

Abstract
AIM
To explore the effects of αA-crystallin in astrocyte gliosis after optic nerve crush (ONC) and the mechanism of α-crystallin in neuroprotection and axon regeneration.
METHODS
ONC was established on the Sprague-Dawley rat model and αA-crystallin (10−4 g/L, 4 µL) was intravitreously injected into the rat model. Flash-visual evoked potential (F-VEP) was examined 14d after ONC, and the glial fibrillary acidic protein (GFAP) levels in the retina and crush site were analyzed 1, 3, 5, 7 and 14d after ONC by immunohistochemistry (IHC) and Western blot respectively. The levels of beta Tubulin (TUJ1), growth-associated membrane phosphoprotein-43 (GAP-43), chondroitin sulfate proteoglycans (CSPGs) and neurocan were also determined by IHC 14d after ONC.
RESULTS
GFAP level in the retina and the optic nerve significantly increased 1d after ONC, and reached the peak level 7d post-ONC. Injection of αA-crystallin significantly decreased GFAP level in both the retina and the crush site 3d after ONC, and induced astrocytes architecture remodeling at the crush site. Quantification of retinal ganglion cell (RGC) axons indicated αA-crystallin markedly promoted axon regeneration in ONC rats and enhanced the regenerated axons penetrated into the glial scar. CSPGs and neurocan expression also decreased 14d after αA-crystallin injection. The amplitude (N1-P1) and latency (P1) of F-VEP were also restored.
CONCLUSION
Our results suggest α-crystallin promotes the axon regeneration of RGCs and suppresses the activation of astrocytes.
Keywords: αA-crystallin, axonal regeneration, astrocyte, glial scar, chondroitin sulfate proteoglycans, optic nerve crush
INTRODUCTION
Acute optic nerve injury, which is caused by trauma, ischemia or glaucoma, often leads to retinal ganglion cell (RGC) damage accompanied by the activation of astrocytes, microglial cells and oligodendrocytes[1]–[4]. The cellular response to injury includes migration to the injury site, proliferation and the secretion of inhibitory molecules and proteins to form an unfavorable environment for axon regeneration. Astrocytes are a type of cells that mainly respond to injury to form glial scar at the crush site; they also secrete inhibitory extracellular matrix (ECM) molecules, such as chondroitin sulfate proteoglycan (CSPG), which had been thought play an important role in axon regeneration[5]–[6]. Some methods were previously reported to rescues the RGC survival and promote axon regeneration, including enhancing the neurotrophic factor support[7], interfering with the apoptotic signaling through caspase-3 and RhoA/Rho-kinase (RhoA/Rock) pathway to reduce the RGC loss[8]–[9], promoting the intrinsic capability for axon regeneration, and manipulating the inhibitory physical and chemical barrier, which was also thought to be an ideal strategy to enhance neurite outgrowth[4],[9]–[10]. All of the above strategies were limited in improving RGC survival or short-distance axon regeneration; therefore, it is necessary to identify multi-target molecules and comprehensive interventions to promote highly effective axon regeneration.
It is known that the heat shock protein (HSP) α-crystallin consists of noncovalently associated A and B subunit, and plays a crucial role in RGC survival and axon regeneration[11]–[13]. α-crystallin acts as a therapeutic protein through its anti-apoptotic, anti-inflammatory, anti-aggregation and other activities[14]. It has previously been reported that lens injury could stimulate axon regeneration in the optic nerve cut model and that the growth cone might reach the retinoreceptive layer of the superior colliculus at 5wk after optic nerve lesion[15]. α-crystallin promotes axon outgrowth by regulating the RhoA/Rock signaling pathway[16], α-crystallin also promotes rat retinal neurite growth on myelin substrates in vitro[17], promotes RGCs survival and inhibits microglial activation in vivo[3]. These studies showed that α-crystallin might be a multi-target molecule. However, it remains unclear why the axon could significantly penetrate the physical and chemical barrier following α-crystallin treatment. Our hypothesis is that α-crystallin can directly influence the astrocyte response to injury. We previously observed that αA-crystallin could suppress the activation and proliferation of astrocytes in vitro, and the astrocyte cell scratch assay also showed that a higher concentration, 10 µg/mL, could inhibited astrocyte migration. These results suggested that αA-crystallin could interact with astrocytes and influence astrocyte activation.
αA-crystallin shares approximately 55% sequence identity at the amino acid level with αB-crystallin, which is abundantly expressed in the ocular system. In this study, we investigated whether αA-crystallin could enhance axon regeneration by suppressing astrocyte activation and secretion of inhibitory factors.
MATERIALS AND METHODS
Adult (150-200 g) female Sprague-Dawley rats were used for the experiments. All experimental protocols were approved by the Institutional Animal Care and Use Committee of the Third Military Medical University, Chongqing, China. All procedures were conducted in accordance with the Institutional Animal Care and Use Committee of the Third Military Medical University for the use of animals in ophthalmic and vision research. The animals were housed with standard chow and water ad libitum, and sustained on a 12h:12h light and dark cycle at a temperature of 21°C-25°C.
Optic Nerve Crush and Intravitreous Injection
The optic nerve crush (ONC) injury model was performed as previously reported[13],[16]. Briefly, the eight-week-old adult rats (150-200 g) were anesthetized by an intraperitoneal injection of 10% chloral hydrate (0.4 g/kg, it was allowed to anesthetize rodent animals in China). A 0.5-1 cm incision was made in the temporal conjunctiva of each eye under a microscope, and 3-5 mm optic nerve was bluntly exposed. The optic nerve was clamped at 2 mm behind the eyeball for 10s using an atraumatic artery clamp to cause moderate injury. By avoiding the injury site, the appearance of the ophthalmic artery and the vascular integrity of the retina were verified by funduscope examination; the cases in which the retinal vascular integrity was in question were excluded from the group. For the sham operation, only 3-5 mm of the optic nerve was exposed. αA-crystallin (10−4 g/L; 4 µL) [Recombinant Human Crystallin Alpha A, Cellsciences, USA, Lot no. 3172602, purity: >95% as determined by reversed phase high-performance liquid chromatography and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analyses], which was dissolved in sterile phosphate buffer solution (PBS), was injected into vitreous cavity using a 10 µL microinjector (made in Ninbo, China) by a posterior approach to the left eye, taking care not to injure the lens. The same volume (4 µL) of sterile PBS was injected in the right eye after ONC. A topical anti-inflammatory ointment was embrocated on the eye surface after operation. Food and water were provided ad libitum post-injury.
Visual Electrophysiology Investigation
The visual electrophysiology method was performed according to previously reported protocol[18]–[19]. Briefly, the rats were maintained with a 12h:12h light dark cycle (7:00 a.m.-7:00 p.m.). The flash-visual evoked potential (F-VEP) was recorded before the operation, 12h after the operation and 14d after the operation. Each rat was adapted to a dark room and prepared under long-wave form red light when the F-VEPs were being recorded. The animals were anesthetized and immobilized on a special supporter, and the pupils were dilated with one drop of tropicamide. Three needle electrodes were inserted subcutaneously, one was inserted at that middle point of the two eyes at a 0.5 cm distance close to the nasal side and served as the reference lead, another was inserted in the sagittal suture near the visual cortex and served as a recording lead, and the last one was placed in the tail and served as the ground lead. The stimulus was a LED flash intensity of 3.93 cd/m2•s with 1 Hz frequency and a band pass width of 1-100 Hz, and the stable waveforms were superimposed 100 times using the Reti-scan system (Roland, Germany). When one eye was recorded, the other one was covered. Each eye was recorded three times with a 5min interval. The recorded F-VEP waveform was labeled with the latencies N1, P1 and N2 (response time, ms) and amplitudes N1-P1 (delta between trough N1 and peak P1, µv).
Tissue Preparation and Fixation
The rats were anesthetized by an intraperitoneal injection of 10% chloral hydrate (0.4 g/kg) and perfused through the heart with 0.9% normal saline followed by 4% paraformaldehyde (PFA). The eye cups and optic nerves were carefully isolated and post-fixed for 2h with 4% PFA before being incubated in 30% sucrose/PBS overnight, followed by freezing in opti-mum cutting temperature compound (O.C.T. Compound) (Sakura, USA) cryopreservation medium and storage at -80°C. The O.C.T. Compound embedded eyecups generate 10 µm-thick frozen sections as well as longitudinal optic nerve sections. The sections were preserved at -20°C until further use.
Immunofluorescence
The sections were immunofluorescently stained as previously described[20]–[21]. The sections were dried in air, washed in PBS three times for 5min each, and subsequently incubated in 0.3% Triton X-100 (Sigma-Aldrich, USA) for 10min before incubation in blocking solution (8% goat serum, diluted in PBS) for 60min at room temperature. The sections were then incubated with the following primary antibodies in 3% goat serum overnight at 4°C: rabbit anti-glial fibrillary acidic protein (GFAP) (Abcam, Cambrige, MA, USA, 1:500), mouse anti-beta Tubulin (TUJ1) monoclonal antibody (Earthox, San Francisco, USA, 1:300), mouse anti-feline CSPG Brain core protein (US Biological, US, 1:400), and mouse anti-neurocan antibody (EMD Millipore, 1:500). The sections were subsequently washed four times for 5min each and then incubated with the Alexa Fluor-568-conjugated (1:500) or -488-conjugated (1:300) IgG secondary antibodies (Molecular Probes, USA) for 1h in a dim room at 37°C. The nuclei were stained with 40, 6-diamidino-2-phenylindole (DAPI, Beyotime Institute of Biotechnology, Shanghai, China, and diluted 1:5 in PBS) for 10min. The images were observed using and Olympus OP70 microscope (Olympus Microscopy, Japan) or a Leica TCS SP50 confocal microscope (Leica Microsystems, Wetzlar, Germany).
Immunohistochemistry (IHC) was used to observe the growth-associated membrane phosphoprotein-43 (GAP-43) -positive regenerating axons. The rabbit anti-GAP43 (Abcam, Cambridge, USA, 1:300) primary antibody was diluted in 8% goat serum and 0.1% Triton X-100 in PBS. Subsequently, the sections were incubated with an Alexa Fluor-488-conjugated secondary antibody IgG (Molecular probes, 1:300) for 2-3h in a dark room at 37°C.
Western Blot Analysis
To examine the protein expression in the tissues, a Western blot (WB) analysis was performed as previously reported[3],[16]. The retinas or optic nerves were homogenized in ice-cold protein lysis buffer containing protease and phosphatase inhibitors (150 mmol/L NaCl, 1% Triton X-100, 50 mmol/L of Tris-HCl buffer, pH 7.4, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), sodium orthovanadate, sodium fluoride, Leupeptin and Ethylenediaminetetraacetic acid) for approximately 30min. The homogenate was centrifuged at 15 000 g for 10min at 4°C, and the supernatants were collected and stored at -80°C until use. A bicinchoninic acid kit (Beyotime Institute of Biotechnology, Shanghai, China) was used to investigate the concentration of the samples. The samples (50 µg of protein/lane) were separated on a 10% SDS-PAGE gel and transferred to an Immobilon membrane (Millipore) for 60min at 100 V. The membranes were probed with the following primary antibodies overnight at 4°C: rabbit anti-GFAP (Abcam, Cambridge, USA, 1:2000), mouse anti- TUJ1 monoclonal antibody (Earthox, San Francisco, USA, 1:1000), rabbit anti-GAP43 antibody (Abcam, Cambridge, USA, 1:500), mouse anti-feline CSPG Brain core protein (US Biological, USA, 1:1000), and mouse anti glyceraldehyde-3-phosphate dehydrogenase monoclonal antibody (GAPDH, CWbio, China, 1:2000). After washing, the membranes were incubated with different HRP-conjugated secondary antibodies for 2h at 37°C. The membranes were observed using the Odyssey infrared imaging system (Bio-Rad Laboratories, Hercules, CA, USA). The relative of amounts of the proteins were calculated as the ratio of GFAP, TUJ1, GAP-43, or CSPG to GAPDH, respectively. The experiments were repeated three times for each protein.
Axon Regeneration and Quantitation
The regenerate axons were quantified by counting the number of GAP-43-positive axons, as previously described[22]. The number of axons that had extended 50, 100, 200 and 300 µm from the middle crush site was counted in six sections per case. The section width of the optic nerve was measured at the points where the axons were counted and was used to calculate the number of axons per millimeter of nerve, which was then averaged for the six sections per case. The total number of extending axons 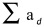 from a distance d of the crush site was calculated by the following formula:
from a distance d of the crush site was calculated by the following formula:
The radius r was calculated by adding all of the sections with a thickness t (10 µm).
Statistical Analysis
The data were expressed as the means±SD. The statistical analyses were performed with the SPSS 13.0 software, and the gray value analyses were performed using the ImageJ software. The statistical differences between groups were analyzed using Student's t-tests or one-way analysis of variance (ANOVA) and considered to be significant with P-value less than 0.05.
RESULTS
αA-crystallin suppressed the activation of gliosis in the retinas of the ONC rats. To study the effect of αA-crystallin on the retinal gliosis response in vivo after ONC, we examined the expression of GFAP at different time points after ONC by IHC and WB. GFAP expression was restricted to the ganglion cell layer in the normal retinas (Figure 1A, a, g, m, and s), and the sham operation did not influence the GFAP expression and distribution in the retinas (Figure 1A. b-f). ONC injury led to obvious GFAP-positive processes that were distributed in the inner plexiform layer (IPL) and inner nuclear layer (INL) at only 1d after ONC, and more intense GFAP-positive processes were observed in all retinal layers after 3, 5, 7 and 14d (Figure 1A, g-l). However, the αA-crystallin (10−4 g/L, 4 µL) treatment effectively attenuated the expression of GFAP. The time period for the processes to extend across the entire width of the retina was delayed to 5d after ONC, and fewer processes appeared (Figure 1A, s-x). The intravitreous injection of PBS (4 µL) did not influence the ONC-induced increase in the GFAP-positive processes. We observed that the GFAP-positive processes were also distributed in the outer layers at 3d after ONC injury (Figure 1A, m-r).
Figure 1. Intravitreous injection of αA-crystallin suppressed the activation of gliosis in the retinas of the ONC rats.

A: Immunofluorescence staining showed the level of GFAP (green) in the retinas of the optic nerve sham injury group (sham) (a-f), ONC injury group (injury only) (g-l), PBS-treated ONC injury group (PBS injection) (m-r) and αA-crystallin-treated ONC injury group (α-A injection) (s-x). B, C: WB analysis showing that the GFAP levels were significantly attenuated by αA-crystallin treatment at 3 (P<0.01), 5 (P<0.05), 7 (P<0.01) and 14d (P<0.01) after ONC. GCL: Ganglion cell layer; IPL: Inner plexiform layer; INL: Inner nuclear layer; OPL: Outer plexiform cell; ONL: Outer nuclear cell. The quantitative data represent the means±SD (n=3). aP<0.05, bP<0.01. Scale bar=100 µm.
Immunoblot analysis confirmed that the ONC injury induced the activation of gliosis in the retinas (Figure 1B, 1C). The retinal GFAP levels were increased approximately 0.7-fold at 3d after ONC injury compared to the sham operation group, and the GFAP levels increased approximately 0.8-fold at 14d after ONC (Figure 1C). A similar result was observed in the PBS-treated group. However, the GFAP expression level was significantly attenuated by αA-crystallin treatment at 3 (P<0.01), 5 (P<0.05), 7 (P<0.01) and 14d (P<0.01) compared to the injury only group (Figure 1C). Notably, the activation of gliosis in the retinas after ONC injury has been described previously[1],[21],[23]–[24]. We observed that the GFAP levels reached a peak at 2wk in the injury only group, which is consistent with the results described by Rong et al[24]. There was no significant difference in the GFAP levels after an additional two weeks after optic nerve injury (at 4wk, data not shown). It has also been reported that α-crystallin pretreatment effectively diminished the systemic inflammation-induced GFAP levels[11] and inhibited the retinal microglial activation induced by ONC[3],[25], but there was no report that described the influence of αA-crystallin in glial cell activation in the total retinas following traumatic optic nerve injury. Our results indicate that alpha-crystallin could suppress the ONC-induced activation of gliosis in the retinas.
Intravitreous injection of αA-crystallin suppressed the activation and morphological remodeling of astrocytes at the ONC site. We next observed whether αA-crystallin treatment could also affect astrocyte activation after ONC. Frozen sections of the optic nerve were stained with GFAP antibody (Figure 2A). After the optic nerve injury, the cellular architecture at the crush site was disrupted, and the astrocytes were degenerated and formed a GFAP-immunofluorescence reactivity (GFAP-IR)-free zone at 3d after the ONC injury (Figure 2A, g-l). At approximately 7d after injury, the astrocytes at the distal side of the optic nerve migrated into this zone and formed a glial scar. The arrangement of the astrocyte processes at the marginal crush site was more arbitrary (Figure 2A, k, Z2). By 14d, the GFAP-IR-free zone was partially filled by GFAP-positive processes (Figure 3A, a-d), which is consistent with previous reports[26]–[27] and suggested that a glial scar had formed. The PBS treatment did not influence the ONC-induced changes in the astrocytes (Figure 2A, m-r). Fortunately, the intravitreous αA-crystallin injection effectively suppressed the astrocyte migration and changed the processes (Figure 2A, s-x, Z4); only a few GFAP-positive processes were present in the GFAP-IR-free gap.
Figure 2. Intravitreous injection αA-crystallin suppressed the astrocyte activation at the ONC site.

A: Immunofluorescence staining showed GFAP (green) activation at the crush site in optic nerve sham injury group (sham) (a-f, and e zoomed image Z1), ONC injury group (injury only) (g-l, and k zoomed image Z2), PBS-treated ONC injury group (PBS injection) (m-r, and q zoomed image Z3) and αA-crystallin-treated ONC injury group (α-A injection) (s-x, and w zoomed image Z4). B, C: The WB analysis of GFAP showed that the GFAP levels were significantly inhibited in the αA-crystallin-treated group compared to the ONC and PBS-treated groups after 5 (P<0.05), 7 (P<0.01) and 14 (P<0.01), respectively. cr: The proximal side of the crush site. The quantitative data represent the means±SD (n=3). aP<0.05, bP<0.01. Scale bars=100 or 50 µm.
Figure 3. Intravitreous injection of αA-crystallin increased the number of TUJ1-positive processes at 14d after ONC.

A: Immunofluorescence staining showed the GFAP-positive fibers (green) and TUJ1-positive processes (red) in the crush site following ONC. The sections were obtained from the ONC injury group (injury only) (a-d), PBS-treated ONC injury group (PBS injection) (e-h) and αA-crystallin-treated ONC injury group (α-A injection) (i-l). B, C: WB analysis showed that the GFAP levels in the optic nerve were significantly decreased in the αA-crystallin-treated group compared to the injury only (P<0.01) and PBS-treated groups (P<0.01). However, the TUJ1 levels were higher in the αA-crystallin-treated group compared to the injury only group (P<0.05). cr: The middle of crush site; ONC: Optic nerve crush. The quantitative data represent the means±SD (n=5). aP<0.05, bP<0.01. Scale bar=100 µm.
WB analysis was used to determine the levels of the GFAP protein to further characterize the influence of astrocyte activation by αA-crystallin treatment. The result showed that GFAP was significantly inhibited in the αA-crystallin-treated group compared to the injury only and PBS-treated groups (Figure 2B, 2C). The GFAP levels increased approximately 0.6-fold (n=3) at only 1d after ONC. Compared to the sham operation, the ONC group reached a 1.23-fold peak increase at 7d and was maintained at approximately 1.2-fold at 14d; PBS treatment did not significantly influence the ONC-induced GFAP expression. A previous report demonstrated that GFAP expression began in the distal part of the ONC site at 1d after crush and was maintained for at least 2wk[28], similar to changes in GFAP expression we observed following the ONC. However, GFAP was suppressed by the αA-crystallin treatment compared to the ONC and PBS treatment groups, respectively, after 5 (P<0.05), 7 (P<0.01) and 14d (P<0.01). Our results indicated that αA-crystallin can suppress the activation and morphological remodeling of astrocytes.
αA-crystallin increased the number of TUJ1-positive process at 14d after ONC. TUJ1 is a marker for microtubules in neuronal cells, and it is a specific marker for RGCs. The number of TUJ1-positive processes indicates the number of surviving axons. After ONC injury, the GFAP-expressing astrocytes progressively invaded the perilesional zone at 14d after ONC (Figure 3A, 3B), and a few TUJ1-positive axons were observed across the crush site (Figure 3A, 3C). Conversely, the αA-crystallin (10−4 g/L, 4 µL) treatment inhibited astrocyte activation and the GFAP-negative region was obviously larger than that in the PBS-treated group. The number of TUJ1-postive processes was obviously increased and some had extended across the crush site (Figure 3A, j, k). The intravitreous PBS injection (4 µL) did not suppress the ONC-induced astrocyte invasion or improve axon survival (Figure 3A, f, g).
Western blotting confirmed the levels of GFAP and TUJ1 in the optic nerve (Figure 3B, 3C). Compared to the sham operation, GFAP expression increased 1.1-fold (n=5) in the injury only group, while αA-crystallin treatment increased the GFAP levels 0.3-fold (n=5) at only 14d after ONC. The GFAP levels were significantly decreased in the αA-crystallin-treated group compared to the injury only (P<0.01) and PBS-treated (P<0.01) groups. The TUJ1 expression level decreased by 30 percent (n=5) in the αA-crystallin-treated group, but was decreased by 50 percent (n=5) in the injury only group. Our results suggested that the αA-crystallin treatment rescued the number of TUJ1 positive processes at 14d after ONC.
Intravitreous injection of αA-crystallin enhanced axon regeneration at 14d after ONC. GAP-43 expression was observed to confirm the hypothesis that αA-crystallin treatment can enhance axon regeneration. Because RGCs express the GAP-43 protein during axon regeneration, probing for the protein could indicate that the RGCs are growing[29]–[31]. Fourteen days after ONC, there were a few regenerating axons in the crush site of the ONC injury only and PBS-treated groups (Figure 4A), and very few axons had extended beyond the crush site. However, in the αA-crystallin-treated group, the axons had regenerated extensively and passed the crush site (Figure 4A, i-l). The quantification of the axons revealed that more axonal fibers had extended to 300 µm past the crush site in the αA-crystallin-treated group than in the other groups (Figure 4B); the number of axonal fibers was significantly higher in the αA-crystallin treated group (mean number of axons per optic nerves±SD: 50 µm, 536±44; 100 µm, 316±61; 200 µm, 206±55; 300 µm, 95±34; n=6) than in the PBS-treated (mean number of axons per optic nerves±SD: 50 µm, 269±104; 100 µm, 151±42; 200 µm, 38±41; 300 µm, 11±17; n=6) and injury only groups (mean number of axons per optic nerves±SD: 50 µm, 286±105; 100 µm, 180±51; 200 µm, 40±34; 300 µm, 12±20; n=6). We also analyzed the levels of the GAP-43 protein by Western blotting (Figure 4C). We found that the level of GAP-43 in the αA-crystallin-treated group increased 1.8-fold compared to the injury only group (n=4, P<0.01); the levels in the PBS-treated group increased 0.5-fold compared to the injury only group, and there was no significant difference. The result indicated that αA-crystallin might play an important role in axon outgrowth after traumatic optic nerve injury.
Figure 4. Intravitreous injection of αA-crystallin enhanced axon regeneration at 14d after ONC.

A: Longitudinal sections through the adult mouse optic nerve showing the regenerating GAP-43-positive axons in ONC injury (injury only) (a-c, and c zoomed image d), PBS-treated ONC injury (PBS injection) (e-g, and g zoomed image h) and αA-crystallin treated ONC injury groups (α-A injection) (i-k, and k zoomed image l). B: The quantification of the axons revealed that more axonal fibers extended to 300 µm past the crush site in the αA-crystallin-treated group than in the other groups (n=6, one-way ANOVA, bP<0.01). C: WB analysis showed that the level of GAP-43 in αA-crystallin-treated group was significantly improved compared to the injury only and PBS treated groups (n=4, bP<0.01). cr: The middle of crush site; n.s: No significance. The quantitative data represent the means±SD. Scale bar=100 µm.
Intravitreous injection of αA-crystallin restored the F-VEP of rats at 14d after ONC. To functionally evaluate the αA-crystallin influence on the optic nerve electrophysiology, flash visual evoked potential investigations were performed using the Roland reimport visual electrophysiology system. The waveforms recorded from the control and treated rats are shown in Figure 5A. At 12h after the ONC injury, the F-VEP waveforms appeared silent and the peaks were dramatically decreased. Two weeks after injury, the latency and peaks were not obviously restored in the injury only and PBS-treated groups, while the waveforms were restored to approximately the pre-ONC level by αA-crystallin treatment. The statistical analysis of the amplitudes (N1-P1) and latency (P1) indicated the changes were specific; in the normal rats, the average amplitudes for N1-P1 were approximately 22.97±5.94 µV (n=27). At 12h after ONC, the amplitudes for N1-P1 were 4.86±2.25 µV (n=8) and the latency of P1 was 117±9ms. At 14d after ONC, the amplitudes for N1-P1 were 5.24±1.67 µV, and PBS treatment did not restore the amplitude. However, the amplitudes for N1-P1 were obviously restored (12.02±1.64 µV) (P<0.01) by αA-crystallin treatment at 2wk after injury (Figure 5B, a). Meanwhile, the latency of P1 was approximately 78±4ms (n=27) in the normal rats, and it was delayed to 126±9ms 14d after ONC; intravitreous injection of αA-crystallin restored the latency to 100±9ms (P<0.01) at 14d after ONC. Our findings indicated that Alpha-crystallin can also improve the electrical functions of F-VEPs in the ONC injury model.
Figure 5. Intravitreous injection of αA-crystallin restored the F-VEP of rats at 14d after ONC.
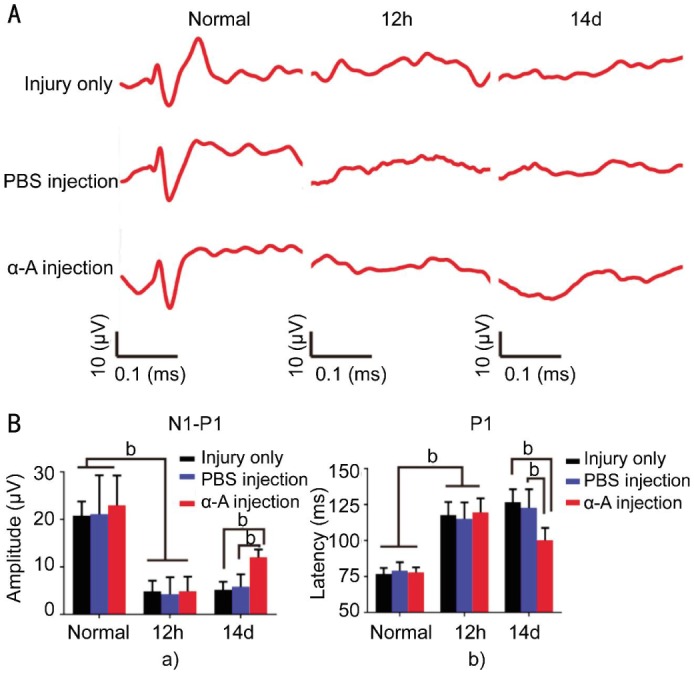
A: Representative waveforms of the ONC injury group (injury only), PBS-treated ONC injury group (PBS injection) and αA-crystallin-treated ONC injury group (α-A injection). B: The amplitudes (N1-P1) and latency (P1) were altered by αA-crystallin treatment after ONC. a: The amplitude (N1-P1) diagram showed a significant increase in the amplitude at 14d after ONC in the αA-crystallin-treated group compared to the ONC injury (P<0.01) and PBS-treated groups (P<0.01). b: The latency (P1) diagram showed that P1 was obviously delayed after the ONC and was significantly restored by αA-crystallin treatment at 2wk after ONC (P<0.01). The data were shown as mean±SD (n=9). aP<0.05, bP<0.01.
Intravitreous injection of αA-crystallin decreased the expression of ECM of the crush site at 14d after ONC. The ECM macromolecules that are deposited by reactive astrocytes in response to injury are one of the main barriers that prevent axon outgrowth. The ECM molecules CSPGs include NG2, neurocan, collagens, fibronectin, tenascins and other classes of proteoglycans. Notably, the CSPGs inhibit neurite outgrowth in vitro[32]–[33]. Two remarkable CSPG proteins, the NG2 proteoglycan and neurocan, were observed to investigate whether αA- crystallin can reduce the deposition of ECM (Figures 6, 7). Two weeks after ONC, substantial CSPG expression was observed around the injury site (Figure 6A, a-d). In particular, the CSPG expression at the distal crush site was much higher than that at the proximal site. PBS treatment did not influence the obvious increase in CSPG expression following ONC injury (Figure 6A, e-h). However, in the αA-crystallin-treated group, CSPG expression was reduced (Figure 6A, i-l). Western blotting confirmed that the expression of CSPGs was obviously increased in the injury only (increased 0.72-fold compared to the sham operation) and PBS-treated (increased 0.77-fold compared to the sham operation) groups. However, in the αA-crystallin-treated group, the CSPGs were significantly suppressed compared to the injury only (P<0.05) and PBS-treated (P<0.05) groups.
Figure 6. Intravitreous injection of αA-crystallin decreased the expression of CSPGs in the crush site at 14d after ONC.

A: Longitudinal sections through the adult mouse optic nerve showing the CSPG levels (green) in the ONC injury (injury only) (a-c, and c zoomed image d), PBS-treated ONC injury group (PBS injection) (e-g, and g zoomed image h) and αA-crystallin-treated ONC injury groups (α-A injection) (i-k, and k zoomed image l). B, C: Western blots showing that the expression of CSPGs was obviously increased in the injury only and PBS-treated groups. However, in the αA-crystallin-treated group, the CSPGs were significantly decreased compared to the injury and PBS-treated (P<0.05) groups. cr: The middle of crush site. The arrows indicate CSPG-positive labeling. The data were shown as means±SD (n=5). aP<0.05.
Figure 7. Intravitreous injection of αA-crystallin decreased the expression of neurocan in the crush site at 14d after ONC.

Immunofluorescence staining showed the neurocan (green) expression in the crush site following ONC in the ONC injury (injury only) (A-C, and C zoomed image D), PBS-treated ONC injury (PBS injection) (E-G, and G zoomed image H) and αA-crystallin-treated ONC injury groups (α-A injection) (I-K, and K zoomed image L). The arrows indicate neurocan-positive labeling. cr: The middle of crush site. The arrows indicate neurocan-positive labeling.
Neurocan is one of the major CSPGs in the nervous tissue, and this axonal extension inhibitor was upregulated in the scar region after stroke (Figure 7). Extensive neurocan expression was observed around the crush site at 14d after ONC (Figure 7A-7D). αA-crystallin treatment obviously decreased the expression of neurocan (Figure 7I-7L). Our findings indicated that the αA-crystallin-enhanced axon regeneration might occur by suppressing astrocyte activation and influencing astrocyte secretions.
DISCUSSION
In this study, we found that the intravitreous injection of αA-crystallin can suppress the activation of gliosis in retinas and influence the cellular architecture of astrocyte remodeling around the crush site in the rats with ONC injury.
Many of the retinal gliosis responses observed in vivo after ONC injury and treatment have been studied using the intravitreous injection method[1],[2],[16],[21]. In traumatic brain and optic nerve injury, astrocytes play an important role in the injury response. Quiescent cells are activated only a few minutes after optic nerve injury[34], and may be sustained for up to three month to form a mature glial scar at the injury site[27]. The activated astrocyte exhibited a hypertrophic soma, the number of processes increased and extended, GFAP expression increased[35], and a glial barrier formed through the retinas and nerves. We found that GFAP expression increased in the retinas at only 1d after ONC and reached a peak level at 14d (increased approximately 0.8-fold). Similarly, the GFAP levels increased significantly after the operation. No significant difference in the GFAP levels was found after an additional two weeks following the optic nerve injury (at 4wk, data not shown).
We further observed that the αA-crystallin (10−4 g/L, 4 µL) treatment decreased the GFAP levels in the retinas and optic nerves and which concentration was used in previous studies[16]–[17],[25]. Furthermore, the arrangement of astrocytes around the crush site was less arbitrary, and the astrocyte migration into the GFAP-IR-free zone was inhibited. It has been reported that HSPs could influence astrocyte activation and proliferation. βA3/A1-crystallin plays an important role in mediating STAT3 signaling to promote GFAP expression and VEGF secretion from optic nerve astrocytes[36]. αB-crystallin takes part in suppressing neuroinflammation by the astrocyte dopamine D2 receptor[37]. GFAP toxicity and aggregation was suppressed by αB-crystallin in an Alexander disease mouse[38]. Our findings suggested that crystallin can also affect the activation and remodeling of astrocytes in the ONC model. However, we cannot maintain the concentration of crystallin in the retinas and nerves at an optimal level for 14d or longer, and it is not clear how αA-crystallin was delivered to the crush site. One explanation was transported via the optic nerve stump to reach the lesion site[12].
ONC injury induces dramatic apoptosis of RGCs, leading to severe axon loss and the failure to regenerate[39]. We found that αA-crystallin treatment improved TUJ1-positive process survival and neurite outgrowth. TUJ1 is known as neuronal β III tubulin and is widely used as a RGCs marker, the number of TUJ1-positive processes reflected RGC survival. Crystallin promoted RGC survival in the retinas, as previously described[3],[12], although the authors have not further investigated whether the RGC survival were functional. We next found that the amplitudes (N1-P1) and latency (P1) of the F-VEP waveforms were restored by the αA-crystallin treatment. The amplitudes (N1-P1), which reflected the number of synaptic contacts between the intact axons and their targets [40], was increased to 12.02±1.64 µV compared to the ONC only group (4.86±2.25 µV). Furthermore, the latency was restored to 100±9ms compared to the ONC only group (126±9ms), which reflected the conduction and axon myelin sheath integrity. We described the protective effect of α-crystallin in electrophysiology, which suggested that crystallin protected the RGCs, the axons' synaptic contacts and the myelin sheath integrity. Previous studies also indicated that Alpha-crystallin treatment could improve optic nerve function, Pangratz-Fuehrer et al[41] found that an intravenous α-crystallin injection led to the acceleration of visual-evoked potential latency over 3wk in ischemic optic neuropathy. While we lacked direct evidence that the F-VEPs were promoted by the rescued axons, it was possible that crystallin protected the moderately damaged axons through the “shock period”, which then recovered their electrical function. Our future studies may uncover more specific evidence to explain the crystallin-mediated rescue of optic nerve electrical potentials.
We used GAP-43 to identify the new growth cones and axons that passed through the crush site, and found that the GAP-43 positive processes had obviously penetrated the crush site, which was accompanied by reduced glial scar formation at 14d after αA-crystallin treatment. The astrocyte architecture was also less arbitrary. The more robust axons that may be regenerated in the Bcl-2tg GFAP−/−Vim−/− mice after ONC indicated the critical role of the glial scar in forbidden axon regeneration[42]. By dismissing the idea that the glial barrier promoted axon regeneration, Robitta et al[43] observed that the neuronal density in co-cultures with GFAP−/−Vim−/− astrocytes was different from those cultured with wild type astrocytes, which was related to attenuated scar formation. Rodriguez et al[44] found that the abrogation of β-catenin signaling could reduce glial scarring and improve axon regeneration. Our hypothesis is αA-crystallin enhances axon regeneration by suppressing astrocyte activation and proliferation. While we cannot exclude the possibility that crystallin intrinsically enhances a neurite-promoting activity, one option is that crystallin acts as a neurite-promoting factor through autocrine and paracrine mechanisms[45].
To study the mechanism by which αA-crystallin enhanced the axon regeneration by suppressing astrocyte activity, we next observed the secretion of CSPG molecules from astrocyte, including NG2 and neurocan. We found that the CSPGs and neurocan were significantly decreased by the αA-crystallin treatment. In response to injury, astrocytes release the inhibitory CSPG molecules, creating a chemical barrier to regeneration[46]. CSPGs' inhibitory activity depends on the glycosaminoglycan components[47]–[48]. Other studies demonstrated that eliminating the inhibition of CSPGs promoted axon regeneration; matrix metalloproteinases and a related protease called ADAMTS-4 degrade CSPGs to promote astrocyte remodeling for axon regeneration[6]. In spinal cord injury, chondroitinase ABC had been used as a strategy to promote axon regeneration and repair plasticity and function[5]. Previous reports also demonstrated that retinal neuron cultures containing α-crystallin could significantly promote increased neurite density and length on myelin-coated dishes via α-crystallin binding to myelin-associated inhibitory molecules or their receptors[17]. Our findings suggest that αA-crystallin could promote neurite outgrowth by suppressing the expression and secretion of CSPGs from astrocytes.
Our findings suggest that α-crystallin is a multi-target molecule that enhances axon regeneration. This molecule might rescue the RGCs following optic nerve injury and also suppress astrocyte activation and the secretion of inhibitory molecules. However, the mechanism is still unclear, and future studies are needed. Our studies provide a strategy to cure optic nerve injury diseases.
Acknowledgments
Authors' Contributions: Shao WY, Xu HW, and Wang Y contributed to data analysis, manuscripts preparation and revised version. Shao WY, Gu XL, and Liu X contributed the experiments Western blots and immunofluorescences, other experiments were finished by Shao WY. Wang Y, Xu HW, Wu N, and Ying X given some good advices in project design.
Foundation: Supported by the National Nature Science Foundation of China (No.81270996).
Conflicts of Interest: Shao WY, None; Liu X, None; Gu XL, None; Ying X, None; Wu N, None; Xu HW, None; Wang Y, None.
REFERENCES
- 1.Chen H, Weber AJ. Expression of glial fibrillary acidic protein and glutamine synthetase by Müller cells after optic nerve damage and intravitreal application of brain-derived neurotrophic factor. Glia. 2002;38(2):115–125. doi: 10.1002/glia.10061. [DOI] [PubMed] [Google Scholar]
- 2.Engelmann R, Dieterich DC, Bien A, Kreutz MR. A different retinal glia response to optic nerve injury/lipopolysaccharide administration in hooded and albino rats. Brain Res. 2001;889(1–2):251–255. doi: 10.1016/s0006-8993(00)03145-0. [DOI] [PubMed] [Google Scholar]
- 3.Wu N, Yu J, Chen S, Xu J, Ying X, Ye M, Li Y, Wang Y. α-Crystallin protects RGC survival and inhibits microglial activation after optic nerve crush. Life Sci. 2014;94(1):17–23. doi: 10.1016/j.lfs.2013.10.034. [DOI] [PubMed] [Google Scholar]
- 4.Yang XT, Huang GH, Feng DF, Chen K. Insight into astrocyte activation after optic nerve injury. J Neurosci Res. 2015;93(4):539–548. doi: 10.1002/jnr.23487. [DOI] [PubMed] [Google Scholar]
- 5.Bradbury EJ, Moon LD, Popat RJ, King VR, Bennett GS, Patel PN, Fawcett JW, McMahon SB. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature. 2002;416(6881):636–640. doi: 10.1038/416636a. [DOI] [PubMed] [Google Scholar]
- 6.Cua RC, Lau LW, Keough MB, Midha R, Apte SS, Yong VW. Overcoming neurite-inhibitory chondroitin sulfate proteoglycans in the astrocyte matrix. Glia. 2013;61(6):972–984. doi: 10.1002/glia.22489. [DOI] [PubMed] [Google Scholar]
- 7.Johnson EC, Guo Y, Cepurna WO, Morrison JC. Neurotrophin roles in retinal ganglion cell survival: lessons from rat glaucoma models. Exp Eye Res. 2009;88(4):808–815. doi: 10.1016/j.exer.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berry M, Ahmed Z, Lorber B, Douglas M, Logan A. Regeneration of axons in the visual system. Restor Neurol Neurosci. 2008;26(2–3):147–174. [PubMed] [Google Scholar]
- 9.Moore DL, Goldberg JL. Four steps to optic nerve regeneration. J Neuroophthalmol. 2010;30(4):347–360. doi: 10.1097/WNO.0b013e3181e755af. [DOI] [PubMed] [Google Scholar]
- 10.Bradbury EJ, Carter LM. Manipulating the glial scar: chondroitinase ABC as a therapy for spinal cord injury. Brain Res Bull. 2011;84(4–5):306–316. doi: 10.1016/j.brainresbull.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 11.Masilamoni JG, Jesudason EP, Baben B, Jebaraj CE, Dhandayuthapani S, Jayakumar R. Molecular chaperone alpha-crystallin prevents detrimental effects of neuroinflammation. Biochim Biophys Acta. 2006;1762(3):284–293. doi: 10.1016/j.bbadis.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 12.Thanos S, Bohm MR, Meyer zu Horste M, Prokosch-Willing V, Hennig M, Bauer D, Heiligenhaus A. Role of crystallins in ocular neuroprotection and axonal regeneration. Prog Retin Eye Res. 2014;42:145–161. doi: 10.1016/j.preteyeres.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 13.Ying X, Zhang J, Wang Y, Wu N, Wang Y, Yew DT. Alpha-crystallin protected axons from optic nerve degeneration after crushing in rats. J Mol Neurosci. 2008;35(3):253–258. doi: 10.1007/s12031-007-9010-1. [DOI] [PubMed] [Google Scholar]
- 14.Reddy VS, Reddy GB. Emerging role for αB-crystallin as a therapeutic agent: pros and cons. Curr Mol Med. 2015;15(1):47–61. doi: 10.2174/1566524015666150114112853. [DOI] [PubMed] [Google Scholar]
- 15.Fischer D, Pavlidis M, Thanos S. Cataractogenic lens injury prevents traumatic ganglion cell death and promotes axonal regeneration both in vivo and in culture. Invest Ophthalmol Vis Sci. 2000;41(12):3943–3954. [PubMed] [Google Scholar]
- 16.Wang YH, Wang DW, Wu N, Wang Y, Yin ZQ. α-Crystallin promotes rat axonal regeneration through regulation of RhoA/rock/cofilin/MLC signaling pathways. J Mol Neurosci. 2012;46(1):138–144. doi: 10.1007/s12031-011-9537-z. [DOI] [PubMed] [Google Scholar]
- 17.Wang YH, Wang DW, Wu N, Wang Y, Yin ZQ. Alpha-crystallin promotes rat retinal neurite growth on myelin substrates in vitro. Ophthalmic Res. 2011;45(3):164–168. doi: 10.1159/000319944. [DOI] [PubMed] [Google Scholar]
- 18.Jiang B, Zhang P, Zhou D, Zhang J, Xu X, Tang L. Intravitreal transplantation of human umbilical cord blood stem cells protects rats from traumatic optic neuropathy. PLoS One. 2013;8(8):e69938. doi: 10.1371/journal.pone.0069938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y, Gong Z, Liu L, Sun H. Combined effect of olfactory ensheathing cell (OEC) transplantation and glial cell line-derived neurotrophic factor (GDNF) intravitreal injection on optic nerve injury in rats. Mol Vis. 2010;16:2903–2910. [PMC free article] [PubMed] [Google Scholar]
- 20.Joly S, Jordi N, Schwab ME, Pernet V. The Ephrin receptor EphA4 restricts axonal sprouting and enhances branching in the injured mouse optic nerve. Eur J Neurosci. 2014;40(7):3021–3031. doi: 10.1111/ejn.12677. [DOI] [PubMed] [Google Scholar]
- 21.Lorber B, Tassoni A, Bull ND, Moschos MM, Martin KR. Retinal ganglion cell survival and axon regeneration in WldS transgenic rats after optic nerve crush and lens injury. BMC Neurosci. 2012;13:56. doi: 10.1186/1471-2202-13-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leon S, Yin Y, Nguyen J, Irwin N, Benowitz LI. Lens injury stimulates axon regeneration in the mature rat optic nerve. J Neurosci. 2000;20(12):4615–4626. doi: 10.1523/JNEUROSCI.20-12-04615.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kirsch M, Trautmann N, Ernst M, Hofmann HD. Involvement of gp130-associated cytokine signaling in Müller cell activation following optic nerve lesion. Glia. 2010;58(7):768–779. doi: 10.1002/glia.20961. [DOI] [PubMed] [Google Scholar]
- 24.Rong XF, Yang S, Miao H, Guo T, Wang Z, Shi W, Mo X, Yuan W, Jin T. Effects of erythropoietin-dextran microparticle-based PLGA/PLA microspheres on RGCs. Invest Ophthalmol Vis Sci. 2012;53(10):6025–6034. doi: 10.1167/iovs.12-9898. [DOI] [PubMed] [Google Scholar]
- 25.Wu N, Wang YH, Zhao HS, Liu DN, Ying X, Yin ZQ, Wang Y. alpha-Crystallin downregulates the expression of TNF-alpha and iNOS by activated rat retinal microglia in vitro and in vivo. Ophthalmic Res. 2009;42(1):21–28. doi: 10.1159/000219681. [DOI] [PubMed] [Google Scholar]
- 26.Frank M, Wolburg H. Cellular reactions at the lesion site after crushing of the rat optic nerve. Glia. 1996;16(3):227–240. doi: 10.1002/(SICI)1098-1136(199603)16:3<227::AID-GLIA5>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 27.Qu J, Jakobs TC. The Time Course of Gene Expression during Reactive Gliosis in the Optic Nerve. PLoS One. 2013;8(6):e67094. doi: 10.1371/journal.pone.0067094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feng DF, Chen ET, Li XY, Liu Y, Wang Y. Standardizing optic nerve crushes with an aneurysm clip. Neurol Res. 2010;32(5):476–481. doi: 10.1179/016164110X12556180206158. [DOI] [PubMed] [Google Scholar]
- 29.Berry M, Carlile J, Hunter A. Peripheral nerve explants grafted into the vitreous body of the eye promote the regeneration of retinal ganglion cell axons severed in the optic nerve. J Neurocytol. 1996;25(2):147–170. doi: 10.1007/BF02284793. [DOI] [PubMed] [Google Scholar]
- 30.Doster SK, Lozano AM, Aguayo AJ, Willard MB. Expression of the growth-associated protein GAP-43 in adult rat retinal ganglion cells following axon injury. Neuron. 1991;6(4):635–647. doi: 10.1016/0896-6273(91)90066-9. [DOI] [PubMed] [Google Scholar]
- 31.Meiri KF, Pfenninger KH, Willard MB. Growth-associated protein, GAP-43, a polypeptide that is induced when neurons extend axons, is a component of growth cones and corresponds to pp46, a major polypeptide of a subcellular fraction enriched in growth cones. Proc Natl Acad Sci U S A. 1986;83(10):3537–3541. doi: 10.1073/pnas.83.10.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmalfeldt M, Bandtlow CE, Dours-Zimmermann MT, Winterhalter KH, Zimmermann DR. Brain derived versican V2 is a potent inhibitor of axonal growth. J Cell Sci. 2000;113(Pt 5):807–816. doi: 10.1242/jcs.113.5.807. [DOI] [PubMed] [Google Scholar]
- 33.Yamada H, Fredette B, Shitara K, Hagihara K, Miura R, Ranscht B, Stallcup WB, Yamaguchi Y. The brain chondroitin sulfate proteoglycan brevican associates with astrocytes ensheathing cerebellar glomeruli and inhibits neurite outgrowth from granule neurons. J Neurosci. 1997;17(20):7784–7795. doi: 10.1523/JNEUROSCI.17-20-07784.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Norenberg MD. Astrocyte responses to CNS injury. J Neuropathol Exp Neurol. 1994;53(3):213–220. doi: 10.1097/00005072-199405000-00001. [DOI] [PubMed] [Google Scholar]
- 35.Sun D, Lye-Barthel M, Masland RH, Jakobs TC. Structural remodeling of fibrous astrocytes after axonal injury. J Neurosci. 2010;30(42):14008–14019. doi: 10.1523/JNEUROSCI.3605-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valapala M, Edwards M, Hose S, Hu JF, Wawrousek E, Lutty GA, Zigler JS, Qian J, Sinha D. βA3/A1-crystallin is a critical mediator of STAT3 signaling in optic nerve astrocytes. Sci Rep. 2015;5:8755. doi: 10.1038/srep08755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shao W, Zhang SZ, Tang M, Zhang XH, Zhou Z, Yin YQ, Zhou QB, Huang YY, Liu YJ, Wawrousek E, Chen T, Li SB, Xu M, Zhou JN, Hu G, Zhou JW. Suppression of neuroinflammation by astrocytic dopamine D2 receptors via αB-crystallin. Nature. 2013;494(7435):90–94. doi: 10.1038/nature11748. [DOI] [PubMed] [Google Scholar]
- 38.Hagemann TL, Boelens WC, Wawrousek EF, Messing A. Suppression of GFAP toxicity by alphaB-crystallin in mouse models of Alexander disease. Hum Mol Genet. 2009;18(7):1190–1199. doi: 10.1093/hmg/ddp013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berkelaar M, Clarke DB, Wang YC, Bray GM, Aguayo AJ. Axotomy results in delayed death and apoptosis of retinal ganglion cells in adult rats. J Neurosci. 1994;14(7):4368–4374. doi: 10.1523/JNEUROSCI.14-07-04368.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maier J, Dagnelie G, Spekreijse H, van Dijk BW. Principal components analysis for source localization of VEPs in man. Vision Res. 1987;27(2):165–177. doi: 10.1016/0042-6989(87)90179-9. [DOI] [PubMed] [Google Scholar]
- 41.Pangratz-Fuehrer S, Kaur K, Ousman SS, Steinman L, Liao YJ. Functional rescue of experimental ischemic optic neuropathy with αB-crystallin. Eye (Lond) 2011;25(6):809–817. doi: 10.1038/eye.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cho KS, Yang L, Lu B, Feng Ma H, Huang X, Pekny M, Chen DF. Re-establishing the regenerative potential of central nervous system axons in postnatal mice. J Cell Sci. 2005;118(Pt 5):863–872. doi: 10.1242/jcs.01658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ribotta MG, Menet V, Privat A. Glial scar and axonal regeneration in the CNS: lessons from GFAP and vimentin transgenic mice. Acta Neurochir Suppl. 2004;89:87–92. doi: 10.1007/978-3-7091-0603-7_12. [DOI] [PubMed] [Google Scholar]
- 44.Rodriguez JP, Coulter M, Miotke J, Meyer RL, Takemaru KI, Levine JM. Abrogation of β-catenin signaling in oligodendrocyte precursor cells reduces glial scarring and promotes axon regeneration after CNS injury. J Neurosci. 2014;34(31):10285–10297. doi: 10.1523/JNEUROSCI.4915-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thanos S, Bohm MR, Schallenberg M, Oellers P. Traumatology of the optic nerve and contribution of crystallins to axonal regeneration. Cell Tissue Res. 2012;349(1):49–69. doi: 10.1007/s00441-012-1442-4. [DOI] [PubMed] [Google Scholar]
- 46.Yiu G, He Z. Glial inhibition of CNS axon regeneration. Nat Rev Neurosci. 2006;7(8):617–627. doi: 10.1038/nrn1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carulli D, Laabs T, Geller HM, Fawcett JW. Chondroitin sulfate proteoglycans in neural development and regeneration. Curr Opin Neurobiol. 2005;15(1):116–120. doi: 10.1016/j.conb.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 48.Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5(2):146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]