

It was the best of times, it was the worst of times, it was the age of wisdom, it was the age of foolishness, it was the epoch of belief, it was the epoch of incredulity, it was the season of Light, it was the season of Darkness, it was the spring of hope, it was the winter of despair, we had everything before us, we had nothing before us, we were all going direct to Heaven, we were all going direct the other way.
Charles Dickens, A tale of two cities, 1859
Haemophilia is probably one of the best examples in medicine where basic scientific discovery has been rapidly translated into clinical practice. Many patients with haemophilia have been enthusiastic to participate in trials of new treatments and although such treatments have prolonged life they have been associated with devastating side effects.
Early history
Many people are aware of this rare sex-linked disorder because Queen Victoria was a carrier. She had two carrier daughters, Alice and Beatrice, and a son with haemophilia, Leopold.1 Alice was the grandmother of Alexis, the Tsarovitch, whose repeated haemophilic bleedings resulted in his mother, Alexandra, coming under the influence of Rasputin. It has been suggested that haemophilia may have had a profound effect on Russian history.2 Leopold was born in 1853 and was only eight years old when his father, Albert, died in 1861. Leopold was recovering in the south of France with a severe knee bleed and returned to find his mother in deep mourning. Leopold was highly intelligent – he was looked after by the physician Legge who wrote a definitive treatise on haemophilia. Queen Victoria relied heavily on Leopold as a secretary and only reluctantly allowed him to become a student at the University of Oxford. He became a friend of the Liddell family whose daughter, Alice, was Charles Dodgson's Alice in Wonderland. It has been speculated that Leopold wanted to marry Alice but neither Mrs Liddell nor Queen Victoria would countenance the match. Leopold also befriended Ruskin and was later able to secure some of Turner's sketches for what was later to become the Ruskin School of Art. Leopold married Helen of Waldeck and fathered a carrier daughter, Alice, and posthumously a son, Charles – he died aged 30 years following a severe knee bleed in the south of France possibly from an overdose of opium used as a pain killer.4
Beatrice was Queen Victoria's last child and born in 1856 she was only four years old when her father died. The remainder of her life, until she died in 1942, was dedicated to her mother's service – as a secretary during her life and then after her death she edited Queen Victoria's voluminous correspondence and diary although much was destroyed. Her daughter, Ena, became Queen of Spain and had two haemophilic sons, Alphonso and Gonzalo. Beatrice also had three sons, two of whom, Leopold and Maurice, were affected with haemophilia. All three sons served in the first world war, when Maurice was killed, and Leopold died in his late 20s following surgery.4
The first published medical description of haemophilia was written by Dr John Otto who was a physician in the New York Hospital from 1796 to 1817. He described a case of a woman carrier and noted that the inheritance of the disorder was sex linked and was often associated with early death.5
Treasury of human inheritance by Bulloch and Fildes includes 1,000 references and case reports and 200 pedigrees of haemophilic families.6 It includes a description of seven generations of the Appleton-Swain family extending from the early part of the 18th century to the later years of the 19th. The family originated from Reading, a small town near Boston, MA. This family was first described by Hay who noted ‘None but males are bleeders … whose daughters only have sons thus disposed’. The family was reinvestigated by Osler in 1885. Many of the haemophilic males died an early death from bleeding.6
The natural history of haemophilia without treatment was vividly reported in a monograph published by Carroll Birch in 1937.7 He recorded the cause of death in 113 patients – many from very trivial injury; 82 died before 15 years of age and only eight survived beyond the age of 40.
The treatment of haemophilia
Haemophilia is a sex-linked disorder, men express the disease and women are carriers. Haemophilia A, factor VIII deficiency, has an incidence of 1/10,000 and haemophilia B, factor IX deficiency, has an incidence one sixth of haemophilia A. The bleeding tendency is severe (no clotting factor), resulting in spontaneous bleeding into joints, muscles and internal organs or non-severe (low clotting factor) resulting in bleeding in relation to trauma or surgery.
The first treatment for haemophilia was reported in 1840 in The Lancet.8 George Firmin, an 11-year-old boy, bled after surgery for squint. He received blood directly from a stout women using the recently developed syringe by Dr Blundell (Fig 1). The paper describes the family history of haemophilia.
Fig 1.

Blundell's syringe for the direct transfusion of blood. Reproduced from The Lancet with permission from Elsevier.8
Fractionation of human plasma was developed in response to the challenges of the second world war. The major components of plasma were separated by the control of their solubility in a multi-variable system. The five variables were salt, protein, alcohol, pH and temperature.9 Cohn's fraction 1 was rich in factor VIII and fibrinogen. In 1961, McMillan pioneered the use of human factor VIII in the USA. Replacement therapy with Cohn's fraction 1 was used in 15 patients with haemophilia presenting with a variety of haemorrhagic and surgical conditions. There was effective haemostasis in every patient however mild and transient hepatitis developed in one patient 35 days after infusion (this was possibly hepatitis C).10
In 1954, in the UK, in Macfarlane had speculated that:
… maintenance therapy would be impracticable if only human AHG(FVIII) were available, since it would need a special panel of about 500,000 donors to treat the 500 haemophiliacs estimated to exist in the country…. Bovine blood has 16 times the anti haemophilic activity (FVIII) of human blood and enough would be available for the continuous treatment of the whole haemophilic population of this country.11
Bovine antihaemophilic globulin (AHG, FVIII) was produced in Oxford and used to cover tooth extractions. The treatment was effective and the rise in FVIII was measured by the newly developed thromboplastin generation test.12 However the material showed some antigenic properties – an early recognition of inhibitor or antibody development. This led the Oxford group to develop an alternative animal source of FVIII – porcine
FVIII.13
The scientist Edith Bidwell led much of the early fractionation work at Oxford and in 1961 the first patient to be treated with human FIX concentrate was reported.14 A four-year-old boy, with severe haemophilia B, had developed a large haematoma in the antecubital fossa following a difficult venepuncture. The resulting haemorrhage had become infected resulting in osteomyelitis of the radius. A through-the-elbow amputation was performed in June 1960 under cover of FIX concentrate. The patient, age 39 in 1995, qualified as an architectural technician, drove, and played golf.15
The life of people with haemophilia was revolutionised by the development of cryoprecipitate. Judith Pool had discovered that if plasma was cooled to a very low temperature in the test tube a ‘cryoprecipitate’ developed which contained fibrinogen and FVIII. She published a method for making cryoprecipitate in a closed bag system from a blood transfusion.16 This meant that people with haemophilia could learn to treat themselves at home. Such treatment is still used in the developing world.
During the 1970s human freeze-dried (lyophilised) FVIII and FIX became available and patients were able to treat themselves conveniently at home. The blood donors were British for the manufacture of NHS concentrates in Oxford (later moved to Bio Products Laboratory, Elstree) and Edinburgh. Commercial products were manufactured from mostly American donor plasma. The donor pool size could be between 10,000 and 20,000 donations and cryoprecipitate was initially produced from the fresh frozen plasma. The FVIII was extracted using ethanol and salt – Cohn's fractionation – and the final product was freeze dried. It was reconstituted by adding water and (self) administered intravenously. Such products were not heated until 1985. The availability of these products resulted in a dramatic increase in treatment. The lives of patients with haemophilia were improved because they could self-treat at home as soon as spontaneous bleeds occurred. However, they resulted in HIV and hepatitis C virus (HCV) epidemics.
HIV 1979–85
The HIV epidemic occurred during the years 1978–85 and was largely caused by US-derived commercial concentrate. The first patient to seroconvert in the UK was treated in 1979 for abdominal bleeding and developed non-A non-B hepatitis (HCV) followed by HIV.17 When an HIV test became available in 1985 it was possible to check stored samples from patients with haemophilia to establish the date of seroconversion. In this way, a cohort of 111 patients with HIV with known dates of seroconversion was identified (Fig 2).18 The median age was 22 years (range 2–77) and the median date of infection January 1983 (range December 1979–July 1985). All these patients were co-infected with HCV either at the time of HIV infection or before. This cohort was closely monitored clinically and serial CD4 counts had been assessed regularly from December 1982. It was established that there was a linear decline of CD4 count from the normal of 800 per microlitre and on average AIDS developed when the CD4 count reached 50.19 It was thus possible to model for each patient in the cohort the time from seroconversion when he would develop AIDS.20 Predictions from this study suggested that one-quarter of people infected with HIV would remain free of AIDS (without treatment) for 20 years or more. This aroused a lot of publicity because previously it had been thought that HIV inevitably resulted in rapid death.
Fig 2.
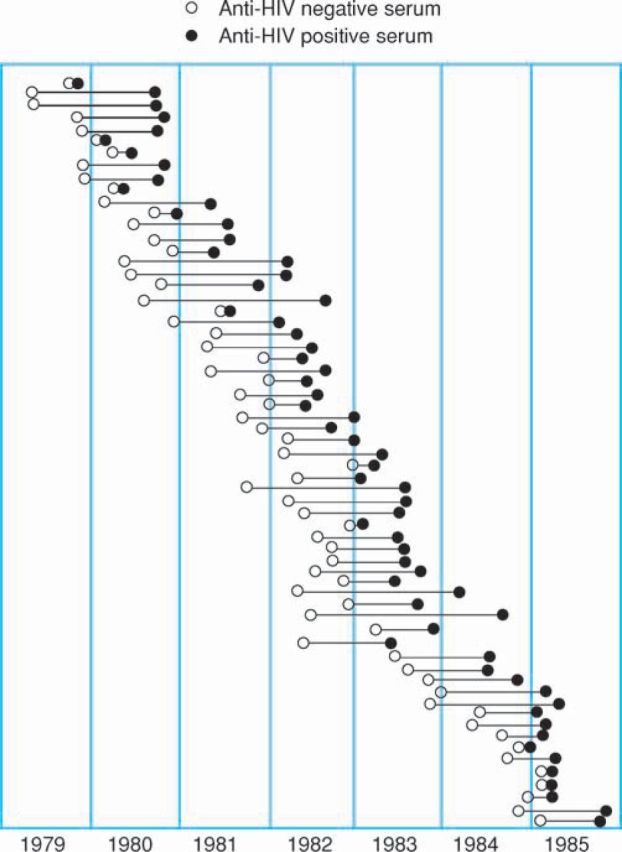
Patients with haemophilia and estimated dates of seroconversion. Reproduced with permission from Wiley Blackwell.18
The epidemic of HIV in haemophilic patients in the USA showed an increase in deaths per million in the 1970s of 0.50 to 60 by 1990.21 In the UK 1,246 of 7,250 patients with haemophilia were infected with HIV. Observations on this wellcharacterised cohort resulted in a series of publications charting the course of the epidemic.22–25 Highly active retroviral therapy became available in the early 1990s and as a result the deaths from HIV were reduced and this was shown very clearly in the patients with haemophilia in the UK (Fig 3).25
Fig 3.

Impact of HIV on mortality rates in the UK haemophilia population. Reproduced with permission from Wolters Kluwer Health.25
Hepatitis C virus 1961–85
The HCV epidemic was a much longer one, occurring between 1961 and 1985. The first patients became infected from the first large pool plasma-derived FIX concentrates and the epidemic ended with the dry heating of concentrates in 1985. Thus all patients with HIV were co-infected with HCV either at the time of HIV infection or before. The natural history of HCV in a population of 310 patients whose date of infection was known showed that 25 years after infection with HCV 19% had progressed to death from liver disease.26
The first recognition that hepatitis was a hazard of blood transfusion was a publication in 1943 reporting seven cases of jaundice which had occurred one to four months after transfusion of whole blood or plasma.27 It was also observed in the London blood transfusion service during the second world war that whereas 7.3% of 2,040 patients transfused pooled plasma or serum developed jaundice, none of those transfused whole blood developed jaundice – it was recommended that ‘to minimise the risk of jaundice only small pools should be used for transfusion’.28 Thus, it was not surprising that large pool clotting factor concentrates should cause hepatitis – however this was difficult to characterise in the absence of a test for HCV until 1991. There was also enthusiasm for the new concentrates among both patients and carers because such treatment transformed the lives of patients. In a historical interview Dr Rosemary Biggs explained:
The next thing that started to crop up was that patients started to get jaundice, and we felt at that time that they were better alive and having jaundice than dead with haemophilia.29
In an anonymous leader published in 1981 it was recognised that:
In some cases early death from liver disease may be the price paid by haemophiliacs for the improved quality of life afforded by the easy availability of clotting factor concentrates.30
The high risk, approaching 100%, of non-A non-B hepatitis following a first exposure to plasma-derived clotting factor concentrate (irrespective of whether the donors were NHS or US (commercial) origin) was demonstrated, although the hepatitis from commercial product was more severe with a shorter incubation period.31 Once testing had become available it was possible to characterise the HCV epidemic in haemophilic patients more clearly.26 Approximately one third of those infected with HCV were also infected with HIV. It was found that the relative hazard of death for those co-infected with HIV and HCV was 19.47 compared with those infected with HCV alone. For those infected with HCV alone and those co-infected the progression rate to liver-related death was 3% and 21% respectively.26
Many patients with haemophilia have been ‘cured’ or ‘cleared’ of HCV with interferon-based therapies, most recently with pegylated interferon and ribavirin. In an international multi-centre cohort study 147 patients maintained a sustained viral response up to 15 years after treatment whereas in 148 unsuccessfully treated patients the cumulative incidence of end-stage liver failure was 13% after 15 years.32
The ultimate cure for end-stage liver failure is liver transplantation and a small number of transplants have been performed in haemophilic patients. A report from Birmingham in 2002 described 11 haemophilic patients who were mono-infected with HCV and who had been successfully transplanted. Since the liver is the site of synthesis of clotting factors, on average 36 hours post-transplant the patients no longer needed treatment with clotting factor concentrate – liver transplantation is essentially ‘gene therapy’ for haemophilia.33
Safer new treatments
The HIV and HCV epidemics were the stimuli to achieve safe plasma-derived products using viral inactivation processes. These were effectively introduced in 1985 and no HIV or HCV transmissions following exposure to clotting factor concentrates have occurred since that time. The first-generation products were conventionally fractionated and heated in lyophilised state (‘dry heated’). These have now been withdrawn. Second generation products involve dry superheating at 80°C for 72 hours; solvent detergent; pasteurisation; and heating in hot vapour. Third generation products are prepared by monoclonal immunoadsorption either directed to FVIII or von Willebrand factor, the carrier protein for FVIII.34
In 1984 a series of landmark papers were published in Nature describing the structure of FVIII and the cloning of the gene.35 This enabled the manufacture of recombinant FVIII and the investigation of such products in worldwide trials. The results of a study in 107 patients including pharmacokinetics, treatment for home therapy, surgery and in previously untreated patients (PUPs), who were mostly children, demonstrated that it had biological activity similar to plasma factor VIII and was safe and efficacious in the treatment of haemophilia.36 This meticulous study showed for the first time the natural history of the treatment in previously untreated patients and the development of inhibitors (antibodies to FVIII) – six of 21 children developed them. It soon emerged that the three recombinant products, two full-length FVIII, and one B-domain depleted, had similar inhibitor incidences of 25%.37,38 Inhibitors now emerged as the biggest challenge in the treatment of haemophilia.
It is perhaps ironic that recombinant products are established as very safe treatment with respect to transfusion transmitted disease but that there is now debate as to whether they pose a greater risk for inhibitor development than plasma-derived products. Thus the relative risk of inhibitor development with recombinant factor VIII versus plasma-derived FVIII was 2.4 (all inhibitors), 2.6 (high inhibitors), and 3.2 (high inhibitors and/or immune tolerance induction in a study of 62 PUPs treated with plasma-derived concentrate and 86 treated with full-length recombinant product).39 The management of inhibitors is now the most challenging issue in haemophilia care. A recombinant porcine factor VIII has been developed and this, together with hybrid constructs of both recombinant human and porcine FVIII, may provide alternative safe and effective treatment for these patients.40,41
‘We were all going direct to heaven’: gene therapy
The ultimate cure of haemophilia could be brought about through gene therapy. The early animal models achieved good results with in vivo production of the coagulant protein. However more recent human studies using adeno-associated virus to deliver FIX to the liver have resulted in antibody developing to the vector itself.42 Haemophilia is no longer a life-threatening disease and in most developed parts of the world, sufferers have access to treatment with recombinant factor VIII or IX concentrate. Thus for the foreseeable future it is likely that gene therapy for haemophilia will remain a research endeavour. Clinical trials may be difficult in the face of constraints laid down by the Declaration of Helsinki, the Council for International Organisations of Medical Sciences (CIOMS) and the World Health Organization.43 The most likely future development of treatment are molecules of clotting factors with longer half lives. Thus a factor VIII has been developed which has specific non-covalent binding to PEGylated liposomes and this has been shown to have prolongation of circulation time as well as haemostatic efficacy and is now in clinical trial.44
‘We were all going direct the other way’: variant CJD
Even though plasma-derived concentrates are very safe with respect to HIV and hepatitis transmission and also recombinant products are used predominantly in the developed world there exists the spectre of variant Creutzfeldt Jakob disease (vCJD), particularly in the UK. The peak exposure of the UK population to vCJD through the food chain was 1998 when nearly 400,000 cattle were infected with bovine spongiform encephalopathy (BSE). There has been almost no BSE since 2000 and the small epidemic of vCJD in humans has peaked with a total 166 cases. However there have now been four cases of transmission by blood from donors incubating the disease.45 Since 1999 all plasma-derived concentrate manufactured in the UK has been manufactured from non-UK plasma and all haemophilic patients are now treated with recombinant products. However there remains concern that haemophilic patients previously treated with UK donor plasma products could carry vCJD especially since there are polymorphisms of the prion protein which could have very long incubation periods.46 An autopsy-based study of 33 haemophilic patients who were treated exclusively with UK donor-sourced concentrates from 1963 to 1995, covering the years of the BSE epidemic, showed no evidence of prion protein using immunochemistry applied to brain samples.47 There have also been no cases of vCJD in the UK haemophilic population.48 However, abnormal prion protein (PRPSC) has been demonstrated in the spleen of an adult haemophilic patient in the UK: the cause of death was unrelated.49
Conclusion
The outlook for people with haemophilia is now very good. In a study of 6,018 people with haemophilia in the UK the 1,977–1,998 who were not infected with HIV have a median life expectancy of 63 years for those with severe haemophilia and 75 years for those with non-severe haemophilia. This approaches that for the normal male population (Fig 4).48
Fig 4.

Survival in 6,018 men with haemophilia not infected with HIV between 1977 and 1998 and in the general male UK population. Reproduced with permission from the American Society of Hematology.48
References
- 1.Ingram GIC. The history of haemophilia. J Clin Path 1976;29:469–79. 10.1136/jcp.29.6.469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Massie RK. Nicholas and Alexandra. London: Gollanz, 1968. [Google Scholar]
- 3.Zeepvat C. Prince Leopold: the untold story of Queen Victoria's youngest son. Gloucester: Sutton publishing, 1998. [Google Scholar]
- 4.Dennison M. The last princess. London: Weidenfeld and Nicolson, 2007. [Google Scholar]
- 5.Otto JC. Review of American publications in medicine, surgery and the medical repository. New York: Auxillary Branches of Sciences, 1803;VI(I):1–4. [Google Scholar]
- 6.Bulloch W, Fildes P. Treasury of human inheritance, Parts V and VI, Section XIVa, Haemophilia. London: Dulau & Co, 1911. [Google Scholar]
- 7.Birch C, La F. Haemophilia, clinical and genetic aspects. Urbana: University of Illinois, 1937. [Google Scholar]
- 8.Lane S. Successful transfusion of blood. Lancet 1840;(i):185–8.
- 9.Cohn EJ, Strong LE, Hughes WL. et al Preparation and properties of serum and plasma proteins. J Am Chem Soc 1946;68:459–75. [DOI] [PubMed] [Google Scholar]
- 10.McMillan CW, Diamond LK, Surgenor DM. Treatment of classical haemophilia: The use of Fibrinogen Rich in FVIII for haemorrhage and surgery. N Engl J Med 1961;265:224–30 and 277–83. [DOI] [PubMed] [Google Scholar]
- 11.Macfarlane RG, Biggs R, Bidwell E. Bovine anti haemophilic globulin in the treatment of haemophilia. Lancet 1954;266:1316–9. [DOI] [PubMed] [Google Scholar]
- 12.Biggs R, Douglas AS. The thromboplastin generation test. J Clin Path 1953;6:23. 10.1136/jcp.6.1.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bidwell E. The purification of antihaemophilic globulin from animal plasma. Br J Haematol 155;1:35–45 and 386–9. 10.1111/j.1365-2141.1955.tb05527.x [DOI] [PubMed] [Google Scholar]
- 14.Biggs R, Bidwell E, Handley DA. et al The preparation and assay of Christmas factor (factor IX concentrate and its use in the treatment of two patients. Br J Haematol 1961;7:349–64. 10.1111/j.1365-2141.1961.tb00345.x [DOI] [Google Scholar]
- 15.Rizza CR. Historical annotation: the first patient to receive factor IX cconcentrate in the UK: a recollection. Haemophilia 1995;1:210–2. [DOI] [PubMed] [Google Scholar]
- 16.Pool JGP, Shannon AE. Production of high-potency concentrates of antihaemophilic globulin in a closed-bag system. N Engl J Med 1965;273:1443–7. 10.1056/NEJM196512302732701 [DOI] [PubMed] [Google Scholar]
- 17.Lee CA, Webster A, Griffiths PD, Kernoff PBA. Symptomless HIV infection after more than ten years. Lancet 1990;335:426. 10.1016/0140-6736(90)90273-8 [DOI] [PubMed] [Google Scholar]
- 18.Lee CA, Phillips A, Elford J. et al The natural history of human immunodeficiency virus infection in a haemophilic cohort. Br J Haematol 1989;73:228–34. 10.1111/j.1365-2141.1989.tb00257.x [DOI] [PubMed] [Google Scholar]
- 19.Phillips AN, Lee CA, Elford J. et al Serial CD4 lymphocyte counts and the development of AIDS. Lancet 1991;337:389–92. [DOI] [PubMed] [Google Scholar]
- 20.Phillips AN, Sabin CA, Elford J. et al Use of CD4 lymphocyte count to predict long term survival free from AIDS after HIV infection. BMJ 1994;309:309–13. 10.1136/bmj.309.6950.309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chorba TL, Holman RC, Strine TW. Changes in longevity and causes of death among persons with haemophilia A. A J Haem 1991;37:243–6. 10.1002/ajh.2830450204 [DOI] [PubMed] [Google Scholar]
- 22.Darby SC, Rizza CR, Doll R. et al Incidence of AIDS and excess of mortality associated with HIV in haemophiliacs in the United Kingdom: report on behalf of the Directors of Haemophilia Centres in the UK. BMJ 1989;298:1064–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Darby SC, Ewart W, Giangrande PLF. et al On behalf of the UK Haemophilia Directors Organisation. Mortality before and after HIV infection in the complete UK population of haemophiliacs. Nature 1995;377:79–82. [DOI] [PubMed] [Google Scholar]
- 24.Darby SC, Ewart DW, Giangrande PLF, Spooner RJD, Rizza CR. For the UK Haemophilia Directors Organisation Importance of age at infection with HIV-1 for survival and development of AIDS in the UK haemophilia population. Lancet 1996;347:1573–80. [DOI] [PubMed] [Google Scholar]
- 25.UKHCDO. The impact of mortality rates in the complete UK haemophilia population. AIDS 2004;18:525–33. 10.1097/00002030-200402200-00020 [DOI] [PubMed] [Google Scholar]
- 26.Yee TT, Griffioen A, Sabin CA, Dusheko G, Lee CA. The natural history of HCV in a cohort of haemophilic patients infected between 1961 and 1985. Gut 2000;47:845–51. 10.1136/gut.47.6.845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beeson PB. Jaundice occurring one to four months after transfusion of blood or plasma. JAMA 1943;121:1332–4. 10.1001/jama.1943.02840170016005 [DOI] [Google Scholar]
- 28.Spurling N, Shone J, Vaughan J. The incidence, incubation period and symptomatology of homologous serum jaundice. BMJ 1946;2:409–11. [PubMed] [Google Scholar]
- 29.Historical Annotation. Witnessing medical history: an interview with Dr Rosemary Biggs. Haemophilia 1998;4:769–77. 10.1046/j.1365-2516.1998.00199.x [DOI] [PubMed] [Google Scholar]
- 30.Leader. BMJ 1981;283:1. 10.1136/bmj.283.6283.16788240 [DOI] [Google Scholar]
- 31.Kernoff PBA, Lee CA, Karayiannis P, Thomas HC. High risk of non-A non-B hepatitis after a first exposure to volunteer or commercial clotting factor concentrates: effects of prophylactic immune serum globulin. Br J Haematol 1985;60:469–79. 10.1111/j.1365-2141.1985.tb07444.x [DOI] [PubMed] [Google Scholar]
- 32.Postwouwer D, Yee TT, Makris M. et al Antiviral therapy for chronic hepatitis C in patients with inherited bleeding disorders: an international, multicentercohort study. J Thromb Haemost 2007;5:1624–9. [DOI] [PubMed] [Google Scholar]
- 33.Wilde J, Teixeira P, Bramhall SR. et al Liver transplantation in haemophilia. Br J Haematol 2002;117:952–6. 10.1046/j.1365-2141.2002.03528.x [DOI] [PubMed] [Google Scholar]
- 34.Lee CA. Coagulation factor replacement therapy. In: Hoffbrand AV, Brenner MK. (eds), Recent advances in haematology. Edinburgh: Churchill Livingstone, 1992:73–88. [Google Scholar]
- 35.Vehar GA, Key B, Eaton D. et al Structure of human FVIII. Nature 1984;312:337–42. [DOI] [PubMed] [Google Scholar]
- 36.Scharz RS, Abildgaaard CF, Aledort LM. et al Human recombinant DNA-derived antihaemophilic factor (factor VIII) in the treatment of haemophilia. N Engl J Med 1990;323:1800–5. [DOI] [PubMed] [Google Scholar]
- 37.Bray GL, Gompertz ED, Courter S. et al A multicenter study of recombinant FVIII (Recombinant): safety, efficacy, and inhibitor risk in previously untreated patients with haemophilia A. Blood 1994;83:2428–35. [PubMed] [Google Scholar]
- 38.Lusher JM, Lee CA, Kessler CM, Bedrosian CL. For the Refacto Phase 3 Study Group. The safety and efficacy of B-domain deleted recombinant factor VIII concentrate in patients with severe haemophilia A. Haemophilia 2003;9:38–49. [DOI] [PubMed] [Google Scholar]
- 39.Goudemand J, Rothschild C, Demiguel V. et al Influence of the type of factor VIII concentrate on the incidence of factor VIII inhibitors in previously untreated patients with severe haemophilia A. Blood 2006;107:46–51. 10.1182/blood-2005-04-1371 [DOI] [PubMed] [Google Scholar]
- 40.Healey JF, Lubin IM, Lollar P. The cDNA and derived amino acid sequence of porcine factor VIII. Blood 1996;88:4209–14. [PubMed] [Google Scholar]
- 41.Barrow RT, Healey JF, Gailini D, Scandella D, Lollar P. Reduction of the antigenicity of factor VIII toward complex inhibitory antibody plasmas using multiply-substituted hybrid human/porcine factor VIII molecules. Blood 2000;95:564–8. [PubMed] [Google Scholar]
- 42.High KA. Update on progress and hurdles in novel genetic therapies for hemophilia. Hematol Educ Program 2007;2007:466–72. 10.1182/asheducation-2007.1.466 [DOI] [PubMed] [Google Scholar]
- 43.Kimmelman K. Staunch protections: the ethics of haemophilia gene transfer research. Haemophilia 2008;14:5–14. 10.1111/j.1365-2516.2007.01567.x [DOI] [PubMed] [Google Scholar]
- 44.Baru M, Carmel-Goren L, Barenholz Y. et al Factor VIII efficient and specific non-covalent binding to PEGylated liposomes enables prolongation of its circulation time and haemostatic efficacy. Thromb Haemost 2005;93:1061–8. 10.1160/TH04-08-0485 [DOI] [PubMed] [Google Scholar]
- 45.Llewelyn CA, Hewitt PE, Knight RS. et al Possible transmission of variant Creutzfeldt-Jkob disease by blood transfusion. Lancet 2004;363:417–21. [DOI] [PubMed] [Google Scholar]
- 46.Head MW, Ironside JW. Mad cows and monkey business: the end of vCJD. Lancet 2005;365:781–3. [DOI] [PubMed] [Google Scholar]
- 47.Lee CA, Ironside JW, Bell JE. et al retrospective neuropathological review of prion disease in UK haemophilic patients. Thromb Haemost 1998;80:909–11. [PubMed] [Google Scholar]
- 48.Darby SC, Kan SW, Spooner RJ. et al Mortality rates, life expectancy, and causes of death in people with haemophilia A or B in the United Kingdom who were not infected with HIV. Blood 2007;110:815–25. 10.1182/blood-2006-10-050435 [DOI] [PubMed] [Google Scholar]
- 49.Ironside JW, Peden A, Head MW, Keeling DM, Millar CM. Detection of PRPSC in the spleen of an adult haemophilic patient in the UK. J Thromb Haemost 2009;7 (Suppl 2) Abstract OC-MO-031. [Google Scholar]