

ABSTRACT
Severe cutaneous adverse reactions (SCARs) can present in a multitude of ways including Stevens–Johnson syndrome, toxic epidermal necrolysis, drug reaction with eosinophilia and systemic symptoms, and acute generalised exanthematous pustulosis. While the prognosis and therapy for these conditions may vary, it is crucial that the culprit drug is identified and withheld early as this can influence patient outcome. Mainstay of management is by supportive therapy. In all SCARs, long-term sequelae which may not be apparent initially can be debilitating and cause lasting impact on the quality of life of survivors.
KEYWORDS: Severe adverse drug reactions, Stevens–Johnson syndrome, toxic epidermal necrolysis, acute generalised exanthematous pustulosis, drug reaction with eosinophilia and systemic symptoms
Key points
Severe cutaneous adverse reactions (SCARs) include Stevens–Johnson syndrome, toxic epidermal necrolysis, drug reaction with eosinophilia and systemic symptoms and acute generalised exanthematous pustulosis.
Possible genetic factors causing susceptibility to SCARs.
Treatment involves removal of the causative drug, supportive care and infection prevention.
Multi-system complications can impact quality of life of survivors.
Education of patient on avoidance of re-exposure is crucial.
Introduction
‘Drug reaction’ is an umbrella term encompassing all adverse events related to administration of a drug, regardless of immunological or non-immunological aetiology.1 The majority of reactions are not true allergies, occurring to any individual exposed to the drug in sufficient quantities, and are usually predictable, such as diarrhoea or development of candida following a course of antibiotics. On the other hand, allergic reactions to medications occur following an unpredictable immunological response to drug ingestion. These reactions cover a large spectrum of severity from mild and transient exanthems, to severe life-threatening systemic responses.
Serious adverse drug reactions (ADRs) account for 6.7% of drug reactions, with 0.32% of these being fatal in hospitalised patients,2 and can have lasting impact on the patient with multiple long-term complications following resolution of the acute episode. This article will discuss severe cutaneous adverse reactions (SCARs) – Stevens–Johnson Syndrome (SJS), toxic epidermal necrolysis (TEN), acute generalised exanthematous pustolosis (AGEP) and drug reaction with eosinophilia and systemic symptoms (DRESS).
Stevens–Johnson syndrome and toxic epidermal necrolysis
SJS and TEN, otherwise known as Lyell's syndrome, are terms used to describe a spectrum of delayed-type hypersensitivity phenotypes involving at least two mucosal surfaces, usually triggered by drugs, but can also less commonly occur secondary to infections. Presentation is usually with an acute febrile illness and target lesions, which rapidly evolve into widespread epithelial sloughing including the oral cavity, lips, conjunctivae and genitals (Figs 1 and 2).3 SJS is used to describe the less severe spectrum in which there is <10% body surface area (BSA) detachment; TEN for cases with >30% BSA detachment; and SJS-TEN overlap for those with 10–30% BSA detachment.4
Fig 1.

Female patient with toxic epidermal necrolysis to amoxicillin. Note the large haemorrhagic areas of denuded skin.
Fig 2.

A close-up of incipiently detached epidermis in TEN. Gentle lateral pressure on normal skin produces bullae or detachment, known as Nikolsky sign.
A prodromal phase of malaise, fever, arthralgia and stinging sensation of mucosal surfaces may precede the appearance of tender maculopapular, urticarial or petechial rash, with rapid spread over 3–4 days leading to desquamation. Detachment of the epidermis on application of gentle lateral pressure on the skin (Nikolsky's sign) is usually present. Commonly implicated drugs include antibacterials, anticonvulsants, non-steroidal anti-inflammatory agents and allopurinol (Table 1).5 Antibiotics are the most common cause, followed by non-steroidal anti-inflammatory agents.6,7
Table 1.
Drugs commonly causing SJS/TEN.
| Group | Drugs |
|---|---|
| Antibacterials | Sulphonamides, penicillins, cephalosporins, quinolones, vancomycin |
| Anticonvulsants | Phenytoin, carbamazepine, phenobarbitone, valproate, lamotrigine |
| NSAIDs | Aspirin, piroxicam, diclofenac, phenylbutazone |
| Antiretrovirals | Nevirapine, protease inhibitors, abacavir |
| Antituberculous | Isoniazid, ethambutol |
| Antigout | Allopurinol |
| Antimalarials | Chloroquine |
NSAIDs = non-steroidal anti-inflammatory drugs; SJS = Stevens–Johnson syndrome; TEN = toxic epidermal necrolysis.
The causative drug or its metabolites is thought to stimulate cytotoxic T cells and soluble factors which induce keratinocyte apoptosis, of which several factors, including soluble Fas ligand, granzyme and granulysin, have been postulated as being key in triggering cell death.8,9 Several HLA genotypes (HLA-B*1502, HLA-B5801, HLA-B*5701) have been implicated, largely in Asian populations, with increasing susceptibility to TEN with usage of antiepileptics and allopurinol.9 Incidence of SJS/TEN is reportedly 2/million/year,10 occurring more frequently in older individuals, with a mortality of 9% for SJS and 29% for TEN.11
Treatment is primarily supportive, with focus on prompt withdrawal of the culprit drug and symptom management, ideally in a burns unit or intensive care setting experienced in management of SJS/TEN. This would include pressure relief with air fluidised beds, anti-shear handling, lubrication particularly of mucosal surfaces to prevent adhesions, application of amniotic membranes to cornea, fluid and electrolyte replacement, and enteral nutrition, as well as thermoregulation support in view of the compromise in skin barrier. Detached epidermis acts as a natural dressing for the underlying denuded dermis and debridement of infected areas remains controversial.11 Sedation as part of pain management may be necessary.
The main cause of mortality is sepsis, with areas of epithelial detachment at risk of becoming infected. Patients should be monitored closely for development of infections in reverse isolation, and commenced on antimicrobial therapy should signs of infection arise. Approximately 10–20% of patients may require ventilation in the acute phase due to alveoli infiltration and bronchopneumonia.11 To date, there is no clear evidence to indicate definite benefit or harm from systemic therapies of corticosteroids, cyclosporine or intravenous immunoglobulin.12
A scoring system based on seven prognostic markers (SCORTEN) has been devised to predict prognosis in patients with epidermal necrolysis,13 based on age, presence of malignancy, heart rate, percentage of epidermal detachment, and serum levels of urea, glucose and bicarbonate. Hyper- or hypopigmentation frequently occurs following regeneration of epithelium. Chronic sequelae from SJS/TEN have been reported, including respiratory, gastrointestinal, oral, gynaecological, genitourinary and renal.14 However, ocular sequelae have been the most widely described. Chronic conjunctivitis, pseudomembrane formation and corneal scarring, may ensue leading to loss of vision. A list of further organ involvement is listed in Table 2. The multitude of complications can impact significantly on quality of life of patients, and clinicians need to be vigilant in initial preventative care and involvement of a variety of specialists to prevent further deterioration.
Table 2.
Complications of SJS/TEN.4
| Organ system | Complications |
|---|---|
| Cutaneous | Pigmentation abnormalities, nail deformities, alopecia |
| Ocular | Chronic conjunctivitis, pseudomembrane formation, trichiasis, corneal damage, cataracts, blindness |
| Respiratory | Bronchiolitis obliterans, bronchiectasis, chronic bronchitis |
| Gastrointestinal/hepatic | Oesophageal stricture, intestinal ulcer, chronic cholestasis, ischaemic hepatitis, vanishing bile duct syndrome |
| Oral | Dental hypoplasia |
| Gynaecologic | Vulval and vaginal adenosis, fusion of labia minora and majora, labial synechiae |
| Otolaryngologic | Hypopharyngeal stenosis, nasal septal synechiae, pinna synechiae |
| Renal | Glomerulonephritis |
| Haematological | Idiopathic thrombocytopaenic purpura |
SJS = Stevens–Johnson syndrome; TEN = toxic epidermal necrolysis.
Drug reaction with eosinophilia and systemic symptoms
Multiple terminologies have been used to refer to the collection of symptoms of lymphadenopathy with haematological derangement and systemic illness, including anticonvulsant hypersensitivity syndrome, allopurinol hypersensitivity syndrome, dapsone syndrome, febrile mucocutaneous syndrome, drug-induced hypersensitivity syndrome, drug-induced delayed multiorgan hypersensitivity syndrome or more simply as hypersensitivity syndrome,15 before the term DRESS was introduced in 1996 by Bocquet et al.16 Typical presentation of an affected patient would be with fever of >38°C, rash, lymphadenopathy, leucocytosis, eosinophilia and abnormal liver function tests. Cutaneous changes are most commonly of an urticated, maculopapular eruption but also can present with vesicles, bullae, pustules or purpura (Fig 3). Facial oedema is often present, particularly in the periorbital distribution,17 as is interstitial inflammation involving the liver and kidneys.18 Notably the extent of skin involvement can vary from patient to patient, with a long latent period between introduction of a medication and the initial cutaneous changes of 3–8 weeks,19 therefore requiring a high index of suspicion in order to arrive at a diagnosis of DRESS. Drugs most commonly associated with DRESS include anticonvulsants, allopurinol, minocycline, sulfasalazine and abacavir.18–19 Mortality from DRESS is significant at 10%, the majority of which are due to liver failure.15,20
Fig 3.
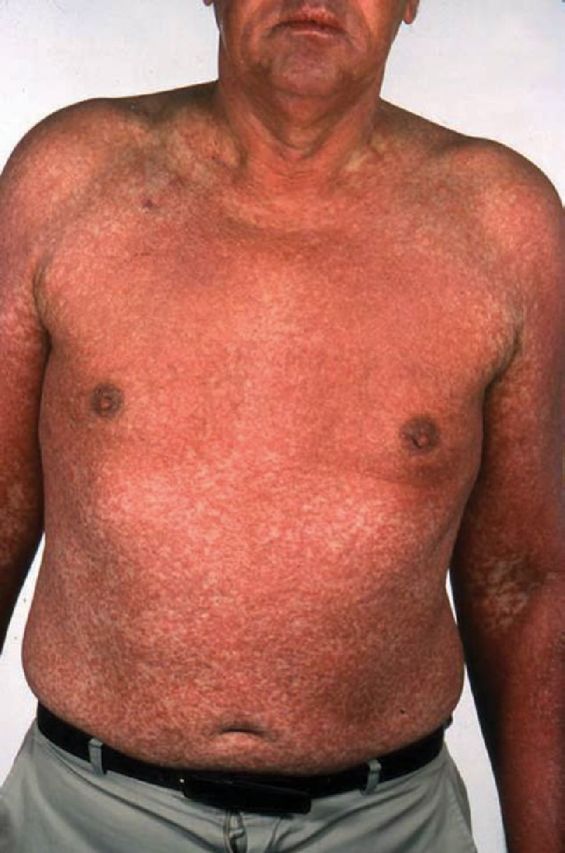
A male patient with drug reaction with eosinophilia and systemic symptoms to allopurinol. Note the widespread maculopapular urticated eruption, classic of this disease.
Inter-individual variation is felt to be a reflection of genetic and environmental factors resulting in the spectrum of severity seen. It has been postulated that accumulation of anticonvulsant metabolites as a result of failure of drug detoxification pathways results in a predisposition to ADRs. A strong association between allopurinol-DRESS or SJS and presence of HLA-B*5801 was demonstrated in a study of a population of Han Chinese, although there has been no definitive data on function of the implicated alleles.21 In order to aid diagnosis in view of the varied presentations, the RegiSCAR group have recommended a set of diagnostic criteria, the inclusion criteria as listed in Box 1; however, these are based on application to hospitalised patients.22 A separate group in Japan have included human herpesvirus 6 activation as a diagnostic criterion, based on previous postulation of transient drug-induced hypogammaglobulinaemia enabling virus reactivation in the acute phase of DRESS.23
Box 1.
Inclusion criteria for potential cases of DRESS.22
| > hospitalisation |
| > reaction suspected to be drug related |
| > acute skin rash |
| > fever >38°C |
| > enlarged lymph nodes at ≥2 sites |
| > involvement of ≥1 internal organ |
| > blood count abnormalities |
| > lymphocytes above or below the laboratory limits |
| > eosinophils above the laboratory limits |
| > platelets below the laboratory limits. |
When there is involvement of internal organs recommended treatment is with systemic corticosteroids with gradual tapering in addition to topical treatment, supportive care and withdrawal of the offending drug, alongside monitoring of haematological, hepatic and renal parameters.17 Data from other forms of therapy have been inconclusive.17
Acute generalised exanthematous pustulosis
AGEP is characterised by an eruption consisting of sheets of sterile, non-follicular pustules arising on an erythematous backdrop, particularly in flexural areas (Figs 4 and 5). It can be distinguished from the other SCAR syndromes both from the morphology of the rash, but also by the short latency between ingestion of the culprit medication, and the onset of the rash, usually no longer than 5 days and often as short as 48 hours.24 Mucosal involvement is rare, and if present, is usually limited to one site, typically cheilitis of the lips. Mortality tends to be <5% in AGEP, with poorest outcome linked to those with multiple comorbidities.24,25 Drugs implicated in AGEP include aminopenicillins, macrolides, quinolones, terbinafine, carbamazepine and antimalarials; however unlike SJS/TEN, allopurinol and antiepileptic drugs do not appear to play a major trigger for AGEP.24–26 Unlike in SJS/TEN where there appears to be low sensitivity and high risk of systemic reactions, patch testing appears to be useful in AGEP to identify the responsible drug.27,28
Fig 4.
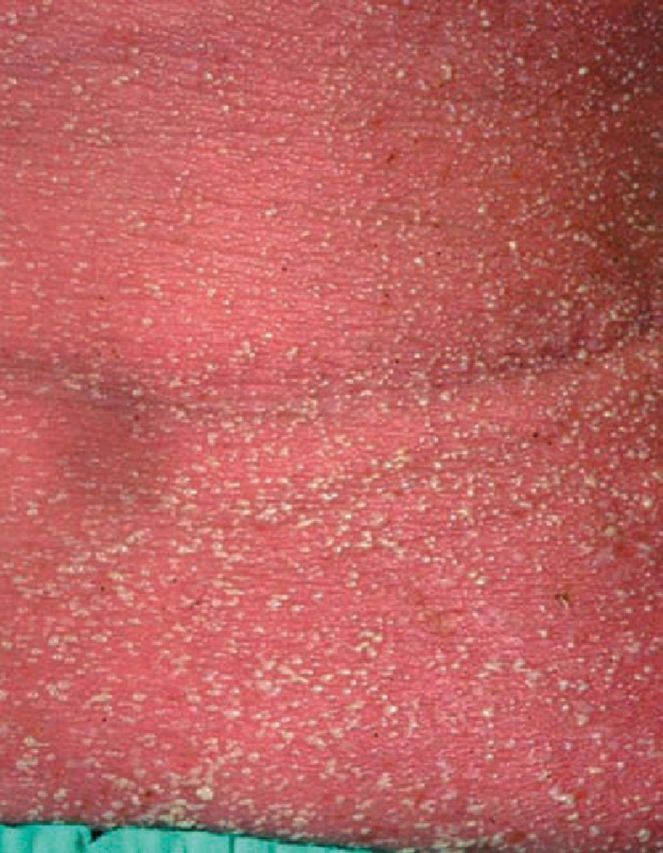
Acute generalised exanthematous pustulosis secondary to flucloxacillin. Note the sheets of monomorphic pustules on an erythematous base.
Fig 5.

The pustules of acute generalised exanthematous pustulosis are classically found at flexural sites.
Drug-specific T cells activated by the causative agent infiltrate the epithelium, induce blistering through perforin/granzyme B and Fas ligand mechanisms to induce apoptosis, and express interleukin-8 causing subsequent neutrophil migration which form pustules. A leucocytosis, typically a neutrophilia may be present. Eosinophils are less commonly elevated. A recent study of 58 patients with AGEP suggested that involvement of internal organs may be present in up to 17% of patients with AGEP, including hepatic, renal and pulmonary dysfunction.29 Agranulocytosis has also been described, suggesting that bone marrow may rarely be affected. Systemic involvement of this kind appear to be self-limiting and resolve spontaneously, while cutaneous features resolve within a few days with desquamation of the affected area.24 Mild cases can be treated with topical corticosteroids and oral antihistamines, while severe cases may necessitate systemic corticosteroids.
Conclusion
Diagnosis of severe ADRs requires detailed history taking, including medications commenced several months ago, as continued administration of the culprit drug can lead to worsening of symptoms and poorer prognosis. Due to the rigorous skin management regimes required, patients are best managed in an intensive care setting or specialist burns units, with particular focus on fluid replacement, nutrition, analgesia and management of mucosal involvement. Given that the sequelae of the disease is impossible to predict, patients should be followed up and monitored for signs of complications following resolution of the acute phase, as well as provided with education on avoidance of the causative medication.
References
- 1.Riedl MA, Casillas AM. Adverse drug reactions: types and treatment options. Am Fam Physician 2003;68:1781–91. [PubMed] [Google Scholar]
- 2.Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998;279:1200–5. [DOI] [PubMed] [Google Scholar]
- 3.Letko E, Papatiodis DN, Papaliodis GN, et al. Stevens–Johnson syndrome and toxic epidermal necrolysis: a review of the literature. Ann Allergy Asthma Immunol 2005;94:419–36. [DOI] [PubMed] [Google Scholar]
- 4.Bastuji-Garin S, Rzany B, Stern RS, et al. Clinical classification of cases of toxic epidermal necrolysis, stevens-johnson syndrome, and erythema multiforme. Arch Dermatol 1993;129:92–6. [PubMed] [Google Scholar]
- 5.Verma R, Vasudevan B, Pragasam V. Severe cutaneous adverse drug reactions. Med J Armed Forces India 2013;69:375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schopf E, Stuhmer A, Rzany B, et al. Toxic epidermal necrolysis and Stevens–Johnson syndrome: an epidemiologic study from West Germany. Arch Dermatol 1991;127:839–42. [DOI] [PubMed] [Google Scholar]
- 7.Roujeau JC, Guillaume JC, Fabre JP, et al. Toxic epidermal necrolysis (Lyell syndrome): incidence and drug etiology in France, 1981–1985. Arch Dermatol 1990;126:37–42. [DOI] [PubMed] [Google Scholar]
- 8.Khalili B, Bahna SL. Pathogenesis and recent therapeutic trends in stevens-johnson syndrome and toxic epidermal necrolysis. Ann Allergy Asthma Immunol 2006;97:272–80. [DOI] [PubMed] [Google Scholar]
- 9.Chung WH, Hung SI. Genetic markers and danger signals in Stevens–Johnson syndrome and toxic epidermal necrolysis. Allergol Int 2010;59:325–32. [DOI] [PubMed] [Google Scholar]
- 10.Rzany B, Mockenhaupt M, Baur S, et al. Epidemiology of erythema exsudative multiforme majus, Stevens–Johnson syndrome, and toxic epidermal necrolysis in Germany (1990-1992): structure and results of a population-based registry. J Clin Epidemiol 1996;49:769–73. [DOI] [PubMed] [Google Scholar]
- 11.Mockenhaupt M. Stevens–Johnson syndrome and toxic epidermal necrolysis: clinical patterns, diagnostic considerations, etiology and therapeutic management. Semin Cutan Med Surg 2014;33:10–6. [DOI] [PubMed] [Google Scholar]
- 12.Creamer D, Walsh SA, Dziewulski P, et al. Draft UK guidelines for the management of Stevens–Johnson syndrome/toxic epidermal necrolysis in adults 2015. London: BAD, 2015. [Google Scholar]
- 13.Bastuju-Garin S, Fouchard N, Bertocchi B, et al. SCORTEN: a severity-of-illness score for toxic epidermal necrolysis. J Invest Dermatol 2000;115:149–53. [DOI] [PubMed] [Google Scholar]
- 14.Saeed H, Mantagos IS, Chodosh J. Complications of Stevens–Johnson syndrome beyond the eye and skin. Burns 2015;S0305–4179. [DOI] [PubMed] [Google Scholar]
- 15.Walsh SA, Creamer D. Drug reaction with eosinophilia and systemic symptoms (DRESS): a clinical update and review of current thinking. Clin Exp Dermatol 2011;36:6–11. [DOI] [PubMed] [Google Scholar]
- 16.Bocquet H, Bagot M, Roujeau JC. Drug-induced pseudolymphoma and drug hypersensitivity syndrome (drug rash with eosinophilia and systemic symptoms:DRESS). Semin Cutan Med Surg 1996;15:250–7. [DOI] [PubMed] [Google Scholar]
- 17.Paulmann M, Mockenhaupt M. Severe drug-induced skin reactions: clinical features, diagnosis, etiology and therapy. J Dtsch Dermatol Ges 2015;13:625–45. [DOI] [PubMed] [Google Scholar]
- 18.Kardaun SH, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and sytemic symptoms (DRESS): an original multisystem adverse drug reaction. Results from prospective RegiSCAR study. Br J Dermatol 2013;169:1071–80. [DOI] [PubMed] [Google Scholar]
- 19.Eshki M, Allanore L, Musette P, et al. Twelve-year analysis of severe cases of drug reaction with eosinophilia and systemic symptoms: a cause of unpredictable multiorgan failure. Arch Dermatol 2009;145:67–72. [DOI] [PubMed] [Google Scholar]
- 20.Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review. Am J Med 2011;124:588–97. [DOI] [PubMed] [Google Scholar]
- 21.Hung SI, Chung WH, Liou LB, et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci USA 2005;102:4134–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kardaun SH, Sidoroff A, Valeyrie-Allanore L, et al. Variability in the clinical pattern of cutaneous side-effects of drugs with systemic symptoms: does a DRESS syndrome really exist? Br J Dermatol 2007;156:609–10. [DOI] [PubMed] [Google Scholar]
- 23.Shiohara T, Iljima M, Ikezawa Z, et al. The diagnosis of DRESS syndrome has been sufficiently established on the basis of typical clinical features and viral reactivations. Br J Dermatol 2007;156:1045–92. [DOI] [PubMed] [Google Scholar]
- 24.Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP): a review and update. J Am Acad Dermatol 2015;73:843–8. [DOI] [PubMed] [Google Scholar]
- 25.Fernando SL. Acute generalised exanthematous pustulosis. Australas J Dermatol 2012;53:87–92. [DOI] [PubMed] [Google Scholar]
- 26.Sidoroff A, Dunant A, Viboud C, et al. Risk factors for acute generalised exanthematous pustulosis (AGEP) – results of a multinational case-control study (EuroSCAR). Br J Dermatol 2007;157:989–96. [DOI] [PubMed] [Google Scholar]
- 27.Wolkenstein P, Chosidow O, Fletchet ML, et al. Patch-testing in severe cutaneous adverse drug reactions including Stevens–Johnson syndrome and toxic epidermal necrolysis. Contact Dermatitis 1996;35:234–6. [DOI] [PubMed] [Google Scholar]
- 28.Barbaud A, Collet E, Milpied B, et al. A multicentre study to determine the value and safety of drug patch tests for the three main classes of severe cutaneous adverse drug reactions. Br J Dermatol 2013;168:555–62. [DOI] [PubMed] [Google Scholar]
- 29.Hotz C, Valeyrie-Allanore L, Haddad C, et al. Systemic involvement of acute generalised exanthematous pustulosis: a retrospective study on 58 patients. Br J Dermatol 2013;169:1223–32. [DOI] [PubMed] [Google Scholar]