

Ehlers–Danlos syndrome (EDS) is a heterogeneous group of inherited connective tissue disorders. These are separate and specific conditions that are distinct in features and, where it is known, genetic basis. The skin, joints, blood vessels and internal organs are variably affected (Table 1).
Table 1.
Clinical presentations that suggest Ehlers–Danlos syndrome as a diagnosis.

Presentation
With various organ systems potentially affected, the initial presentation can be to one or more medical specialties (Table 2).
Table 2.
Examples of Ehlers–Danlos syndrome presentation to different specialties.

Classification
The 1997 Villefranche classification of EDS was based on the identification of genetic alterations affecting the synthesis and structure of type I, III and V collagen.1 Since then, the molecular genetic basis of other types of EDS has been delineated and a further classification is due. Aside from genetic alterations in collagen, genetic defects affecting the biosynthesis of other components of the extracellular matrix, as well as signalling pathways and intracellular trafficking, can contribute to EDS pathogenesis.2
The Villefranche nomenclature with six subtypes is still widely used. Of these, the classical, vascular and hypermobility types are most commonly seen and are discussed in this review. The arthrochalasic, dermatosparactic and kyphoscoliotic types are rare. Modern molecular techniques have identified specific genes relating to most of the Villefranche subtypes (Table 3).
Table 3.
Current classifications of Ehlers–Danlos syndrome. Reproduced with permission from Sobey (2014).9

Classical Ehlers–Danlos syndrome
The triad of joint hypermobility, marked skin hyperextensi-bility (Fig 1a) and widened atrophic scars (Fig 1b) is the hallmark of this condition. Further cutaneous signs include easy bruising with staining from haemosiderin deposition, subcutaneous spheroids (subcutaneous fat lobules that have lost blood supply and calcified), molluscoid pseudotumours (thickened fleshy lesions particularly over elbows and knees associated with scars) and the absence of stretch marks (striae).
Fig 1.
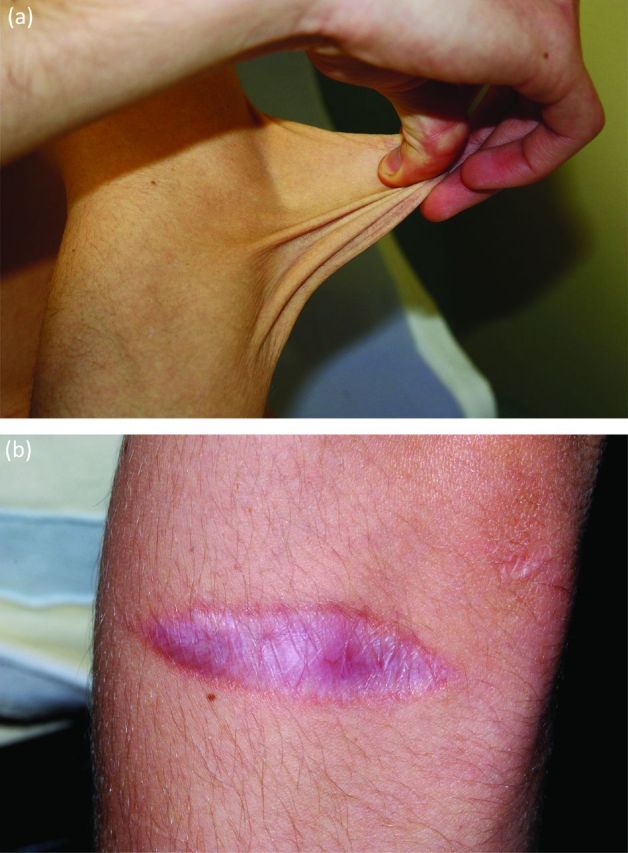
Hallmarks of classical Ehlers–Danlos syndrome. (a) Skin hyperextensibility and (b) atrophic scars.
Electron microscopy of the skin shows typical ‘collagen flowers’ or ‘cauliflowers’ (Fig 2), which result from abnormal fibrillogenesis of the collagen fibrils comprising type I and V collagen. Mutations in type V collagen are known to cause classical EDS.3 Many different mutations in type V collagen have been identified. However, there is as yet no obvious genotype–phenotype correlation. Classical EDS is a dominantly inherited condition, although the severity can vary significantly within the same family. A clinical cameo is presented in Box 1.
Fig 2.

Collagen flowers on electron microscopy of skin in classical Ehlers–Danlos syndrome.
Box 1. Clinical cameo – classical Ehlers–Danlos syndrome.

Cardiology assessment, including echocardiography, is recommended for classical EDS specifically assessing for aortic root dilatation and mitral valve prolapse.4 Joint hypermobility in classical EDS is best managed by rheumatologists together with physiotherapy and occupational therapy. Shin pads can be helpful to reduce injury, particularly during childhood, and can be custom-made through local appliance departments.
Patients with classical EDS should be known to the plastic surgeons at their local hospital so that any wounds can be expertly sutured in the first instance. They should wear a medical warning bracelet inscribed ‘classical EDS’.
Vascular Ehlers–Danlos syndrome
The potential for arterial and hollow organ rupture at a young age with the risk of sudden death makes this the most anxiety-provoking form of EDS for patients, their families and physicians. Easy bruising, which can be at sites not prone to trauma, is often the presenting feature, particularly in childhood. It is not uncommon for these families to have had contact with child protection agencies because of such bruising. Congenital talipes can also be a feature.
The skin is thin and translucent with visible underlying vessels and, although it is not hyperextensible, can be fragile. Some patients with vascular EDS have a typical facial appearance with a thin, pinched nose, prominent eyes and lobeless ears (Fig 3). Joint hypermobility associated with vascular EDS is usually limited to the small joints of the hands. It is important to note that these typical clinical features can be subtle or absent, thus making clinical diagnosis difficult, particularly when there is no contributory family history.
Fig 3.

Lobeless ears in vascular Ehlers–Danlos syndrome.
Where an unexplained arterial rupture or bowel rupture has occurred, the diagnosis of vascular EDS needs to be excluded. Some patients with vascular EDS also present initially with repeated pneumothoraces. Sudden death during the third or fourth decade of life can be the presenting feature.
Arterial (including aortic) dissection, rupture and aneurysm represent the major risks for individuals with vascular EDS. The sigmoid colon is the most common site for bowel rupture. Obstetric complications include uterine and arterial rupture as well as severe lacerations from vaginal delivery.
Vascular EDS is caused by mutation in the COL3A1 gene, which encodes type III collagen. Molecular genetic testing is highly sensitive and specific for this condition. Genotype–phenotype correlations do not yet allow for accurate prognostication.
The optimal ongoing care of patients with vascular EDS is challenging. Surgical interventions are discouraged and, where possible, conservative medical management is preferred. When essential, surgical procedures should be performed by a highly experienced surgical team. It is thought that open surgery is safer than laparoscopic procedures. Good supplies of cross-matched blood should be readily available. A clinical cameo is presented in Box 2.
Box 2. Clinical cameo – vascular Ehlers–Danlos syndrome.

Patients with vascular EDS should wear a medical warning bracelet with information on their emergency care supplied to the organisation providing the bracelet.
Patients with vascular EDS also need to be seen regularly by a cardiologist. The role of vascular imaging is controversial because there is no safe method for treating aneurysms in vascular EDS. The absence of aneurysms is no definite reassurance because these patients might rupture blood vessels at nonaneurysmal sites. Vessel fragility means that any invasive procedure is best avoided because of the risk of inducing vascular rupture by catheterisation.
In 2010 a multicentre randomised trial suggested that celiprolol decreased the incidence of arterial rupture or dissection in patients with a clinical diagnosis of vascular EDS. Further trials are expected.5
Clinicians should be aware that patients with vascular EDS might need psychological support following this diagnosis. The needs of children and adolescents merit specific attention in this area, including transitional care.
Hypermobile Ehlers–Danlos syndrome and/or joint hypermobility syndrome
Much controversy surrounds the naming and diagnosis of hypermobile EDS. There is no diagnostic test for this condition and the genetic basis remains unknown. Moreover, joint hypermobility is a common finding in the general population (approximately 10%) and, within the same family, members might be variably affected. It is no surprise that accurate diagnosis remains a challenge. It is uncertain whether the group of conditions named hypermobile EDS and joint hypermobility syndrome have a similar genetic cause. Inheritance appears to be autosomal dominant.
The Beighton score is used to assess the presence of a generalised joint hypermobility,6 with a score of 5 or more being regarded as indicative of hypermobility.
Although this group of patients is not prone to the life-threatening complications of some other types of EDS, the effects can be debilitating. Severe chronic joint pain coupled with symptoms of autonomic dysfunction can severely impair quality of life and limit opportunities for education and employment. It is more common for women to present with this condition than men, which is still unexplained.7
Assessment in rheumatology is recommended for patients in this group, both to exclude other rheumatological conditions and to advise on management through specialised physiotherapy and occupational therapy. This multidisciplinary approach, together with the assistance of a specialised pain clinic, is helpful.
Postural orthostatic tachycardia syndrome (POTS) is well recognised in this group of patients. POTS is a syndrome of dysautonomia, characterised by multiple symptoms, many of which occur on postural change or standing. These include transient loss of consciousness, presyncope, dizziness, palpitations, chest pain and shortness of breath.
In patients with the autosomal recessive condition of tenascin X-deficient EDS, the clinical features include marked skin laxity, pronounced joint hypermobility and severe bruising. These patients do not show the abnormal scarring of classical EDS. The carrier parents of affected patients, with a single tenascin-X mutation, can have features of joint hypermobility (particularly their mothers).8 There is no evidence to suggest that this is a widespread cause for hypermobile EDS and/or joint hypermobility syndrome.
Rare types of Ehlers–Danlos syndrome
Details on the rare types of EDS mentioned in Table 2 are beyond the scope of this brief review. If these diagnoses are considered, it is recommended that specialist assistance with diagnosis is requested.
The UK EDS National Diagnostic Service
The UK EDS National Diagnostic Service accepts referrals at all ages for clinical assessment and diagnostic testing. The referral criteria are detailed in Box 3.
Box 3. Referral criteria to Ehlers–Danlos syndrome National Diagnostic Service.

Summary
All clinicians need to be aware of EDS and its variable presentations that, in some types, can be life threatening. Recognition of EDS can allow accurate diagnosis by genetic testing, genetic counselling and specific, appropriate management and follow-up.
Key points.
The different types of Ehlers–Danlos syndrome (EDS) are separate conditions with monogenic inheritance
Accurate molecular genetic testing is available for many types of EDS
Vascular EDS is potentially life threatening with a risk of sudden death
There is a UK National Specialist Diagnostic Service for complex EDS
EDS is a group of conditions with multisystem involvement and it can present to all specialties
References
- 1.Beighton P, De Paepe A, Steinmann B, et al. Ehlers–Danlos syndrome: revised nosology, Villefranche, 1997. Am J Med Genet 1998;77:31–7. 10.1002/(SICI)1096-8628(19980428)77:13.0.CO;2-O [DOI] [PubMed] [Google Scholar]
- 2.DePaepe A, Malfait F. The Ehlers–Danlos syndrome, a disorder with many faces. Clin Genet 2012;82:1–11. 10.1111/j.1399-0004.2012.01858.x [DOI] [PubMed] [Google Scholar]
- 3.DePaepe A, Nuytinck L, Hausser I, et al. Mutations in the COL5A1 gene are causal in the Ehlers–Danlos syndrome I and II. Am J Hum Genet 1997:60: 547–54. [PMC free article] [PubMed] [Google Scholar]
- 4.Atzinger CL, Meyer RA, Khoury PR, et al. Cross-sectional and longitudinal assessment of aortic root dilation and valvular anomalies in hypermobile and classic Ehlers–Danlos syndrome. J Pediatr 2011;826–30. 10.1016/j.jpeds.2010.11.023 [DOI] [PubMed] [Google Scholar]
- 5.Ong KT, Perdu J, DeBacker J, et al. Effect of celiprolol on -prevention of cardiovascular events in Ehlers–Danlos syndrome: a prospective, randomised, open, blind-endpoints trial. Lancet 2010;1476–84. 10.1016/S0140-6736(10)60960-9 [DOI] [PubMed] [Google Scholar]
- 6.Beighton P, Solomon L, Soskolne CL. Articular mobility in an African population. Ann Rheum Dis 1973;32:413–8. 10.1136/ard.32.5.413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rombaut L, Malfait F, Cools A, et al. Musculoskeletal complaints, physical activity and health- related quality of life among patients with the Ehlers–Danlos syndrome hypermobility type. Disabil Rehabil 2010;32:1339–45. 10.3109/09638280903514739 [DOI] [PubMed] [Google Scholar]
- 8.Zweers MC, Bristow J, Steijlen PM, et al. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers–Danlos Syndrome. Am J Hum Genet 2003;73:214–7. 10.1086/376564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sobey G. Ehlers–Danlos syndrome: how to diagnose and when to perform genetic tests. Arch Dis Child 2014;doi:10.1136/-archdischild-2013-304822. [DOI] [PubMed] [Google Scholar]