and growth of the pathogen in its host, due to its capacity to protect the bacterium against NO, a key molecule of the innate immune defense. As Mfd is widely conserved in the bacterial kingdom, these data highlight a mechanism that may be used by a large spectrum of bacteria to overcome the host immune response and especially the mutagenic properties of NO.
The host defense against bacteria is predominantly mediated by cellular immune mechanisms. Phagocytic neutrophils and macrophages infiltrate inflamed areas, where they produce an extensive array of toxic substances. The chemicals secreted include reactive oxygen and nitrogen species, which cause DNA damage and gene mutations1,2. Nitric oxide (NO) is synthesized by an enzymatic reaction involving NO synthases (NOS). NOS are expressed both as constitutive enzymes, which contribute to vasorelaxation and neurotransmission, and as inducible isoforms (iNOS). Various cells including macrophages, neutrophils and epithelial cells express iNOS, and an excess of NO is produced during most types of infections. NO can damage biological molecules including proteins and nucleic acids3. NO is cytotoxic and mutagenic both for various pathogens and for host cells2,4,5. Thus, NO plays an important and complex role during infections, limiting microbial proliferation within host cells and contributing to microbial clearance.
Bacteria express sensor proteins able to detect NO, and switch on the expression of enzymes that detoxify NO before it reaches lethal levels6. In E. coli, a mutant deficient for the nucleotide excision repair (NER) pathway is sensitive to HNO2 treatment7 and the base excision repair (BER) pathway protects Salmonella from the genotoxic effects of the host NO8. RecBCD-dependent recombinational repair also plays a role in preventing the genotoxic effects of NO. The NO sensitivity in the absence of RecBCD-dependent homologous recombination indicates that NO toxicity is due, at least partially, to the formation of DNA double-strand breaks (DSBs)9.
Damage to DNA can affect transcription fidelity and processivity and thereby threatens cell viability. DNA lesions that block RNA polymerase (RNAP) prevent transcription. In bacteria, RNAP stalling triggers a specialized DNA repair mechanism, called transcription coupled repair (TCR) pathway. Mfd (Mutation Frequency Decline) is an evolutionarily conserved bacterial protein involved in TCR10. Mfd removes RNAP stalled by DNA damage. Mfd utilizes ATP to translocate along DNA, most likely forcing RNAP forward and ultimately dissociating it from the DNA template10. In E. coli, Mfd also contains binding domains that may recruit UvrA and trigger the associated NER pathway11. It was recently shown in B. subtilis that Mfd and UvrA act in the same pathway in the context of transcription12. Mfd also decreases the efficiency with which RNAP bypasses abasic sites, possibly reducing the level of transcriptional mutagenesis caused by these DNA lesions13. Mfd is required for DNA repair in E. coli following experimental UV irradiation14 and in Helicobacter pylori following exposure to mitomycin C15. Mfd was also found to be important for processing the genetic damage during Bacillus subtilis stationary phase associated mutagenesis16 and sporulation17,18 and during DNA recombination19. Furthermore, Mfd has been associated with the development of antibiotic resistance in Campylobacter jejuni and H. pylori15,20. Thus, the mechanism of adaptive mutagenesis and DNA repair by Mfd has been thoroughly studied in the context of both artificially induced and spontaneous mutations14,16,21,22. However, to our knowledge its involvement in bacterial pathogenesis has never been reported.
In this study, we show that the bacterial Mfd protein is essential to survive the deleterious effect of the nitrogen response. We use two different human pathogens, the Gram-positive Bacillus cereus and the Gram-negative Shigella flexneri and show that, in both cases, Mfd plays an important role in bacterial survival in the context of NO-induced stress. We provide insights into the role of Mfd during bacterial pathogenesis and into the mechanisms of DNA damage and repair after NO stress.
Results
Mfd is required for virulence and bacterial growth in vivo
B. cereus is an emerging pathogenic bacteria, which can infect hosts as diverse as insects and humans23,24,25,26,27. We used this remarkable property of wide host spectrum to develop a direct approach to identify new B. cereus virulence factors. A library of mutated genes of the B. cereus 407 (Bc 407) strain was constructed28 and over 1500 mutants were individually tested for their capacity to kill Bombyx mori insect larvae. A strain mutated in the trcf gene was identified as avirulent. This gene (Bc0058 in the B. cereus reference strain ATCC 14579) encodes for the Mfd protein.
A stable mfd deletion strain was constructed by insertion of a kanamycin-resistance cassette into the mfd gene. B. cereus growth of wild-type and ∆mfd mutant strains did not differ in LB medium (Fig. S1). The pathogenicity of the wild type and mutant strains was tested in B. mori (Fig. 1A). Deletion of mfd induced a drastic loss of virulence compared to the wild-type strain. To confirm that the chromosomal deletion did not induce a polar effect, the mfd mutant strain was genetically complemented by introduction of a plasmid carrying a functional mfd gene and the corresponding promoter region. Complementation of the ∆mfd strain by the mfd+ gene restored the wild-type virulence phenotype (Fig. 1A). In addition, a plasmid carrying the spoVT+ gene situated downstream of mfd was also introduced into the ∆mfd mutant (Fig. 1C). The spoVT+ gene did not restore the virulence capacity of the mutant, which remained similar to that of the single ∆mfd mutant, showing that the downstream spoVT gene is not involved in the mfd mutant phenotype (Fig. S2). This finding in addition to the complement data shows that Mfd is required for B. cereus virulence in an insect model of infection.
Figure 1.

(A) Various doses of wild-type (Bc 407), mfd mutant (Bc ∆mfd) and the complemented Bc ∆mfd/mfd+ strains were injected into the hemocoel of B. mori larvae. Insect mortality was recorded 24 h post infection. The results are mean values of at least three independent experiments. (B) B. mori were infected with 50 cfu of B. cereus wild-type (Bc 407) or mfd mutant (Bc ∆mfd) strains. After the indicated times, larvae were crushed in PBS medium and cfu were counted by plating serial dilutions on agar plates. The results reported are mean values of at least five independent experiments. (C) mfd gene environment in Bacilli is schematically represented. The promoter regions as defined in http://genome.jouy.inra.fr/cgi-bin/seb/viewdetail.py?id=mfd_60430_63963_1 is indicated as black arrow.
We assessed the survival of the strains during infection by crushing the insects and dilution plating. In sharp contrast to the wild type stain, the ∆mfd mutant was unable to grow in vivo and no bacteria were recovered 24 h after infection (Fig. 1B). Thus, Mfd is required for bacterial survival during the infectious process.
Mfd is required for B. cereus resistance to NO stress produced in vitro or by phagocytic cells
We tested whether the failure of the ∆mfd mutant to survive in the insect host was due to its sensitivity to host cellular defenses. The human monocytic-like cell line PLB was infected with the two strains and bacterial survival was assessed (Fig. 2A). The wild-type strain survived in the PLB cells whereas the mutant did not (P < 0.02), implying that a cellular response was preventing growth of the mutant. Host defense against bacteria is predominantly mediated by cellular immune mechanisms. Phagocytic neutrophils and macrophages infiltrate inflamed areas, where they produce an extensive array of toxic substances. Among the secreted chemicals are oxygen (ROS) and nitrogen (NOS) species, which are known to induce DNA damage and mutations3. To test the effect of ROS on the mfd mutant growth, the wild type cell line PLB and its mutated counterpart deficient in ROS production (gp91phox-KO) were infected with the strains (Fig. 2B). The B. cereus wild type strain grew equally in both cell lines. The mfd mutant grew neither in the wild type nor in the ROS deficient cells, suggesting that suppression of ROS was not sufficient to restore growth of the mfd mutant. This is consistent with another finding showing that Mfd is not required for transcription recovery following oxidative stress in E. coli29. We measured similar nitrite production in the supernatant of cells infected with wild-type or mutant strains (Fig. 2C), suggesting that in both cases, infection of PLB cells induced a NO response. To test the role of Mfd during resistance to NO produced by cells, the survival rates of ∆mfd and wild-type strains were assessed after incubation of PLB cells in the presence of an inhibitor of the nitrogen response, NG-monomethyl-L-arginine (NMMLA), a competitor of the iNOS enzyme (Fig. 3A). The ∆mfd mutant did not survive following incubation of cells in the absence of NMMLA. By contrast, in the presence of the inhibitor, the survival rates of ∆mfd and wild-type strains were similar (P > 0.26) strongly suggesting that Mfd plays a role in NO resistance.
Figure 2.

Human monocytic-like cells PLB (A) and PLB deficient for the ROS response (PLB phox-KO) (B) were incubated with B. cereus wild-type and mfd mutant for 4 h and bacterial survival was calculated by plating serial dilutions on agar plates. The cfu counts were normalized to the initial cfu (t0). The results reported are mean values of at least three independent experiments each in triplicate. P values are calculated using the Student test. (C) Nitrite concentration was measured in the PLB cell supernatant collected after each time of infection by the Griess method. This graph represents a set of representative data out of three independent experiments done in triplicate.
Figure 3.

(A) Human monocytic-like cells, PLB were infected with B. cereus wild-type and the mfd mutant for 4 h in the presence or absence of 1 mM NMMLA (an NO inhibitor). Bacterial survival was calculated by plating serial dilutions on agar plates. Cfu counts were normalized to initial cfu. The results reported are mean values of at least three independent experiments each in triplicate. P values are calculated using the Student test. (B) Bacteria were exposed directly to chemically generated NO (500 μM sodium nitrite) for 4 h in a cell-free system. Bacteria were then harvested and plated on agar plates to evaluate bacterial survival. Cfu counts were normalized to initial cfu. The results reported are mean values of at least five independent experiments. P values are calculated using the Student test. (C) NO production in the insect B. mori was silenced by RNA interference (RNAi). Larvae injected with either 1μg of double-stranded NOS RNA (dsRNA) or water only (control) were infected with either wild-type B. cereus or the ∆mfd mutant and insect mortality was recorded after 24 h. (D) C57/Bl6/Sev129 mice (10 Mice wt) and NOS2−/− mice (10 KO Mice) were inoculated intranasally with B. cereus wild-type and ∆mfd mutant strains (5.106 cfu/mice). Mortality was recorded daily for 7 days.
In a second type of experiment, bacteria were exposed directly to chemically generated acidified nitrite in a cell-free system (Fig. 3B). The wild-type strain survived, whereas survival of the ∆mfd mutant was impaired (P < 0.005). The concentration of NO able to reduce by 50% the survival of the strains (IC 50) was measured by dose-response experiments. The NO-IC 50 required for the wild type strain was of 4.12 ± 1.2 mM and was thus 2.5 times higher that the IC 50 for the mfd mutant, which was 1.61 ± 1.3 mM. These data implicate Mfd in the resistance to NO, a critical mediator of the host innate immune response. We tested the impact of other mutagenic agents on wild type and mfd mutant strain survival. The mfd mutant was not sensitive to UV or mitomycin treatments (not shown), consistent with previous reports showing in the ∆mfd mutant a high UV mutability despite a minimal UV or mitomycin sensitivity30,31.
It has been previously reported that the expression of mfd in B. subtilis is upregulated after ethanol or H2O2 exposure (Basysbio, http://genome.jouy.inra.fr/cgi-bin/seb/index.py). We measured the impact of NO on B. cereus mfd gene regulation by measuring the β-glucoronidase activity of a transcriptional fusion between the mfd promoter and the gus gene (Fig. 4). The mfd promoter showed a constitutive activity that increased throughout bacterial growth in the absence of NO. In the presence of NO, mfd gene expression was significantly upregulated.
Figure 4. mfd gene expression was assessed by measuring the ß-glucoronidase activity of the Bc407 [pHT304G-pmfd’gus] strain grown in LB medium at 37 °C with or without NO.
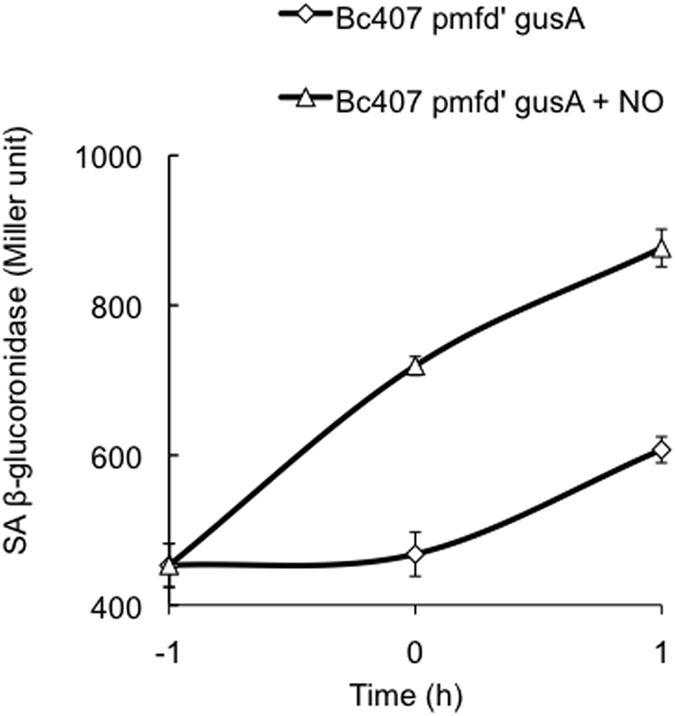
Samples were taken every hour from one hour before to one hour after the entry of the bacteria in the stationary growth phase. Specific activity is expressed in units of β-glucuronidase per milligram of protein (Miller units). The results reported are mean values of three independent experiments.
Mfd is required for B. cereus resistance to the host immune NO response
To determine whether the mfd mutant growth was inversely correlated with the host NO response in vivo, we used two animal models: B. mori larvae and mice. NO production in insects was shut down by injection of NOS-siRNA into the insect hemocoel prior to infection with wild-type and mfd mutant strains. Inhibition of NO production had no significant effect on insect mortality after infection by the wild-type strain, indicating that this strain counteracts the NO host defense (Fig. 3C). In contrast, inhibition of NO production significantly increased the virulence of the ∆mfd strain compared with insects producing an NO-response (P < 0.03). Thus, Mfd is essential for virulence in the context of NO-stress in vivo. The mortality induced by the mfd mutant in the context of NO inhibition was intermediate and did not reach the level of mortality induced by the wild type strain (P > 0.067) indicating that some other lesions, independent of NO, may also be dealt with by Mfd.
Wild-type and ∆mfd strain virulence was also assessed in wild-type mice and in mice deficient for NO production (iNOS-KO) (Fig. 3D). The percentage of surviving wild-type mice infected with the B. cereus wild type strain decreased sharply following infection, while the survival of wild-type mice infected with the ∆mfd mutant was significantly higher. These results show that Mfd makes a substantial contribution to B. cereus pathogenesis in a mammalian model of infection. The survival of iNOS-KO mice infected with wild type and ∆mfd mutant strains was similar to the survival of the wild-type mice infected with the wild-type strain. Thus, in the absence of NO production, the mfd deletion had no effect on virulence. These findings imply that Mfd counteracts the deleterious effect of NO on bacteria in vivo.
Our study as a whole provides significant insights into the previously undescribed role of Mfd during bacterial pathogenesis and, in particular, demonstrates its involvement in the mechanisms of defense against NO stress in vivo.
Mfd as a universal virulence factor implicated in NO stress resistance
To determine whether mfd inactivation affects virulence in another bacterial species, we studied the effect of mfd deletion in Shigella flexneri. S. flexneri is a Gram-negative Bacillus and is the major etiological agent of dysentery in developing countries32,33. NO is produced following S. flexneri infection but does not result in clearance of S. flexneri from infected mice, suggesting that bacteria escape the NO response34. To study the role of Mfd in the resistance of Shigella to NO stress, we evaluated bacterial growth after incubation with phagocytic PLB cells, with and without the NO inhibitor NMMLA (Fig. 5A). In the absence of NMMLA, the wild-type strain was able to grow, whereas growth of the Shigella ∆mfd mutant was severely impaired (P < 0.004). Addition of the NO inhibitor increased the survival of the ∆mfd mutant (P < 0.009) to a level similar to that of the wild-type strain (P > 0.13).
Figure 5.

(A) PLB cells were infected with S. flexneri wild-type (Shig) and the mfd mutant (Shig ∆mfd) in the presence or absence of 1 mM NMMLA for 24 h. Bacterial survival was calculated by plating dilutions on LB agar plates. Cfu counts were normalized to initial cfu. The results reported are mean values of at least three independent experiments each in triplicate. P values are calculated using the Student test. (B) Bacteria were exposed to chemically generated NO (500 μM sodium nitrite) for 4 h in a cell-free system. Bacteria were harvested and plated on agar plates to evaluate bacterial survival. Cfu counts were normalized to initial cfu at t0. The results reported are mean values of at three independent experiments. P values are calculated using the Student test. (C) C57/Bl6/Sev 129 mice (9 Mice wt) and NOS2−/− mice (9 Mice KO) were inoculated intranasally with S. flexneri wild-type and ∆mfd mutant bacteria (107 cfu/mice). Mortality was recorded daily for 9 days.
Survival of the ∆mfd mutant was further tested in NO stress conditions in a cell-free system (Fig. 5B). The growth of the Shigella ∆mfd mutant in the presence of nitrite was much weaker than that of the wild-type strain (P < 0.02). The survival of the wild type and mutant S. flexneri strains in the cell free system without nitrite exposure was identical (Fig. 5B).
The role of Mfd in resistance to NO stress was also tested in vivo in wild type and iNOS-KO mice (Fig. 5C). Survival of wild-type mice infected with the ∆mfd mutant was significantly higher than survival of these mice infected with the wild-type Shigella strain. Thus, Mfd is required for full Shigella virulence. In mice deficient for NO production, both wild type and ∆mfd mutant strains were highly virulent, and mouse survival was even lower than for wild-type mice.
These experiments demonstrate that Mfd plays an important role in promoting bacterial survival in the context of NO-stress during the infection process of two very divergent bacteria.
NO stress results in DNA damage in the ∆mfd mutant
The role of NO during infection clearance has been described in several bacteria and several molecular mechanisms involved in the repair of NO-induced DNA lesions have also been identified7,8,9,35,36. However, the involvement of Mfd during the repair of NO-induced lesions has never been reported. Mfd is involved in DNA repair, and we report that Mfd contributes to the resistance of B. cereus and Shigella to NO, therefore, Mfd may be required to repair DNA damage resulting from NO stress during infection. Pulse field gel electrophoresis (PFGE) has been widely used to detect the formation of linear chromosomal fragments. Indeed, cell lysis performed in the plugs ensures that only linear chromosomes enter the gels, while circular molecules remain in the wells37,38.
In the absence of NO, linear DNA was not detected, neither in the wild type nor in the ∆mfd strains (Fig. 6). However, addition of NO to the ∆mfd mutant strain resulted in the appearance of linear DNA reflecting chromosome breakage. Moreover, NO treatment induced DNA degradation in the ∆mfd mutant, visualized as a smear during electrophoresis. Thus, the presence of NO induces DNA fragmentations and Mfd is required to prevent or repair them.
Figure 6. Chromosomal linear DNA was visualized in B. cereus by pulsed field gel electrophoresis.
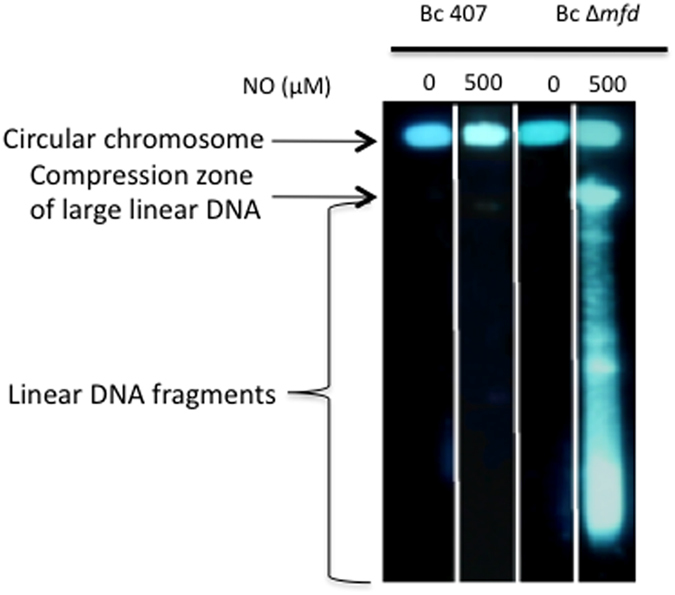
Wild-type and ∆mfd mutant strains were grown until mid exponential growth phase and treated with 500 μM NO in a cell free system. DNA corresponding to 108 bacteria was incorporated in low melting agar gel plots and analyzed by pulsed field electrophoresis (PFGE). Circular chromosomal DNA are blocked in the loading well, whereas linear DNA migrate through the gel. DNA degradation is visible as a smear below the linear DNA band. This shows representative data out of 5 independent experiments.
Mfd helps to counteract the mutagenic effect of NO
No specific pattern of mutations has yet been attributed to NO stress. We aimed to identify the rate and type of mutations and the implication of Mfd in the repair of these DNA damages. To assess the rate and type of NO-induced mutations, we constructed B. cereus strains expressing gusA and measured the percentage of strains loosing their blue color on X-glucuramidase plates after sub-lethal in vitro NO treatment. In the presence of tetracycline, which ensures that all colonies carry a plasmid, no white colonies were found with the wild-type B. cereus strain, whereas in the ∆mfd mutant, 1±0.4 colonies per 2.8 × 103 were white. The PaphA3-gusA region of the plasmids (including the promoter region and the gusA coding sequence) from 16 independent non-clonal colonies was sequenced. Three types of mutation were identified, all of them being transversions (G:C to T:A, A:T to C:G and C:G to G:C), mapping at four different sites of the cloned region (Table 1).
Table 1. Bc 407 {pHT1618’P aphA3 -gusA} and Bc 407 ∆mfd {pHT1618’P aphA3 -gusA} were subjected to sublethal NO treatment (125 μM) for 2 h, then plated on LB-X-glucuramidase plates supplemented with tetracycline (10 μg/mL).
| Clone number | Position# | Transition+ | Amino acid change in the GUS protein* |
|---|---|---|---|
| 1 | 643 | G:C to T:A | Gly to Val |
| 2 | 643 | G:C to T:A | Gly to Val |
| 3 | 1646 | C:G to G:C | Tyr to Stop |
| 4 | 1646 | C:G to G:C | Tyr to Stop |
| 5 | 1646 | C:G to G:C | Tyr to Stop |
| 6 | 1646 | C:G to G:C | Tyr to Stop |
| 7 | 1646 | C:G to G:C | Tyr to Stop |
| 8 | 1646 | C:G to G:C | Tyr to Stop |
| 9 | 1646 | C:G to G:C | Tyr to Stop |
| 10 | 1646 | C:G to G:C | Tyr to Stop |
| 11 | 1651 | A:T to C:G | Tyr to Ser |
| 12 | 1651 | A:T to C:G | Tyr to Ser |
| 13 | 1651 | A:T to C:G | Tyr to Ser |
| 14 | 1651 | A:T to C:G | Tyr to Ser |
| 15 | 2377 | G:C to T:A | Arg to Leu |
| 16 | 2377 | G:C to T:A | Arg to Leu |
| Control plasmid | No mutation | ||
| Control plasmidfrom ∆mfd in LB | No mutation | ||
| Control bluecolonies | No mutation |
#Base position on the gus gene.
+Change in base pair compared to non mutated gene.
*Corresponding change in amino acid in the Gus protein.
The pHT1618’PaphA3-gusA plasmid was isolated from white colonies (only the ∆mfd {pHT1618′PaphA3-gusA} strain gave white colonies), used to transform E. coli TG1, and isolated from the E. coli transformants; the PaphA3-gusA region was then sequenced. Position of mutations (from 1 to 2537 bp, corresponding to the sequenced fragment), base pair changes and the corresponding amino acid changes are indicated for each sequenced region. As controls, the corresponding regions in the initial plasmid, in the plasmid isolated from the ∆mfd {pHT1618′PaphA3-gusA} strain grown for 2 h in LB medium, and in plasmids isolated from blue colonies after NO treatment were also sequenced.
To ensure that the mutations were not generated during replication in E. coli we performed several controls, including the sequencing of plasmids issuing from blue colonies after NO treatment, and control plasmids from ∆mfd strains grown in LB medium without NO treatment. In both cases, we observed no mutation despite replication in E. coli.
Taken together our data show that in the absence of Mfd, NO stress causes at least three types of base substitutions that occur preferentially at specific positions.
Discussion
This study reports the crucial role of the Mfd protein during pathogenesis of bacteria as different as B. cereus and S. flexneri in both invertebrate and mammal hosts. Despite the numerous publications addressing the importance of Mfd in DNA repair and adaptive mutagenesis, to our knowledge its importance during bacterial pathogenesis has not been previously reported.
We demonstrate that Mfd is essential for specific resistance to the deleterious effect of nitrogen stress imposed by host phagocytes. We observed that in B. cereus, in addition to its basal constitutive expression, mfd gene expression is upregulated in the presence of NO. This is in agreement with previous findings using a tiling DNA microarray covering the entire chromosome of B. subtilis and showing a multiple regulatory system of mfd gene expression (http://subtiwiki.uni-goettingen.de/wiki/index.php/Mfd): mfd expression is controlled by SigA and SigB, two regulators implicated in “house keeping” and stress-induced response, respectively39,40. A SigB-dependent regulation is in agreement with the higher expression of mfd in NO stress conditions.
Production of reactive nitrogen species, such as NO is an important component of the host immune defense against bacteria. NO has toxic effects on several molecules including nucleic acids, thereby inducing DNA damage and strand breaks. We demonstrate a link between NO-induced bacterial DNA damage and DNA repair by Mfd. The appearance of linear DNA during the PFGE experiments suggests that NO induces DNA double-strand breaks (DSBs) in the ∆mfd mutant37,38. DSBs are readily detected by PFGE in cells deficient for RecBCD-dependent homologous recombination, owing to the absence of DSB repair38. This is in agreement with previous findings showing the implication of RecBC in NO resistance of Salmonella enterica9. We hypothesize that Mfd could remove a RNAP arrested by a DNA lesion, or repair protein complexes, from the path of recombination-dependent replication forks after fork breakage due to replication blockage.
Previous findings on Salmonella typhimurium showed that NO induces DNA damage targeted by the Base Excision Repair (BER) pathway8. BER is involved in the recognition of modified bases by specific DNA glycosylases, which action on NO-induced DNA damage can induce homologous recombination35. As the formation of DSBs is usually linked to the activation of RecBC-dependent homologous recombination38, it would be interesting to study the link between Mfd, RecBC and BER following NO genotoxic bacterial DNA damage.
Absence of mfd reveals three types of mutation after NO treatment, all of them being transversions (G:C to T:A, A:T to C:G and C:G to G:C). G:C to T:A and A:T to C:G transversions are usually induced by loss of MutT, Fpg and MutY enzymes, which all deal with G oxidation. C:G to G:C probably also involves NO-induced G lesion35,36. Thus, NO treatment in a mfd mutant generates a high level of DNA fragmentation and increases point mutation frequency. These two phenomena might occur independently of each other, or RecBC-dependent DNA replication may be mutagenic in our conditions.
We speculate that the NO damages occur rapidly and throughout the bacterial chromosome. Indeed, B. cereus does not possess any pathogenicity island and we observed that the mfd mutant survival is completely impaired in vivo, suggesting a rapid and considerable extent of damage. Alternatively, Mfd may also have a general impact on virulence by indirectly controlling the production of virulence genes. For instance, in Clostridium difficile, Mfd controls the level of toxin gene expression maybe by relieving RNAP stalled at roadblocks created by the toxin repressors CodY and CcpA41.
Taken together, our findings demonstrate an important role of the DNA repair enzyme Mfd during bacterial resistance to NO stress, by limiting the genotoxic effects of NO generated by the host inflammatory response. These findings allow better defining the mechanistic pathway of bacterial resistance to NO and the efficient contribution of Mfd to bacterial survival during pathogenesis. Mfd is well conserved among bacterial species and these observations reveal a mechanism that may be used by a large spectrum of bacteria to overcome host immune responses, and in particular the damaging properties of reactive nitrogen species.
Materials and Methods
Ethics Statement
All experimental protocols were conducted in compliance with French legislation and approved by the author local ethic committee, named Comethea under the agreement number: B78–720. The experiments were performed by INRA-UIERP animal facilities in Jouy en Josas, France (N° SIRET INRA Jouy en Josas: 18 0070039 00078) in conformity with ethics regulations as established by EU legislation (2010/63/EU).
No samples were obtained from human patient for this study.
Genomic mutant bank screening and identification of avirulent mutants
The B. thuringiensis strain 407 Cry− (Bc 407) was used as a model for B. cereus. This strain has been cured of its plasmid, is acrystalliferous, and shows high phylogenic and phenotypic similarity with the B. cereus reference strain ATCC 14579 and is therefore considered as a B. cereus strain42. A random insertion mutagenesis library was constructed in Bc 407 by using the mini-Tn10 transposon as previously described43. Insertion mutants were screened for loss of virulence in Bombyx mori larvae, following their inoculation into the hemocoel28,44. Chromosomal DNA regions flanking the insertion loci were cloned, and their nucleotide sequences were determined as previously described43.
Bacterial strain and mutant construction
The Bc 407 ∆mfd mutant was constructed as follows. The mfd gene was disrupted through double homologous recombination using the thermosensitive vector pMAD. BamHI-XbaI (515 bp) and PstI-EcoRI (512 bp) DNA fragments corresponding to upstream and downstream regions of the mfd gene were generated from the Bc 407 chromosome by PCR using the primer pairs:
mfd-1 (5′-CGCGGATCCGTAGGCTCCATTAACGCAG-3′),
mfd-2 (5′-GCTCTAGACACCAAGTAACGCCACTAAATC-3′), and
mfd-3 (5′-AACTGCAGGAGGTGTTTCTGCAATTGAGG-3′),
mfd-4 (5′-CCGGAATTCGCTGCCTCATTTCTACTG-3′).
A KanR cassette carrying a kan gene was purified from pDG78345 as a 1.6 Kb XbaI-PstI fragment. The amplified DNA fragments and the KanR cassette were digested with the appropriate enzymes and assembled together by ligation to produce a “mfd-upstream”-“KanR cassette”-“mfd-downstream” BamHI-EcoRI fragment, which was then inserted between the BamHI and EcoRI sites of pMAD. The resulting plasmid was introduced into Bc 407 by electroporation46 and the mfd gene was deleted by a double crossover event as previously described47. Chromosomal allele exchange was confirmed by PCR with oligonucleotide primers located upstream from mfd-1, mfd-5 (5′-AACTGCAGGCAGACACTGCGGAGG-3′) and downstream from mfd-4, mfd-6 (5′-TGCTCTAGACCTTCGGGATTACTACCCTGCC-3′), and in the KanR cassette (5′-CGGGTCGGTAATTGGGTTTG-3′), (5′-GCAGCTGCACCAGCCCCTTG-3′). The insertion mutant strain was designated Bc 407 ∆mfd.
To complement the Bc 407 ∆mfd mutant, the mfd gene, including the coding sequence and the promoter region (Fig. 1C and http://subtiwiki.uni-goettingen.de/wiki/index.php), was obtained using the primer pair mfd-5 and mfd-6, and the sequence of the obtained fragment was verified. The pHT315 cloning vector46 containing the complete mfd gene and its promoter was used to transform Bc 407 Δmfd strain by electroporation. Transformants were selected for resistance to erythromycin. The resulting new strain was designated Bc 407 Δmfd/mfd+.
In Bacilli, the mfd gene is located between a gene coding for a peptidyl tRNA hydrolase and a gene coding for an AbrB-like transcription regulator (spoVT). The spoVT gene situated immediately downstream of mfd was amplified using the primers (5′-AACTGCAGGAGGTGTTTCTGCAATTGAGG-3′), and (5′TGCTCTAGACCCCACGCCAAAAGGCTTG-3′). This fragment contained the spoVT gene with its own promoter as defined in http://subtiwiki.uni-goettingen.de/wiki/index.php, and was inserted into the PstI and XbaI sites of the pHT315 cloning vector. The pHT315 vector containing the spoVT gene with its own promoter was introduced into the ∆mfd strain by electroporation. This new strain was designated Bc 407 Δmfd/spoVT+.
Transcriptional mfd′gusA fusion was constructed using DNA fragments corresponding to the promoter region of mfd generated by PCR using the primer pair pmfd-1 (5′-AGAAACGCTCGCATCTTATCCTG-3′) and pmfd-2 (5′-AGACGTTGCCATCCCTGATATAAG-3′). The promoter region was then inserted in the pHT304-18GusA plasmid48. The recombinant plasmid was introduced into Bc 407 by electroporation. Transformant was named Bc407 [pHT304G-pmfd’gus].
Strains Bc 407 {pHT1618′ PaphA3-gusA} and Bc 407 ∆mfd {pHT1618′ PaphA3-gusA} were constructed as follows. To construct pHT1618′ PaphA3-gusA, the constitutive promoter of the KanR cassette (PaphA3) was amplified as a 571 bp PstI-XbaI fragment from pDG783 using the primers papha3-1 (5′-GCATGCCGTCAGGTGATAAACC-3′) and papha3-2 (5′-GCTCTAGACAATTCCGGTGATATTCTCATTTTAGCC-3′), and the gusA gene was amplified by PCR from pTUM17749 with primers GUS1 (5′-TGCTCTAGATAAAGGAGAAAATTTTATGTTACGTCC-3′) and GUS2 (5′-CCGGAATTCGGTGCGCCAGGAGAGTTG-3′) as a XbaI-EcoRI fragment. These fragments were inserted between the PstI and EcoRI sites of pHT1618, which was used to transform the strains Bc 407 and Bc 407 ∆mfd.
The Shigella flexneri 2a M90T strain was used as the wild-type reference strain. A ∆mfd mutant of this strain was constructed as follows. The mfd gene (S1198) was interrupted by double recombination as previously described50. Upstream and downstream regions of the mfd gene were generated from the S. flexneri 2a M90T chromosome by PCR using the primer pairs: S-mfd-1 (5′-GCTCTAGAGCGCTCCACCAGCCTGCTG-3′) and S-mfd-2 (5′-CGGGGTACCCCGCGCGTGGCGTATTCGCCG-3′), and S-mfd-3 (5′-CGCGGATCCCGGCCTGCTGCCAGATCCGGC-3′) and S-mfd-4 (5′-CCGGAATTCCGGGCGCGGATGTTTGCCG-3′). A KanR cassette carrying a kan gene was excised from pUC18 K2 as a 839 bp fragment. The DNA fragments were digested with the appropriate enzymes and inserted into pGP70450. The resulting plasmid was introduced into M90T by electroporation and the mfd gene was deleted by a double crossover event. Chromosomal allele exchange was confirmed by PCR with oligonucleotide primers located upstream from S-mfd-1, S-mfd-5 (5′-CCCAAGCTTAAGAGGTGCCGTTGCGCCGCC-3′) and downstream from S-mfd-4, S-mfd-6 (5′-TCCCCCGGGGGGTTATCCGGTCCAGCCGGC-3′), and in the KanR cassette (5′-GCAAGGCGATTAAGTTGGGTAACGCCAGGG-3′), (5′-CCCCGCGCGTTGGCCGATTCATTAATGCAG-3′). The insertion mutant strain was designated S. flexneri ∆mfd. Growth curves of wild type and mutant strain were similar in LB or RPMI medium at 37 °C.
NO inducer and inhibitor
Bacteria were exposed directly to chemically generated NO + in a cell-free system as described by Miyagi et al.51 with modifications. Briefly, bacteria were grown in LB medium until mid exponential growth phase at 37 °C with shaking. Cultures were then diluted to 104 cfu/ml in RPMI-1640 medium (Invitrogen) at acidic pH (pH 5.4) in the presence of 0 to 10 mM sodium nitrite (Walco Pure Chemical Industries Ltd); under these acidic conditions, NO+ is generated from the sodium nitrite by a chemical reaction51. The bacteria were then cultured at 37 °C for 0 to 4 h, harvested and plated on agar plates to evaluate bacterial survival. Using this cell free assay, the NO+ concentration required to decrease by 50% the survival rate of each strain (IC50) was calculated using the GraphPad PRISM software (version 6.0, GraphPad Software, San Diego, CA) with non-linear regression. Data are means of at least 5 independent experiments.
To inhibit the nitrogen response in cellular assay, the NO inhibitor NMMLA NG-monomethyl-L-arginine (Sigma) was used at 1 mM. NMMLA competes with L-arginine for the site of action of the iNOS enzyme.
UV and mitomycin treatment
B. cereus wild type and mutant strains were grown at 37 °C under agitation until mid exponential growth phase and diluted to 5. 107 cfu/ml. Serial dilutions were plated as spots of 10 μl on agar plates and exposed to UV light (5J/m2) for 0 to 15 seconds. Alternatively, serial dilutions were plated on agar plates containing 50 ng/mL mitomycin C. Plates were incubated ON at 37 °C and bacterial survival was assessed by observing the size of the spot colonies on the plates for each dilution.
β-glucuronidase assays
The Bc407 [pHT304G-pmfd’gus] was grown in LB medium, pH 5.4 at 37 °C until one hour before the bacterial entry into stationary phase (t-1). At t-1, 0.4 mM NO was added to the culture and samples were taken every hour from one hour before to one hour after the entry of the bacteria in the stationary growth phase. Determination of β-glucoronidase activity was achieved as previously described48. Specific activities are expressed in units of β-glucuronidase per milligram of protein (Miller units).
Cell culture and infection
The human monocytic-like cells, PLB-985 and PLB deficient for Gp91phox, one of the multicomponents of the enzyme NADPH oxidase responsible for ROS production (gp91phox−/−), were used (kindly provided by Dr. Benaroch, Institut Curie, Paris, originating from M. Dinauer laboratory52). Cells were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS, Invitrogen). Cells were incubated at 37 °C under a 5% CO2 atmosphere and saturating humidity. The day before use, cells were counted with a hematocytometer and seeded at a density of 3.105 cells per well into multiwell disposable trays. Cells were infected with bacteria at a multiplicity of infection of 10 at 37 °C. After 4 h or 24 h bacteria and cells were removed from the flask and bacteria were counted by plating serial dilutions on agar plates.
Nitrite concentrations were measured by the Griess method: supernatant collected after each time of infection tested, and a standard, were mixed with an equal volume of Griess reagent (Sigma) and incubated for 10 min in the dark. The absorbance was then measured at 550 nm. The standards were obtained by serial dilution of NO.
Insects and in vivo experiments
Bombyx mori larvae were infected by injection into the hemolymph as described elsewhere44,53. Groups of 20 last-instar larvae (same weight) were injected at the base of their last proleg with 10 μL of suspensions containing various doses of vegetative bacteria. Insect mortality was recorded after 24 h at 25 °C. To estimate the number of bacteria in living or dead larvae, the insect larvae were crushed and homogenized in sterile water; dilutions were plated onto LB agar plates54,55.
RNAi in insects
NO production can be silenced at the transcription level by using RNA interference (RNAi)56. The silencing of gene expression by double-stranded RNA molecules is very efficient in B. mori57, and the gene coding for the inducible nitric oxide synthase-like protein (iNOS-LP) is present in only a single copy (in contrast to other invertebrate immune mechanisms for which many genes are present in several copies). Thus, RNAi is a powerful tool for manipulating NO levels in infected B. mori58. Double-stranded RNA (dsRNA) was produced from genomic DNA of B. mori using the primers 5′-ATTATGCTGAGTGATATCCCTCGAAGTTCTCGTCGTGAGCTA-3′ and 5′-TAATACGACTCACTATAGGGGAGAACCTCAGGAAGATGGATC-3′ and the Megascript RNAi Kit (Ambion). Larvae injected with either 1 μg of double-stranded NOS RNA (dsRNA) or water only (control) were infected with either wild-type Bc 407 or the ∆mfd mutant (50 cfu/larvae) and mortality was recorded after 24 h.
Mouse experiments
Mice experiments were performed by INRA-UIERP animal facilities in Jouy en Josas, France. C57/BL6/Sev129 mice, aged 6- to 8-weeks old were used for infections. Wild-type mice were obtained from Elevage Janvier, France. Mice with a targeted disruption of the NOS-2 gene, generated as previously described59 were generously supplied by Drs. J. D. MacMicking, C. Nathan (Cornell University Medical College, New York) and Jean Claude Jeanny, Institut des Cordeliers, animalerie centrale de la faculté de Pharmacie de Paris (health monitoring report from Harlan UK Technical service). PCR with DNA from tail biopsies was used for genotyping to verify the presence or absence of the NOS-2 gene60. Groups of 9 to 10 mice were inoculated intranasally with wild type or mutant bacteria (5.106cfu/mouse for B. cereus, and 107cfu/mouse for Shigella strains) as described in34,61. Mortality was recorded daily for 7–9 days.
Pulsed-field gel electrophoresis (PFGE)
DNA damage was evaluated by pulsed field electrophoresis as described62 with slight modifications. Bc 407 and Bc 407 ∆mfd strains were grown until mid exponential growth phase at 37 °C with agitation. Bacteria where then submitted to NO stress in the cell free system with 500 μM NO for 4 h at 37 °C. For each condition, 108 bacteria were harvested by centrifugation, incorporated into low-melting agar gel plugs, and lyzed. The DNA released was analyzed by electrophoresis in 1% agarose gels in 0.5X TBE at 6 V/cm for 24 h, with a linear pulse ramp of 3 to 52 s and a switching angle of 120°. Cell lysis performed in the plugs ensures that only linear chromosomes enter the gels, while circular molecules remain in the wells. All large linear fragments (usually >3 Mb) are comprised in the compression zone and thus appear as a singe band. DNA fragmentation and degradation appears as several bands or smear below this single band of linear DNA63.
Mutation rate and type
Bacterial strains were grown until mid exponential growth phase at 37 °C with agitation. Bacteria where then submitted to sublethal NO treatment (125 μM) for 2 h at 37 °C. To assess the rate and type of mutations caused by NO, we constructed B. cereus strains expressing gusA: Bc 407 {pHT1618′PaphA3-gusA} and Bc 407 ∆mfd {pHT1618’PaphA3-gusA}. These strains grow as blue colonies on LB-X-glucuramidase plates. The rate of non-neutral mutations specifically in the PaphA3-gusA gene was measured by scoring the appearance of white colonies on LB X-gluc plates supplemented with tetracycline (10 μg/mL) to maintain the pHT1618-derived plasmid. The pHT1618′ PaphA3-gusA plasmid was isolated from the white B. cereus ∆mfd colonies, and used to transform E. coli TG1. The plasmid was isolated from the E. coli transformants, and the PaphA3-gusA region was sequenced to identify the mutations obtained. To verify that the mutations observed led to a non-functional PaphA3-gusA gene, several plasmids isolated from white colonies were used to transform E. coli, re-isolated from E. coli and reintroduced into the Bc ∆mfd strain. In all cases, the plasmids conferred a white phenotype on LB X-gluc plates as expected (not shown). The original pHT1618′ PaphA3-gusA plasmid was used as control, and conferred a blue phenotype on LB X-gluc plates as expected.
Additional Information
How to cite this article: Guillemet, E. et al. The bacterial DNA repair protein Mfd confers resistance to the host nitrogen immune response. Sci. Rep. 6, 29349; doi: 10.1038/srep29349 (2016).
Supplementary Material
Acknowledgments
We are indebted to the Unité d’Infectiologie Experimentale des Rongeurs et Poissons (IERP), INRA, Jouy en Josas, France, for performing the experiments with mice and to J.C. Jeanny for generously providing the NOS-KO mice. We thank A. Moris and Dr. Benaroch for the gift of the PLB cells. We thank S. Perchat for helpful discussions and technical assistance. We thank R. Dervyn for helpful technical assistance. We thank E. Dervyn and N. Jacques for their help in the pulsed field experiments. We are indebted to B. Michel, MA. Petit and I. Matic for very helpful discussions and proofreading of the manuscript.
Footnotes
Author Contributions E.G., A.L., S.-L.T., C.R., I.B. and N.R. performed experiments; P.S., D.L. and N.R. set up experiments and analyzed the data; N.R. wrote the manuscript; all authors reviewed the manuscript.
References
- Amoroso A., Crespan E., Wimmer U., Hubscher U. & Maga G. DNA polymerases and oxidative damage: friends or foes? Curr Mol Pharmacol 1, 162–170 (2008). [DOI] [PubMed] [Google Scholar]
- Zaki M. H., Akuta T. & Akaike T. Nitric oxide-induced nitrative stress involved in microbial pathogenesis. J Pharmacol Sci 98, 117–129 (2005). [DOI] [PubMed] [Google Scholar]
- Akuta T., Zaki M. H., Yoshitake J., Okamoto T. & Akaike T. Nitrative stress through formation of 8-nitroguanosine: insights into microbial pathogenesis. Nitric Oxide 14, 101–108 (2006). [DOI] [PubMed] [Google Scholar]
- Yoshitake J. et al. Nitric oxide as an endogenous mutagen for Sendai virus without antiviral activity. J Virol 78, 8709–8719 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang J. C., Lin C., Lin D. & Wogan G. N. Mutagenesis associated with nitric oxide production in macrophages. Proc Natl Acad Sci USA 95, 8286–8291 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laver J. R. et al. Bacterial nitric oxide detoxification prevents host cell S-nitrosothiol formation: a novel mechanism of bacterial pathogenesis. FASEB J 24, 286–295 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorkina O., Saparbaev M. & Laval J. Effects of nitrous acid treatment on the survival and mutagenesis of Escherichia coli cells lacking base excision repair (hypoxanthine-DNA glycosylase-ALK A protein) and/or nucleotide excision repair. Mutagenesis 12, 23–28 (1997). [DOI] [PubMed] [Google Scholar]
- Richardson A. R. et al. The Base Excision Repair system of Salmonella enterica serovar typhimurium counteracts DNA damage by host nitric oxide. PLoS Pathog 5, e1000451 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapiro J. M., Libby S. J. & Fang F. C. Inhibition of bacterial DNA replication by zinc mobilization during nitrosative stress. Proc Natl Acad Sci USA 100, 8496–8501 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts J. & Park J. S. Mfd, the bacterial transcription repair coupling factor: translocation, repair and termination. Curr Opin Microbiol 7, 120–125 (2004). [DOI] [PubMed] [Google Scholar]
- Deaconescu A. M. et al. Structural basis for bacterial transcription-coupled DNA repair. Cell 124, 507–520 (2006). [DOI] [PubMed] [Google Scholar]
- Million-Weaver S. et al. An underlying mechanism for the increased mutagenesis of lagging-strand genes in Bacillus subtilis. Proc Natl Acad Sci USA 112, E1096–1105 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. J. & Savery N. J. Effects of the bacterial transcription-repair coupling factor during transcription of DNA containing non-bulky lesions. DNA Repair (Amst) 7, 1670–1679 (2008). [DOI] [PubMed] [Google Scholar]
- Savery N. J. The molecular mechanism of transcription-coupled DNA repair. Trends Microbiol 15, 326–333 (2007). [DOI] [PubMed] [Google Scholar]
- Lee G. H. et al. The Helicobacter pylori Mfd protein is important for antibiotic resistance and DNA repair. Diagn Microbiol Infect Dis 65, 454–456 (2009). [DOI] [PubMed] [Google Scholar]
- Ross C. et al. Novel role of mfd: effects on stationary-phase mutagenesis in Bacillus subtilis. J Bacteriol 188, 7512–7520 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pybus C. et al. Transcription-associated mutation in Bacillus subtilis cells under stress. J Bacteriol 192, 3321–3328 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Guadiana F. H. et al. Transcriptional coupling of DNA repair in sporulating Bacillus subtilis cells. Mol Microbiol 90, 1088–1099 (2013). [DOI] [PubMed] [Google Scholar]
- Ayora S., Rojo F., Ogasawara N., Nakai S. & Alonso J. C. The Mfd protein of Bacillus subtilis 168 is involved in both transcription-coupled DNA repair and DNA recombination. J Mol Biol 256, 301–318 (1996). [DOI] [PubMed] [Google Scholar]
- Han J., Sahin O., Barton Y. W. & Zhang Q. Key role of Mfd in the development of fluoroquinolone resistance in Campylobacter jejuni. PLoS Pathog 4, e1000083 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz R. T. & O’Donnell M. Direct restart of a replication fork stalled by a head-on RNA polymerase. Science 327, 590–92 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges B. A. Starvation-associated mutation in Escherichia coli strains defective in transcription repair coupling factor. Mutat Res 329, 49–56 (1995). [DOI] [PubMed] [Google Scholar]
- Ramarao N. et al. Two unrelated episodes of Bacillus cereus bacteremia in a neonatal intensive care unit. Am J Infect Control 42, 694–695 (2014). [DOI] [PubMed] [Google Scholar]
- Decousser J. et al. Bacillus cereus and severe intestinal infections in preterm neonates: putative role of pooled breast milk. Am J Infect Control 41, 918–921 (2013). [DOI] [PubMed] [Google Scholar]
- Cadot C. et al. InhA1, NprA and HlyII as candidates to differentiate pathogenic from non-pathogenic Bacillus cereus strains. J Clin Microbiol 48, 1358–1365 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamar R. et al. Pathogenic potential of B. cereus strains as revealed by phenotypic analysis. J Clin Microbiol. 51, 320–323 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran S. L. & Ramarao N. Bacillus cereus immune escape: a journey within macrophages. FEMS Microbiol Lett 347, 1–6 (2013). [DOI] [PubMed] [Google Scholar]
- Fedhila S., Guillemet E., Nel P. & Lereclus D. Characterization of two Bacillus thuringiensis genes identified by in vivo screening of virulence factors. Appl Environ Microbiol 70, 4784–4791 2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalow B. J., Courcelle C. T. & Courcelle J. Mfd is required for rapid recovery of transcription following UV-induced DNA damage but not oxidative DNA damage in Escherichia coli. J Bacteriol 194, 2637–2645 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epshtein V. et al. UvrD facilitates DNA repair by pulling RNA polymerase backwards. Nature 505, 372–377 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- George D. L. & Witkin E. M. Ultraviolet light-induced responses of an mfd mutant of Escherichia coli B/r having a slow rate of dimer excision. Mutat Res 28, 347–354 (1975). [DOI] [PubMed] [Google Scholar]
- Zychlinsky A., Prevost M. C. & Sansonetti P. J. Shigella flexneri induces apoptosis in infected macrophages. Nature 358, 167–169 (1992). [DOI] [PubMed] [Google Scholar]
- Sansonetti P. J. & Di Santo J. P. Debugging how bacteria manipulate the immune response. Immunity 26, 149–161 (2007). [DOI] [PubMed] [Google Scholar]
- Way S. S. & Goldberg M. B. Clearance of Shigella flexneri infection occurs through a nitric oxide-independent mechanism. Infect Immun 66, 3012–3016 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spek E. J., Vuong L. N., Matsuguchi T., Marinus M. G. & Engelward B. P. Nitric oxide-induced homologous recombination in Escherichia coli is promoted by DNA glycosylases. J Bacteriol 184, 3501–3507 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano T. et al. Repair activity of base and nucleotide excision repair enzymes for guanine lesions induced by nitrosative stress. Nucleic Acids Res 33, 2181–2191 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzminov A. & Stahl F. W. Double-strand end repair via the RecBC pathway in Escherichia coli primes DNA replication. Genes Dev 13, 345–356 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel B., Ehrlich S. D. & Uzest M. DNA double-strand breaks caused by replication arrest. EMBO J 16, 430–438 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber T. M. & Gross C. A. Multiple sigma subunits and the partitioning of bacterial transcription space. Annu Rev Microbiol 57, 441–466 (2003). [DOI] [PubMed] [Google Scholar]
- van Schaik W. & Abee T. The role of sigmaB in the stress response of Gram-positive bacteria – targets for food preservation and safety. Curr Opin Biotechnol 16, 218–224 (2005). [DOI] [PubMed] [Google Scholar]
- Willing S. E. et al. Increased toxin expression in a Clostridium difficile mfd mutant. BMC Microbiol 15, 280 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lereclus D., Arantes O., Chaufaux J. & Lecadet M.-M. Transformation and expression of a cloned ∂-endotoxin gene in Bacillus thuringiensis. FEMS Microbiol Lett 60, 211–218 (1989). [DOI] [PubMed] [Google Scholar]
- Gominet M., Slamti L., Gilois N., Rose M. & Lereclus D. Oligopeptide permease is required for expression of the Bacillus thuringiensis PlcR regulon and for virulence. Mol Microbiol 40, 963–975 (2001). [DOI] [PubMed] [Google Scholar]
- Ramarao N., Nielsen-LeRoux C. & Lereclus D. The insect Galleria mellonella as a useful infection model to investigate bacterial pathogenesis. J Vis Exp 70, 4392 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchis V., Agaisse H., Chaufaux J. & Lereclus D. Construction of new insecticidal Bacillus thuringiensis recombinant strains by using the sporulation non-dependent expression system of cryIIIA and a site specific recombination vector. J. Biotechnol. 48, 81–96 (1996). [DOI] [PubMed] [Google Scholar]
- Arantes O. & Lereclus D. Construction of cloning vectors for Bacillus thuringiensis. Gene 108, 115–119 (1991). [DOI] [PubMed] [Google Scholar]
- Lereclus D. & Arantes O. spbA locus ensures the segregational stability of pHT1030, a novel type of Gram-positive replicon. Mol. Microbiol. 7, 35–46 (1992). [DOI] [PubMed] [Google Scholar]
- Bouillaut L. et al. FlhA influences Bacillus thuringiensis PlcR-regulated gene transcription, protein production, and virulence. Appl Environ Microbiol 71, 8903–8910 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani N. & Dupuy B. Regulation of toxin synthesis in Clostridium difficile by an alternative RNA polymerase sigma factor. Proc Natl Acad Sci USA 98, 5844–5849 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menard R., Sansonetti P. & Parsot C. The secretion of the Shigella flexneri Ipa invasins is activated by epithelial cells and controlled by IpaB and IpaD. EMBO J 13, 5293–5302 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagi K., Kawakami K. & Saito A. Role of reactive nitrogen and oxygen intermediates in gamma interferon-stimulated murine macrophage bactericidal activity against Burkholderia pseudomallei. Infect Immun 65, 4108–4113 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L., Cross A. R., Zhen L. & Dinauer M. C. Functional analysis of NADPH oxidase in granulocytic cells expressing a delta488-497 gp91(phox) deletion mutant. Blood 94, 2497–2504 (1999). [PubMed] [Google Scholar]
- Salamitou S. et al. The plcR regulon is involved in the opportunistic properties of Bacillus thuringiensis and Bacillus cereus in mice and insects. Microbiology 146, 2825–2832 (2000). [DOI] [PubMed] [Google Scholar]
- Guillemet E. et al. The InhA metalloproteases of Bacillus cereus contribute concomitantly to virulence. J Bacteriol 192, 286–294 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran S. L., Guillemet E., Gohar M., Lereclus D. & Ramarao N. CwpFM (EntFM) is a Bacillus cereus potential cell wall peptidase implicated in adhesion, biofilm formation and virulence. J Bacteriol 192, 2638–2642 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novina C. D. & Sharp P. A. The RNAi revolution. Nature 430, 161–164 (2004). [DOI] [PubMed] [Google Scholar]
- Kanginakudru S. et al. Targeting ie-1 gene by RNAi induces baculoviral resistance in lepidopteran cell lines and in transgenic silkworms. Insect Mol Biol 16, 635–644 (2007). [DOI] [PubMed] [Google Scholar]
- Rivero A. Nitric oxide: an antiparasitic molecule of invertebrates. Trends Parasitol 22, 219–225 (2006). [DOI] [PubMed] [Google Scholar]
- MacMicking J. D. et al. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell 81, 641–650 (1995). [DOI] [PubMed] [Google Scholar]
- Thillaye-Goldenberg B., Goureau O., Naud M. C. & de Kozak Y. Delayed onset and decreased severity of experimental autoimmune uveoretinitis in mice lacking nitric oxide synthase type 2. J Neuroimmunol 110, 31–44 (2000). [DOI] [PubMed] [Google Scholar]
- Tran S. L. et al. Hemolysin II is a Bacillus cereus virulence factor that induces apoptosis of macrophages. Cell Microbiol 13, 92–108 (2011). [DOI] [PubMed] [Google Scholar]
- Liu H., Lees P., Abbott J. & Bee J. A. Pulsed electromagnetic fields preserve proteoglycan composition of extracellular matrix in embryonic chick sternal cartilage. Biochim Biophys Acta 1336, 303–314 (1997). [DOI] [PubMed] [Google Scholar]
- De Septenville A. L., Duigou S., Boubakri H. & Michel B. Replication fork reversal after replication-transcription collision. PLoS Genet 8, e1002622 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.