

Key Points
Cutaneous T-cell lymphoma (CTCL) is an uncommon cutaneous malignancy that usually presents with patches and/or plaques, less commonly with tumours and/or haematological involvement
Mycosis fungoides is usually an indolent condition that can be managed with skin-directed therapies
Diagnosis may be difficult and CTCL can be easily mistaken for other skin conditions, so repeated skin biopsies are often necessary
Specialist multidisciplinary team involvement is vital for prompt diagnosis and optimal management
Prognosis is largely influenced by disease stage, but recent evidence reveals that other factors are also important
Ongoing trials of novel agents continue to provide hope for those with refractory disease
Cutaneous lymphomas are a group of lymphoproliferative disorders characterised by the localisation of malignant lymphocytes to the skin. A cutaneous lymphoma may occur as a result of secondary deposits in the skin from a systemic lymphoma or as a primary cutaneous lymphoma. The skin is the second most common extranodal site for primary lymphoma after the gastrointestinal tract. Approximately two-thirds of primary cutaneous lymphomas are of T-cell origin. The incidence of new cases of cutaneous T-cell lymphoma (CTCL) is 0.4 per 100,000 per year, with the highest incidence in the 40–60 year age group. In the US there is a higher incidence in the black population and it is twice as frequent in men.1 This article will focus on the two most common subtypes of primary CTCL, mycosis fungoides (MF) and Sézary syndrome (SS).
Mycosis fungoides
MF accounts for approximately 60% of new cases of CTCL. It is generally an indolent condition, with slow progression over years or even decades. Patients most commonly present with patches and/or plaques which can easily be misdiagnosed as eczema or psoriasis. However, MF lesions are classically well demarcated, erythematous, asymmetrical patches and plaques, with epidermal atrophy, surface wrinkling and fine scale occurring on sun protected sites, predominantly around the pelvic girdle (Fig 1). Less common variants include hypopigmented patches (particularly in those with deeply pigmented, type V or VI skin), folliculotropic disease and poikiloderma (with erythema, telangiectasia, atrophy and increased and/or decreased skin pigmentation). In 10% of cases MF presents as thickened tumours in the presence of patches and/or plaques or erythroderma (Fig 1). MF tumours can often ulcerate and become secondarily infected, thereby causing significant morbidity.
Fig 1.
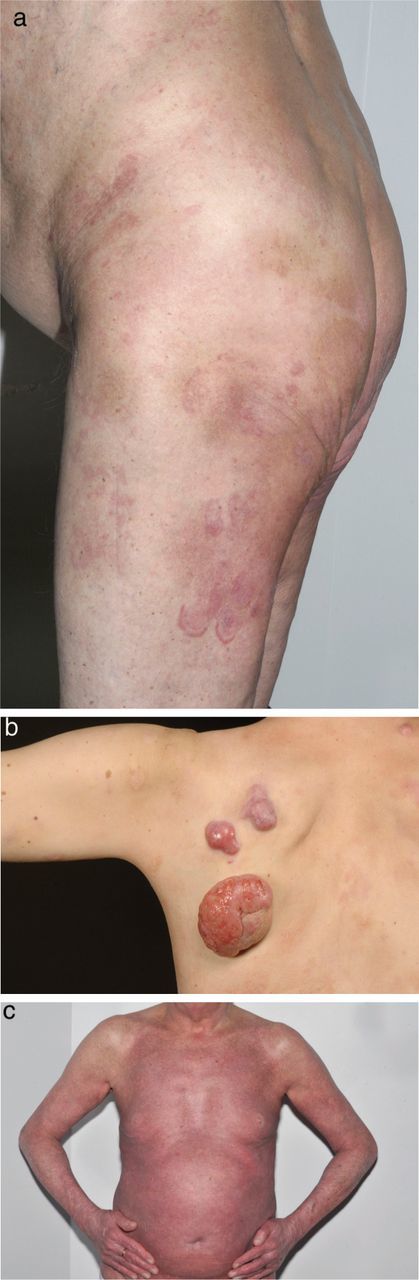
(a) plaque-stage mycosis fungoides (MF), (b) tumour-stage MF and (c) MF with erythroderma.
Sézary syndrome
SS is defined as CTCL with both erythroderma and haematological evidence of leukaemic involvement. These criteria are met in 5% of new cases of CTCL.
Diagnosis and staging
The diagnosis of CTCL is difficult and often requires repeated skin biopsies, with the emphasis on clinicopathological correlation. The following baseline investigations are performed in patients suspected of having CTCL:
- Skin biopsies from lesions of different stages of disease for:
- histological analysis (Fig 2a and 2b)
- immunophenotyping
- T-cell receptor (TCR) analysis (a polymerase chain reaction-based molecular technique to detect a clonal population of T-cells) (Fig 2c).
Blood tests: full blood count, lymphocyte subsets, Sézary cell count (on blood film), lactate dehydrogenase, HTLV-1 and HIV.
Fig 2.

(a) Histology of a mycosis fungoides (MF) plaque showing a dense mononuclear cell infiltrate within the upper dermis. (b) The histology of an MF tumour shows a heavy and extensive dermal mononuclear infiltrate without epidermotropism. (c) Single-strand conformational polymorphism analysis. Polyacrylamide gel demonstrating a T-cell clone in the skin and a polyclonal smear in the blood.
If the patient has skin tumours, erythroderma or palpable lymphadenopathy, a computed tomography (CT) scan of the chest, abdomen and pelvis or a positron emission tomography (PET)/CT scan should also be performed. If lymphadenopathy greater than 1.5 cm is detected on imaging, a lymph node biopsy is recommended in which the entire lymph node is excised and submitted for histological, immunophenotypic and TCR analysis. Excision of the whole node allows representative sampling and assessment of nodal architecture of importance for staging.2 A bone marrow biopsy is recommended in specific cases where there is a high peripheral blood tumour burden.
The classification and staging of CTCL is based on the tumour, lymph node, visceral, blood (TNMB) system developed by the European Organisation for Research and Treatment of Cancer (EORTC) and the International Society for Cutaneous Lymphomas (ISCL). There have been revisions in recent years following advances in investigation and better understanding of variables important in prognosis. The most recent revision is summarised in Tables 1 and 2. All CTCL patients should be reviewed by a specialist multidisciplinary team to confirm the diagnosis and formulate an appropriate management strategy.1,3 This team should comprise dermatologist(s), clinical oncologist(s), radiologist(s), haematologist(s), histopathologist(s), clinical nurse specialist(s), and can also include a clinical psychologist.
Table 1.
ISCL/EORTC revision of the classification of mycosis fungoides and Sézary syndrome.2

Table 2.
ISCL/EORTC revision of mycosis fungoides and Sézary syndrome staging.2

Prognosis
MF has a highly variable prognosis, predominantly dependent on ISCL/EORTC stage. Patients with stage IA disease have a normal life expectancy, but in more advanced stages five-year overall survival ranges from 75% in stage IB/IIA MF to 44% in stage IIB/III disease.4 More recently, other features conferring a worse prognosis have been identified (Table 3), associated with reduced survival and increased risk of disease progression.5,6 Prognosis is poor in SS, with a median life expectancy of only 32 months from diagnosis.3
Table 3.

Treatment
There is no cure for CTCL, so the emphasis of treatment is on clearance or improvement of lesions to produce disease remission. This aims to improve quality of life and optimise disease-free time and overall survival. The choice of treatment agent depends on disease stage. Importantly, there is evidence that aggressive therapy with radiation and chemotherapy in early-stage disease results in a worse prognosis than conservative treatment with sequential topical therapies.7 The several treatment options for CTCL are discussed below.
Skin-directed therapy
In early-stage disease (IA and IB) skin-directed therapies are used including:
Topical high-potency corticosteroids and emollients for localised patches and plaques.
UV phototherapy for more widespread patches and plaques. The safer narrow band UVB (TL-01) can be beneficial in patch or very thin plaque disease, but the more deeply penetrative properties of psoralen UVA range are required for all but the thinnest plaques. Both options provide only temporary relief, and there is the risk of burning and secondary skin cancers with repeated courses of light.
Spot low-dose radiotherapy for localised stubborn plaques or tumours.
Total skin electron beam radiotherapy for widespread plaques or tumours. The electrons used do not penetrate beyond 10 mm below the skin surface, minimising risk of radiotoxicity. Side effects include burning, oedema, alopecia and loss of nails and, longer term, skin atrophy and pigmentary changes.
Systemic therapies
Low-dose interferon-alfa (IFN-a) alters the immune system response and has a role in cell cycle regulation, oncogene suppression and modulation of cell adhesion.1 It is administered as a subcutaneous injection at doses of 3–9 MU three times per week. Patients commonly report flu-like symptoms which limit dose up-titration.
Bexarotene, a retinoic acid derivative, binds to and activates the RXR receptor, interfering with cell proliferation, differentiation and apoptosis. It is administered orally, once daily, with the dose titrated carefully because bexarotene results in systemic side effects of hyperlipidaemia, central hypothyroidism and neutropenia. Concomitant lipid-lowering therapy and thyroxine supplementation are required in most patients.
Extracorporeal photopheresis is a recommended first-line therapy for SS and stage III MF.1 It is a cell- based immunomodulatory therapy involving the separation of leucocyte-rich plasma followed by the ex vivo administration of 8-methoxypsoralen as a photosensitiser and UVA radiation before reinfusion of the plasma to the patient. Treatment is given over two consecutive days every four weeks and continued for at least six months before a decision is made on response. It is often combined with other immunotherapy such as IFN-a or bexarotene.8
Alemtuzumab (Campath), an antibody specific for the CD52 cell surface glycoprotein, is given as an intravenous infusion three times per week for up to 12 weeks. It increases the risk of opportunistic infections.1
Low-dose methotrexate. There are very few studies looking at its use in CTCL. Patients need to be monitored for hepatotoxicity and leucopenia, although these complications are infrequent due to the low doses used.1
Chemotherapy
Chemotherapy is reserved for advanced-stage disease (stage IV). It produces a good initial response, but remission tends to be short-lived so chemotherapy is considered a palliative treatment option. Agents used include:
liposomal doxorubicin
gemcitabine
chlorambucil
multi-agent chemotherapy: a combination of cyclophosphamide, doxorubicin, vincristine and prednisolone.
Allogeneic stem cell transplantation
A relatively small number of patients with refractory disease have received allogeneic stem cell transplantation. Previous studies looking at autologous transplantation have shown poor results. There is a high level of morbidity and mortality, mostly as a result of using high-intensity conditioning and the potentially long wait for a donor.9
Novel agents
Some novel treatments are not currently licensed for use in the UK, although some have Food and Drug Administration (FDA) approval for use in the US. Others remain under clinical trial evaluation. They include:
lenalidomide, a derivative of thalidomide, currently being trialled in the UK
zanolimumab, an anti-CD4 monoclonal antibody
romidepsin and vorinostat, histone deacetylase inhibitors
denileukin diftitox, a recombinant fusion protein combining interleukin-2 and the diphtheria toxin, has FDA approval for use in the US.
References
- 1.Trautinger F, Knobler R, Willemze R, et al. EORTC consensus recommendations for the treatment of mycosis fungoides/Sézary syndrome. Eur J Cancer 2006;42:1014–30 [DOI] [PubMed] [Google Scholar]
- 2.Olsen E, Vonderheid E, Pimpinelli N, et al. Revisions to the staging and classification of mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organisation of Research and Treatment of Cancer (EORTC). Blood 2007;110:1713–22 [DOI] [PubMed] [Google Scholar]
- 3.Whittaker SJ, Marsden JR, Spittle M, et al. Joint British Association of Dermatologists and U.K. Cutaneous Lymphoma Group guidelines for the management of primary cutaneous T-cell lymphomas. Br J Dermatol 2003;149:1095–1107 10.1111/j.1365-2133.2003.05698.x [DOI] [PubMed] [Google Scholar]
- 4.Kim YH, Liu HL, Mraz-Gernhard S, et al. Long-term outcome of 525 patients with mycosis fungoides and Sézary syndrome: clinical prognostic factors and risk for disease progression. Arch Dermatol 2003;139:857–66 [DOI] [PubMed] [Google Scholar]
- 5.Agar NS, Wedgeworth E, Crichton S, et al. Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer staging proposal. J Clin Oncol 2010;28:4730–9 [DOI] [PubMed] [Google Scholar]
- 6.Benton EC, Crichton S, Agar NS, et al. Cutaneous Lymphoma International Prognostic Index (CLIPI). Br J Dermatol 2010;163Suppl 18–11 [Google Scholar]
- 7.Kaye FJ, Bunn PA, Jr, Steinberg SM, et al. A randomized trial comparing combination electron-beam radiation and chemotherapy with topical therapy in the initial treatment of mycosis fungoides. N Engl J Med 1989;321:1784–90 10.1056/NEJM198912283212603 [DOI] [PubMed] [Google Scholar]
- 8.Scarisbrick JJ, Taylor P, Holtick U, et al. U.K. consensus statement on the use of extracorporeal photopheresis for treatment of cutaneous T-cell lymphoma and chronic graft-versus-host disease. Br J Dermatol 2008;158:659–78 10.1111/j.1365-2133.2007.08415.x [DOI] [PubMed] [Google Scholar]
- 9.Duvic M, Donato M, Dabaja B, et al. Total skin electron beam and non-myeloablative allogeneic hematopoietic stem-cell transplantation in advanced mycosis fungoides and Sézary syndrome. J Clin Oncol 2010;28:2365–72 [DOI] [PubMed] [Google Scholar]