

Relevant preclinical benchmarks require rethinking (i.e., identity and purity) or need to be adjusted (i.e., potency and viability) to anticipate safety and efficacy concerns related to the dynamic heterogeneity of human embryonic stem cells (hESCs).This article reviews existing regulatory frameworks for preclinical requirements of first-in-human trials and assesses how their underlying principles may best be applied in the context of hESC-based interventions for treatment of Parkinson's disease.
Keywords: First-in-human trials, Human embryonic stem cells, Preclinical benchmarks, Research ethics, Translational research
Abstract
As research on human embryonic stem cell (hESC)-based therapies is moving from the laboratory to the clinic, there is an urgent need to assess when it can be ethically justified to make the step from preclinical studies to the first protocols involving human subjects. We examined existing regulatory frameworks stating preclinical requirements relevant to the move to first-in-human (FIH) trials and assessed how they may be applied in the context of hESC-based interventions to best protect research participants. Our findings show that some preclinical benchmarks require rethinking (i.e., identity, purity), while others need to be specified (i.e., potency, viability), owing to the distinctive dynamic heterogeneity of hESC-based products, which increases uncertainty and persistence of safety risks and allows for limited predictions of effects in vivo. Rethinking or adaptation of how to apply preclinical benchmarks in specific cases will be required repeatedly for different hESC-based products. This process would benefit from mutual learning if researchers included these components in the description of their methods in publications.
Significance
To design translational research with an eye to protecting human participants in early trials, researchers and regulators need to start their efforts at the preclinical stage. Existing regulatory frameworks for preclinical research, however, are not really adapted to this in the case of stem cell translational medicine. This article reviews existing regulatory frameworks for preclinical requirements and assesses how their underlying principles may best be applied in the context of human embryonic stem cell-based interventions for the therapy of Parkinson's disease. This research will help to address the question of when it is ethically justified to start first-in-human trials in stem cell translational medicine.
Introduction
Research on human embryonic stem cell (hESC)-based therapies for several diseases has reached encouraging results at preclinical stages [1–4], and moves cautiously from the laboratory to the clinic [5–7]. Although some political and religious positions still defend views, according to which hESC-based therapies could be more morally suspect than other therapies, other, arguably more pressing, issues must be examined. In particular, given the novelty and the unique characteristics of hESC-derived products, ethical challenges of human subject research are more complex in this context and there is a need to define appropriate ethical frameworks for the first protocols involving human subjects.
Until recently, most of the literature on bench-to-approval research on hESC-based therapies has focused on how to design first-in-human (FIH) trials [8–17]. This discussion suggests that, provided participants in FIH trials of hESC-based interventions are not exposed to excessive risks, these trials can be designed in an ethically sound way [17]. Less attention has been given to ethical questions regarding the move from the preclinical stage to clinical trials. Although there are detailed regulations on the preclinical requirements to initiate FIH trials, it can be difficult to determine where current standards apply to novel cellular therapies. Early phase I trials in the field of human gene transfer have already raised similar concerns [18]. Hence, it is difficult to determine when to start first phase I trials (i.e., to know when the relevant prerequisites have been met). Moreover, it is likely that this step will be required repeatedly for different hESC-based products.
Similar to discussions on how to design phase I experiments, the question about when to start FIH trials involves balancing uncertainty with potential benefits of research. However, the “when” question takes priority: It aims to determine the relevant prerequisites that have to be met in the transition from preclinical to human experimentation, to evaluate whether to initiate FIH trials. Moreover, the when question has important implications for the design of first phase I trials. For example, defining clinical safety endpoints of FIH trials depends on the assessment of preclinical requirements that can be predictive of safety in humans [19]. Thus, focusing on the when question is to address primary and fundamental ethical issues in translational stem cell research, and to provide key elements to answer the question about the ethical justification of FIH hESC-based interventions.
Materials and Methods
This study was conducted as part of PROMETHEUS (Pluripotent stem cells for therapy of Parkinson's disease: a multidisciplinary translational consortium), a project exploring the preclinical stage of development of an hESC-based therapy for Parkinson's disease (PD), but we have explored the broader issue of research on hESC generally.
Unlike most other discussions on FIH trials of novel therapeutic interventions, we did not proceed deductively from general ethical principles but examined existing regulatory frameworks stating scientific requirements relevant to the move from preclinical to human studies, to explore their ethical justification. We then assessed how these principles may best be adapted or rethought in the context of FIH transplantation of hESC-derived dopaminergic progenitors for the therapy of PD.
We conducted a literature search of guidelines produced by the most important regulatory bodies of new medicinal products in Europe, the U.S., and at the international level, including the European Medicines Agency (EMA), the U.S. Food and Drug Administration (FDA), and the International Conference on Harmonization (ICH). We selected 24 documents, including general ethical guidelines on research with human subjects, procedural guidelines on manufacturing and testing of medicinal products, and specific guidelines on cellular therapies. Within this list, we identified 18 documents (Table 1) that provide guidance on the move from preclinical studies to FIH trials.
Table 1.
List of documents and preclinical benchmarks

Data collection was conducted through a systematic review of the documents to identify the most recurrent preclinical requirements that should be met to make the step from preclinical to clinical studies (Table 1). Collaboration with preclinical and clinical researchers involved in the project was essential to identify requirements for the protection of human subjects, which could have implications in preclinical stages research.
Data analysis consisted of a thorough examination of each preclinical requirement, to consider how those developed for conventional pharmaceuticals and/or biologicals needed to be redefined for application to cellular products.
Results
Relevant Frameworks and the Values Underlying Them
Most guidelines relevant to the move from preclinical to clinical studies apply to classical pharmaceutical products or to biopharmaceuticals (including both living and nonliving biologicals), or to somatic cellular and tissue-based therapies (Table 1). Only a small number of documents cover specific issues associated with the development of hESC-based interventions, often as additional provisions in guidelines on cell-based interventions in general [32, 33, 35].
Current regulatory standards are aimed at ensuring that, before their clinical application, investigational products prove to be safe, effective, and of sufficient quality for their intended medicinal use. Preclinical studies should provide supporting evidence to assess and anticipate possible effects of experimental medicinal products in humans. Specifically, the preclinical stage of development of new medicinal products should assess the stability of safety risks to allow predictions of safety in preparing for risks assessment in clinical trials, and establish proof-of-principle of efficacy to confirm the plausibility of the research hypothesis that will be tested in humans. Ultimately, the background ethical justification of this framework is to ensure that future patients would benefit from new therapies and that human subjects of research are not exposed to excessive or avoidable risk.
The ICH provides general guidance on quality, safety, and efficacy standards for pharmaceutical and biotechnological products through guidelines on good manufacturing practices (GMPs) [36]. Additional provisions on preclinical requirements are detailed in ICH guidelines on test procedures for product-specific properties [20–25]. Although current ICH standards are, to a certain extent, also relevant to novel cellular therapies [38], it can be difficult to determine where they apply to cell-based interventions that only partially meet concepts developed for pharmaceuticals and biologicals. For this reason, regulation on drug development in the U.S. and in Europe is being adapted to address concerns associated with the distinct characteristics of cell-based products [39].
In the U.S., cells and tissue-based products are regulated under Part 1271 of the Code of Federal Regulations [29]. FDA guidelines provide specifications for manufacturing, and preclinical and clinical testing of human somatic-cell therapy investigational products [30]. Because a distinct regulation on hESC-based therapies is not underway, some commentators made specific recommendations on how FDA standards may be applied to preclinical and clinical testing of these products [40, 41]. Moreover, the FDA offers assistance for regulatory compliance in the development of hESC-derived products [35].
In Europe, Directive No. 1394/2007 on advanced therapy medicinal products (ATMPs) provides a framework for specialized guidelines to be developed on how to adapt existing GMP and good clinical practice standards to novel cell therapies [42]. According to the EU directive, ATMPs include gene therapy, tissue-engineered products, and somatic cellular therapy. The rationale for grouping together somatic cell-, gene-, and tissue-based therapies is that they share several common characteristics and represent a distinct class of products, separate from pharmaceuticals or classical biologicals. The EMA made the first tentative steps through the Guideline on Human Cell-Based Medicinal Products [28], which specifies quality requirements for manufacturing, preclinical development, and clinical testing of cellular therapies. In addition, EMA recently adopted a reflection paper to address particular concerns common to different types of stem cell therapies, including those derived from hESCs [32].
The Limits of Existing Regulations as Applied to hESC-Based Products
Existing regulations presume a paradigm of drug development that does not provide clear-cut guidance, or provides misleading guidance when applied to novel interventions such as hESC-based therapies [43]. Compared with conventional small-molecule drugs, biologicals, and somatic cellular therapies, hESC-based interventions have unique characteristics that raise new quality, safety, and efficacy concerns in the transition from preclinical to clinical studies [8, 10, 12, 41, 44, 45].
Common principles of pharmacokinetics and toxicology used in drug development, such as absorption and excretion, are not relevant to assess safety risks associated to hESCs, which can persist and develop over time when transplanted. Based on current knowledge, at least four types of risk need to be further evaluated through preclinical in vitro and animal testing: misdifferentiation, mistargeting, tumor formation, and immune rejection [10, 12, 44]. Specific data from preclinical experiments are required to assess these risks. Moreover, preclinical evaluation of proof-of-principle of efficacy of hESC-based products should be adjusted to assess characteristics such as viability, migration, and differentiation of transplanted cells.
To some extent, safety, and efficacy concerns can be addressed through a multiparametric testing approach for characterization of hESC-based products during manufacturing and in vitro controls to anticipate effects in vivo. However, because of the dynamics of hESC-derived compounds, some pivotal preclinical benchmarks remain necessary; applying them appropriately is not straightforward, and can require clarification or rethinking to adequately address quality, safety, and efficacy concerns in the move from preclinical to FIH studies of hESC-based interventions.
Analysis and Discussion of Preclinical Benchmarks
Established ethical principles of research with human subjects, such as the need for a favorable risk-benefit ratio and the social value of research, underlie preclinical criteria such as the predictability of safety and proof-of-principle of efficacy of investigational products in humans. These criteria form the basis for preclinical “benchmarks” assessed through specific in vitro and in vivo tests. To address the when question (i.e., to evaluate whether to initiate FIH trails), preclinical benchmarks to be met in the transition from preclinical to human experimentation have to be assessed. The most important benchmarks are identity, purity, potency, and viability. Simply stated, these benchmarks aim to answer the following questions: “What is this stuff?,” “Is there other stuff?,” “How strong is this stuff?,” and “Will it stay effective?”
In this report, we analyze the most recurrent preclinical benchmarks (i.e., identity, purity, potency, and viability) in regulatory documents on the development of pharmaceuticals, biologicals, and cellular therapies to clarify their specific function and background ethical justification; and, we discuss their potential application in the context of hESC-based products. Tables 2–5 outline how these benchmarks are described in existing regulations on pharmaceuticals, biologicals, or cellular products. Text boxes highlighted in gray include our considerations on how the benchmarks should be applied to hESC interventions.
Table 2.
Identity: outline of benchmark
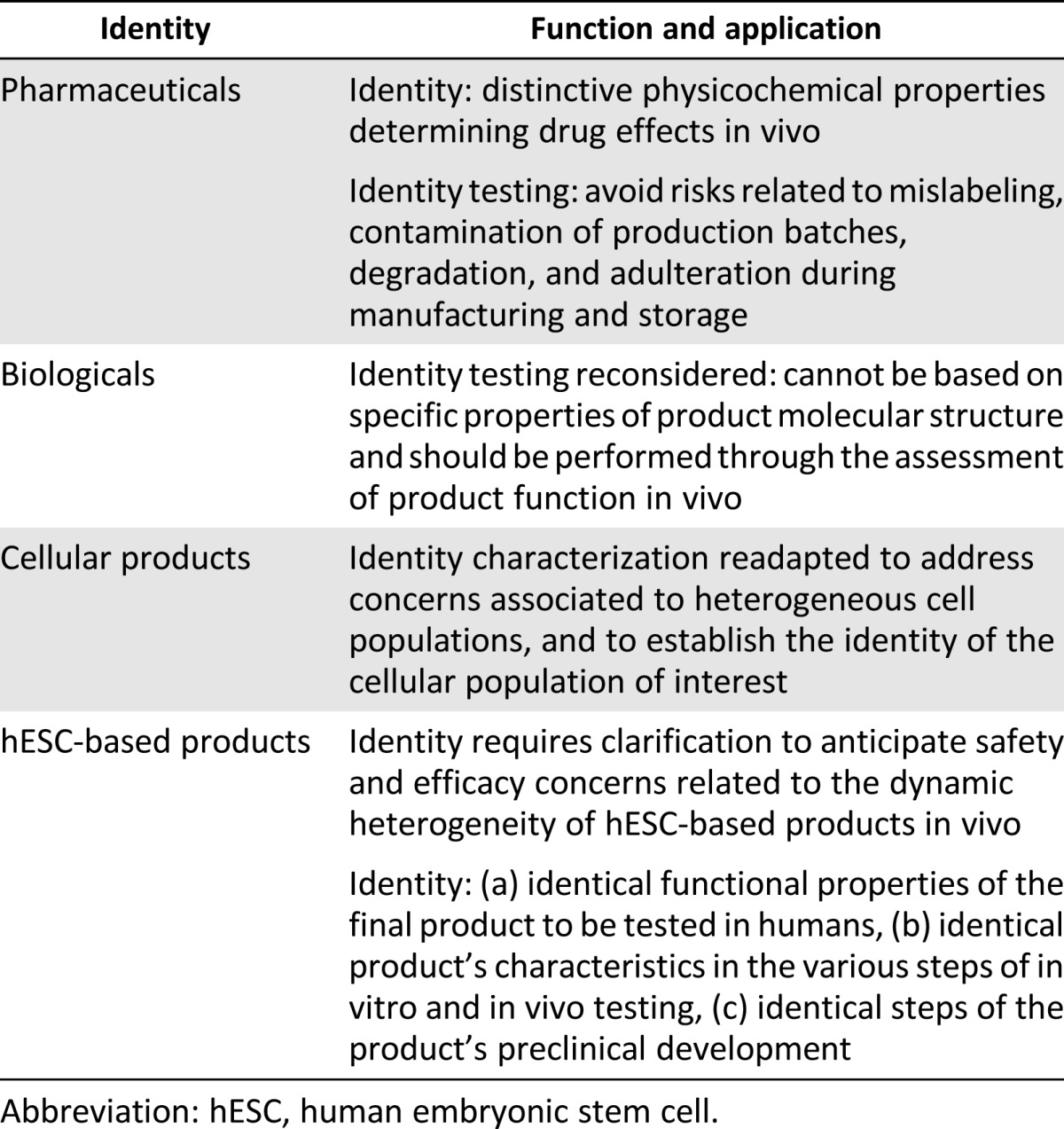
Table 5.
Viability: outline of benchmark
Identity: “What Is This Stuff?”
Identity (Table 2) is a core quality benchmark to assess the suitability of pharmaceutical products for their intended use. The primary aim of identity assays is to discriminate between a particular drug and other pharmaceutical compounds on the basis of the distinctive physicochemical properties that determine drug effects in vivo. Specifically, preclinical identity testing is important to anticipate and evaluate risks related to mislabeling, contamination of production batches, and degradation and adulteration during manufacturing and storage. Because these risks may affect both the safety and the therapeutic effect of a medicinal product, the ethical justification for preclinical identity assessment is to allow predictions of safety risks and proof-of-principle of efficacy in humans.
Biologicals and cell-based products raise unprecedented problems for the application of identity requirements. Unlike conventional pharmaceuticals, identification of biological compounds cannot be based on distinctive properties of a product’s molecular structure, and should be performed through the assessment of the product’s function in vivo. Thus, when compared with chemical drugs, identification of biologicals is reversed because it is necessary to proceed from the product’s effects to its specific properties that are predictive of safety and efficacy.
Identity requirements are further adapted to suit specific characteristics of cellular therapies, where they apply to starting cell materials as well as to noncellular components of chemical or biological origin that may be integral parts of the final product (e.g., reagents and excipients used in manufacture). Because cellular products may consist of heterogeneous cell phenotypes, existing regulations recommend that identity characterization be performed through relevant genotypic, phenotypic, or other markers to distinguish between the multiple cell lines used and to establish the identity of the cell population of interest [28, 30]. For example, for cellular therapies for which identity concerns mainly reflect the variability of the source of cells, identification should include histocompatibility markers for cells of allogeneic origin [28, 33].
In the absence of clear-cut guidance on hESC-derived products, EMA suggests that a combination of markers be used to establish the identity of the cellular population that constitutes the product before release, and that appropriate identity markers be based on the expected function of the product in vivo [32]. The application of identity to hESC-based products, however, should be further clarified and partially reassessed to anticipate safety and efficacy concerns related to the dynamic heterogeneity of hESCs.
A possible approach would be to go beyond a rigorous separation between identity and other preclinical benchmarks. Thus, identity may be considered as a general marker for the assessment of the functional stability of hESC products, because biological markers of all relevant preclinical benchmarks should prove identical from batch to batch. In particular, identity characterization may address three aspects of hESC-based products: identity of functional properties of the final product to be tested in humans, identity of other relevant products’ characteristics in the various steps of in vitro manufacturing and in vivo testing on animal models, and identity of steps of the overall product’s preclinical development. While the first two aspects need to be satisfied, the third is always necessary, in addition to identity characterization of hESC-derived compounds. This would allow the answer to the question “what is this stuff?” to be reliable, despite the differences between stable chemical substances and dynamic biological ones.
As yet, neither of the first two aspects have been assessed for hESC-based products for the therapy of PD. Preclinical studies show that the survival rate of dopamine neurons progenitors derived in vitro decreases when cells are transplanted in vivo [46]. Moreover, evidence of cell surface markers of neural differentiation in vitro associated with cells’ potential to develop into functional midbrain dopaminergic neurons in vivo is still lacking. This indicates that further research on preclinical identity markers, either based on the expected function of the product in vivo or on other cellular characteristics, is needed to prepare to FIH of hESC-derived compounds.
Purity: “Is There Other Stuff?”
According to relevant common usages, the term purity refers to the quality of a substance of being molecularly homogeneous, that is, of not being mixed or adulterated with other substances or extraneous materials (i.e., impurities). The function of purity testing in drug development (Table 3) is to assess the quality of the product for the intended therapeutic use through control of contamination from residual material or microbial agents [36]. The background ethical justification for purity assessment in preclinical studies is to provide supporting evidence of the stability of safety risks and estimate proof-of-principle of efficacy in preparing for clinical studies. General quality standards do not require an absolute assessment of purity but involve the specification of acceptable levels of contamination. Thus, purity is defined as the relative freedom of a pharmaceutical compound from extraneous or residual materials that may be introduced during the manufacturing process. Conversely, impurity is generally defined as any component present in the active pharmaceutical ingredient that is not the desired entity.
Table 3.
Purity: outline of benchmark
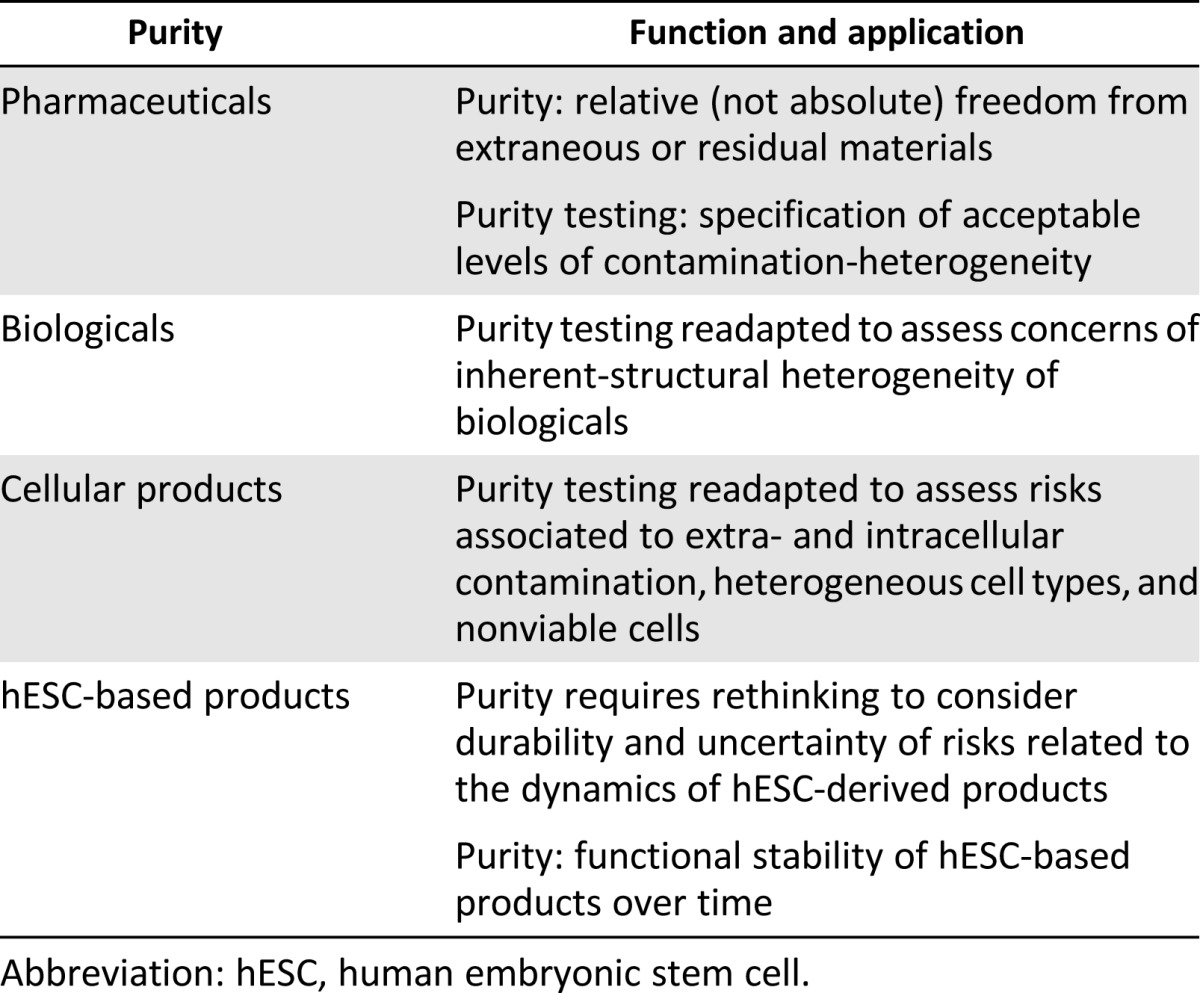
In the context of biological products, the concept of purity developed for pharmaceuticals is partially adapted because of the impossibility of sterilizing compounds. Concerns about contamination are revised and refer to impurities and adventitious contaminants related to the inherent-structural heterogeneity of biologicals, which is due to biosynthetic process used in manufacture [24, 27].
The concept of purity is further readjusted and specified for cellular therapies. Purity concerns are associated with contaminating materials that are product or process related, such as reagents and components used in the manufacture of cell-based products, or adventitious microbial agents that may be introduced in the final product [28, 30]. Moreover, purity characterization of cellular products includes the definition of acceptance criteria for heterogeneous cell phenotypes and nonviable cells that may affect the safety and/or the biological activity of a cell-based product [28].
In the context of hESC-based products, application of purity requirements in terms of acceptable levels of contamination raises concerns because assessing the risk of transmission of unknown diseases will necessarily be imperfect. This risk can be minimized in its likelihood using clinical-grade cells manufactured through animal-product-free maintenance media [47]. However, where hESC-based products integrate quiescent forms of adventitious agents (i.e., retrovirus that cannot be easily detected when inactive), the magnitude of safety risks associated with contamination is intrinsically less manageable. Because cells self-renew and proliferate, hESC-based interventions involve risks that are potentially more durable and cannot be easily addressed in manufacture through point-in-time validation of preclinical benchmarks such as purity.
Moreover, the heterogeneity of the cellular population should not be considered as a merely static attribute of the final product: hESC-based products are characterized by a “dynamic” heterogeneity because the cellular population develops over time. Engineering cells to express suicide genes that can be switched on upon tumor formation is a possible strategy to minimize risks related to the dynamics of stem cell interventions [48]. However, strategies intended to reduce risks related to the dynamics of hESC products would not allow predictions of safety. Likewise, estimations of efficacy cannot be adequately assessed. Even where fluorescence-activated cell-sorting (FACS) techniques would target the cellular population of interest, uncertainty related to the dynamics of hESC-based products cannot be easily assessed through point-in-time identification of the desired cell population.
To successfully address the persistence of risks and the uncertainty of effects related to the dynamic heterogeneity of hESC-based products, testing for purity should include the assessment of the functional stability of the product over time. That is, purity testing should allow the assessment of characteristics of the cellular population that are predictable from batch to batch. The question then becomes: What characteristics of the cellular population would be most predictive of the functional stability of hESC-based product? In the context of hESC-therapy for PD, one possible marker of the functional stability of the cellular product may be the degree of maturation of hESC-derived dopaminergic precursors before transplantation. However, as explained, FACS techniques used to measure in vitro proliferation and pluripotency markers associated to tumor proliferation (e.g., Ki67, Oct3/4, Nanog) do not allow predictions of functional stability in vivo, because of the dynamics of hESC-based compounds.
Potency: “How Strong Is This Stuff?”
Potency as a benchmark in preclinical development of medicinal products appears for the first time in guidelines on biologicals (Table 4). It tries to capture the idea of strength used for conventional small-molecule drugs to indicate the quantitative measure of the active substance, which is related to the desired therapeutic effect.
Table 4.
Potency: outline of benchmark
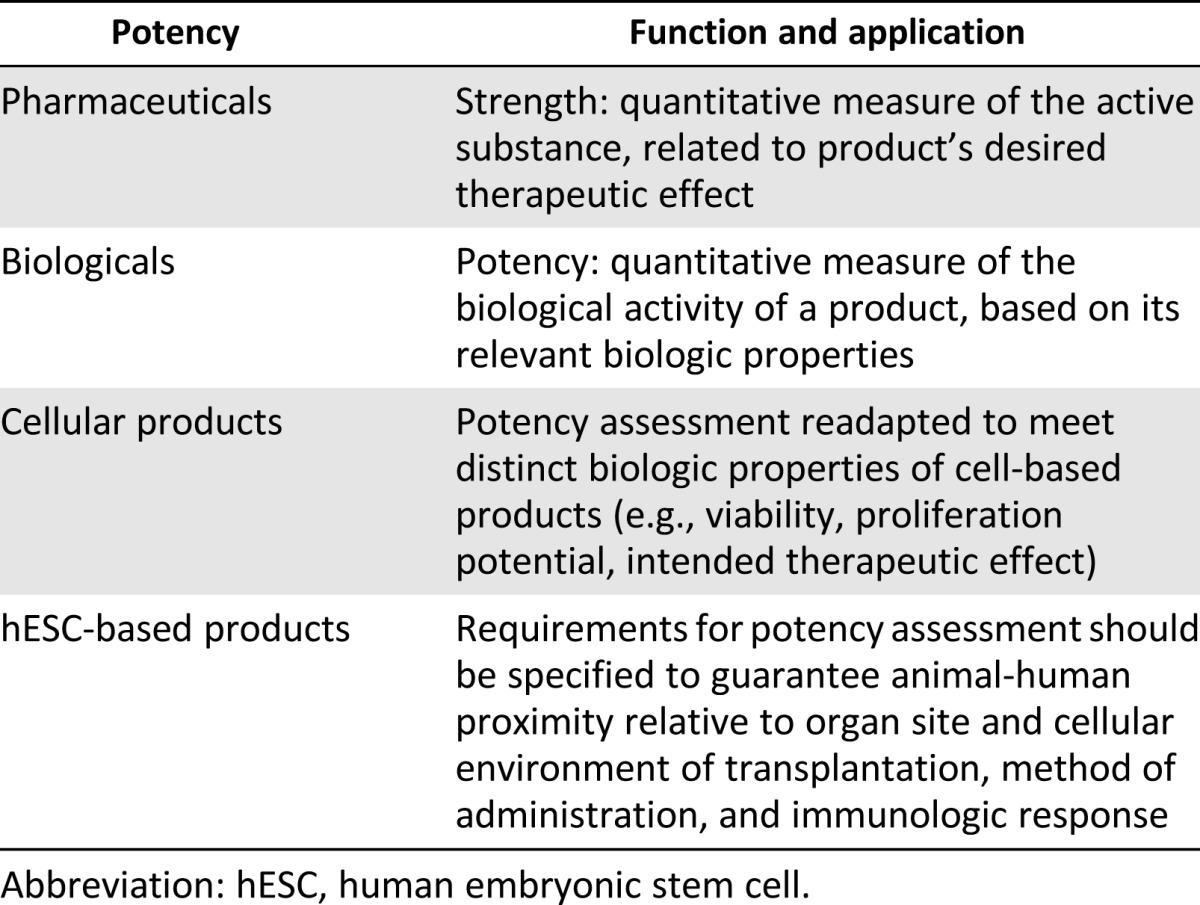
Guidelines on biopharmaceuticals define potency as the quantitative measure of the biological activity of a product, based on its relevant biologic properties [24, 25]. Potency assays apply to the overall manufacturing process, including cultures, product storage, and release, and may include in vitro and/or in vivo tests. The function of preclinical potency testing is to gather information on the correlation between the biological activity of the product and its intended therapeutic outcome. Thus, the ethical justification for potency assessment is to allow predictions of proof-of-principle of efficacy in preparing for FIH trials.
Potency in vitro and in vivo tests are adapted to meet specific properties of cell-based products, such as viability, proliferation potential, and differentiation. In vivo potency tests are recommended where animal models relevant to assess the intended therapeutic use are available. In particular, EMA guidelines on somatic cellular therapies include specific provisions for in vitro and in vivo potency assays that shall be performed depending on product’s clinical use (e.g., repair and regeneration, metabolic or pharmacological activity, immunotherapy) [28].
EMA’s reflection paper on stem cell-based products provides useful insights on how to apply potency benchmarks in the context of hESC-based therapies [32]. It is suggested that potency assays be designed according to the differentiation status of the transplanted cells, and to the mechanism of action relevant to the desired therapeutic effect. Where the product includes heterogeneous cell phenotypes, in vitro potency tests should be able to assess the functional and phenotypic profiles of the different cell populations that constitute the compound. Information gathered through in vitro tests should be correlated to in vivo preclinical functional assays on relevant animal models, performed to obtain complementary data on the biological activity of the product. Results from preclinical studies of hESC-based therapies for brain repair show that further research is needed to validate the correlation between functional properties of transplanted cells in vitro and in vivo [49]. For example, in the context of hESC-based products for the therapy of PD, the stage of differentiation of hESC-derived dopamine progenitors is crucial. Too little differentiation increases the risk of tumor formation, too much increases the risk of apoptosis of transplanted cells.
Preclinical requirements for potency assessment should be further adjusted in preparing for FIH trials of hESC-based therapies. Ideally, protocols for in vivo potency tests should guarantee animal-human proximity relative to the organ site of transplantation, cellular environment, and method of administration. Moreover, because hESC transplantation in xenograft models can only provide limited answers about immune response and about how immunosuppression regimes might affect therapeutic outcomes in humans, preclinical potency testing should include the assessment of properties of the xenograft that are relevant to product functioning in vivo.
Viability: “Will It Stay Effective?”
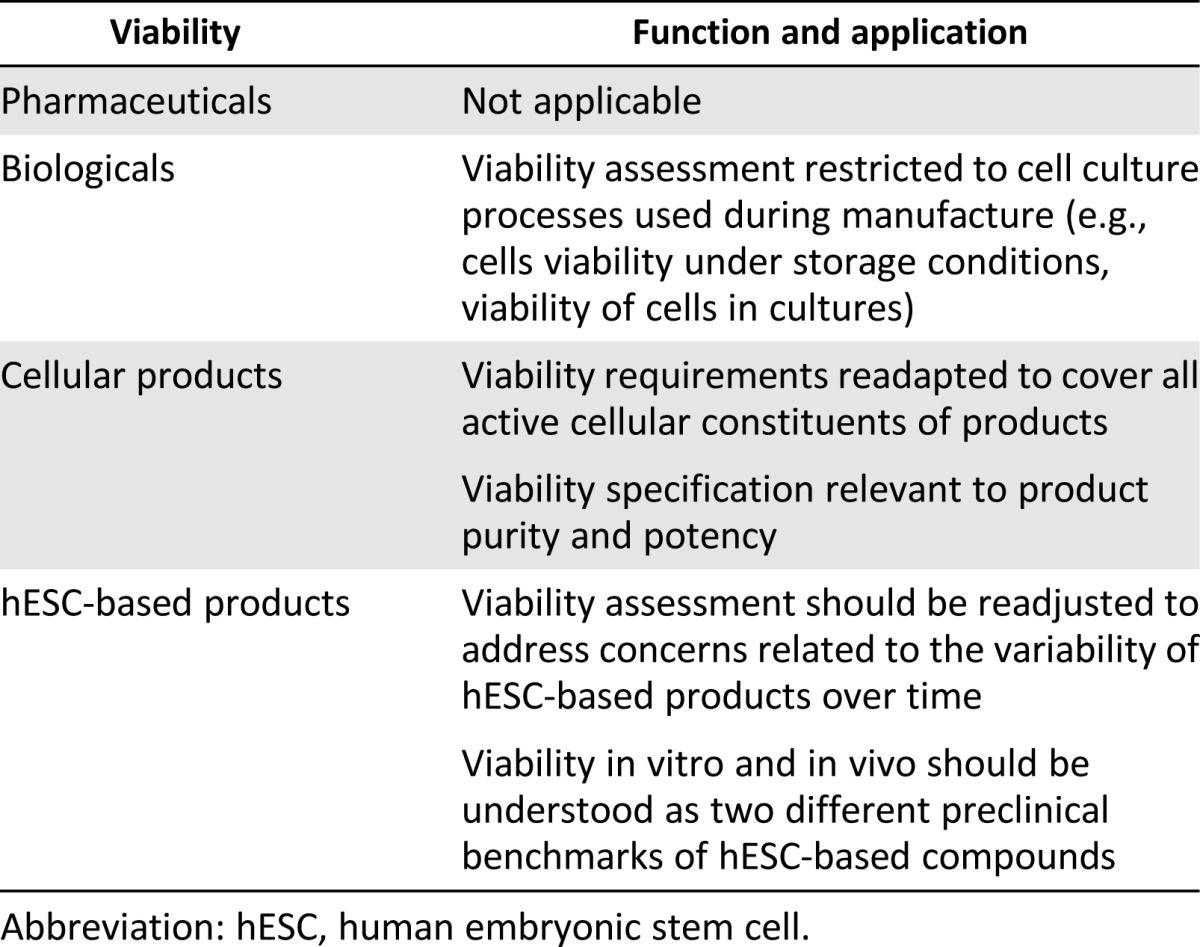
The concept of viability (Table 5) was first introduced in GMP standards with reference to tests on cell culture processes used to manufacture biological products. ICH guidelines recommend that cell banks maintain cell viability under storage conditions, and that viability of cell cultures be monitored during manufacture, where appropriate [36]. The ethical justification for preclinical viability assessment is to estimate both safety risks and proof-of-principle of efficacy, because nonviable cells may affect products’ safety as well as their therapeutic outcome.
Viability is adapted in the context of somatic cellular therapies, where it is applied to all active cellular constituents of products and involves the specification of an acceptable ratio of viable/nonviable cells [30]. Because nonviable cells may contaminate the cell population of interest, viability testing in vitro is correlated to product purity [28]. Moreover, viability is considered essential to test product integrity before release and directly related to the biological activity (i.e., potency) of cellular products. For example, FDA standards require that minimum acceptable-viability in vitro specifications be set as a percentage of viable cells. The association of viability with purity and potency is consistent with preclinical requirements for conventional pharmaceuticals and biologicals, where active compounds are a mixture of therapeutic and nontherapeutic agents. Besides viability testing in vitro, guidelines on somatic cellular therapies recommend in vivo viability testing in relevant animal models to anticipate safety and efficacy concerns related to kinetics, migration, and persistence of cell-based products in the host [28].
When applied to hESC therapies, viability assessment requires further adjustment to address concerns related to the variability of hESC-derived products over time. Indeed, the effectiveness of hESC-based interventions reflects viability, potency, and functional stability of transplanted cells. It is possible that, for heterogeneous and dynamic cell compounds, percentage viability in vitro is not predictive of efficacy in vivo. Because of the dynamics of transplanted cells, viability in vitro and in vivo should be understood as two different preclinical benchmarks of hESC-based compounds. Specifically, viability testing in vitro should be considered as a useful parameter to control quality of cell cultures and to establish product integrity and stability before release, thus allowing for predictions of safety in vivo; whereas viability testing in relevant animal models might inform predictions of efficacy in preparing for FIH experiments. So far, preclinical studies in animal models do not provide clear-cut evidence of the characteristics of hESC-derived cell compounds that would be predictive of product viability in vivo. Based on this benchmark, as well, more research is needed before moving to FIH trials.
Discussion
Our findings show that preclinical benchmarks to be assessed in preparing for FIH trials can be applied to hESC-based interventions. These benchmarks, however, are not clearly set down in existing guidelines relevant to the move from preclinical to clinical studies, which were developed for conventional pharmaceuticals or for biologicals. Our study elucidates that the most important preclinical benchmarks applicable to hESC-based products require adaptation because hESC compounds have specific characteristics. Compared with pharmaceuticals, biologicals, or somatic cellular therapies, hESC-based products involve a distinctive dynamic heterogeneity that increases uncertainty and persistence of safety risks and allows for limited predictions of effects in vivo. To successfully anticipate safety and efficacy concerns of hESC interventions, some preclinical benchmarks require rethinking (i.e., identity, purity), whereas others need to be specified (i.e., potency, viability). Rethinking and specification of these preclinical benchmarks are essential to determine when to start FIH trials of hESC-based interventions.
Because all benchmarks contribute to inform predictions of functional stability of hESC-based products over time, further investigation is needed to clarify whether different preclinical benchmarks indicate distinct products’ properties, or should be considered as different markers for the persistent functional stability of hESC-derived interventions. The best way to answer this question would be for researchers to describe and discuss how they assessed relevant benchmarks in the preclinical phase of product development (e.g., what preclinical markers were tested, through what kind of in vitro and in vivo assays, and so forth). This could be routinely done in publications in which results of preclinical studies are reported. Transparence in dissemination of how preclinical benchmarks are assessed would be an essential contribution by researchers to the application of regulatory requirements, while capitalizing research efforts by pooling experiences from preclinical studies on different hESC-based therapeutic approaches [50]. Ultimately, this strategy would help understanding about whether distinctions between preclinical benchmarks developed for conventional drugs or biologicals are still relevant to cellular interventions.
Moreover, it is likely that actual rethinking or adjustment of preclinical benchmarks will be required repeatedly for different hESC-based products. hESC therapies may differ from one another depending on the protocol for cells’ manufacture and transplantation. Differences in the hESC line used to derive the cellular population of interest, in cell maturation in vitro, or in the time of cell transplantation may affect product safety and efficacy. To best protect participants in FIH trials, further research is needed to assess what differences in cell manufacturing and transplantation would require product-specific rethinking of preclinical benchmarks.
Conclusion
Relevant preclinical benchmarks require rethinking (i.e., identity and purity) or need to be adjusted (i.e., potency and viability) to anticipate safety and efficacy concerns related to the dynamic heterogeneity of hESCs. Rethinking and adaptation of preclinical benchmarks is the first, essential step to address the when question (i.e., to decide whether to move forward from preclinical to FIH studies of hESC-based interventions). Specifically:
Identity should be reassessed as a general marker of functional stability and should address three aspects of hESC-based products: identity of product functional properties to be tested in clinical trials, identity of other relevant products’ characteristics in the various steps of in vitro manufacturing and in vivo animal testing, and identity of steps of the overall product’s preclinical development.
Purity requirements should go beyond a point-in-time validation of acceptable levels of contaminants in manufacturing. Rather, purity should be reconsidered as a benchmark for the functional stability of hESC products over time, through the assessment of the characteristics of the cellular population that are predictable from batch to batch.
Preclinical potency testing in vivo should be specified to guarantee animal-human proximity relative to organ site, cellular environment, and method of hESC transplantation; and xenograft properties relevant to allow predictions about immune response and immunosuppression regimes in humans.
Viability assessment should be adjusted to assess two different aspects of hESC products: integrity and stability in vitro before release, and effectiveness in vivo on relevant animal models.
Moreover, our analyses show that functional stability over time at target sites ought to be considered as a general cross-benchmark for the preclinical assessment of hESC-derived products. Specifically, in the context of hESC-based therapy for PD, preclinical testing of functional stability will likely depend on the stability of parkinsonism in nonhuman primate models [51].
Rethinking or adaptation of how to apply these preclinical benchmarks in specific cases will be required repeatedly for different hESC-based products because distinct protocols for cell manufacture and transplantation may affect product safety and efficacy differently. This process would benefit from sharing of knowledge if researchers included these components in the description of their methods in publications. Clarifying these components, and publishing how they were achieved, could help the assessment of when it is ethically justified to start FIH phase I trials to become increasingly evidence based.
Acknowledgments
This research was supported by Grant CRSI33_125408/1 of the Swiss National Science Foundation. We thank our colleagues in the PROMETHEUS project who provided insight and expertise that greatly assisted the research, including Karl-Heinz Krause (PI), Patrick Brundin, Pierre Burkhard, Michel Dubois-Dauphin, Nathalie Ginovart, Nathalie Ilic, Marisa Jaconi, Anis Feki, Shahan Momjan, Didier Pittet, Eric Rouiller, Jacques Schrenzel, Vannary Tieng-Caulet, Jean Villard, and Walter Zing.
Author Contributions
G.B.: conception and design, collection of data, data analysis and interpretation, manuscript writing, final approval of manuscript; S.A.H. and A.M.: conception and design, data analysis and interpretation, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
G.B. is employed as senior lecturer at the University of Lausanne, has an uncompensated advisory role as a member of the Human Research Ethics Committee at the École Polytechnique Fédérale de Lausanne Research Office, and has uncompensated research funding from the following sources: the Research Commission of the University Department for Preventive and Social Medicine, CHUV Lausanne University Hospital (1 year of funding, from October 1, 2013, to October 31, 2014), and the Public Health Service of the Vaud Canton (16 months of funding, from April 1, 2014, to July 31, 2015). The other authors indicated no potential conflicts of interest.
References
- 1.Doi D, Morizane A, Kikuchi T, et al. Prolonged maturation culture favors a reduction in the tumorigenicity and the dopaminergic function of human ESC-derived neural cells in a primate model of Parkinson’s disease. Stem Cells. 2012;30:935–945. doi: 10.1002/stem.1060. [DOI] [PubMed] [Google Scholar]
- 2.Ratcliffe E, Glen KE, Naing MW, et al. Current status and perspectives on stem cell-based therapies undergoing clinical trials for regenerative medicine: Case studies. Br Med Bull. 2013;108:73–94. doi: 10.1093/bmb/ldt034. [DOI] [PubMed] [Google Scholar]
- 3.Redmond DE, Jr, McEntire CRS, Kingsbery JP, et al. Comparison of fetal mesencephalic grafts, AAV-delivered GDNF, and both combined in an MPTP-induced nonhuman primate Parkinson’s model. Mol Ther. 2013;21:2160–2168. doi: 10.1038/mt.2013.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sundberg M, Bogetofte H, Lawson T, et al. Improved cell therapy protocols for Parkinson’s disease based on differentiation efficiency and safety of hESC-, hiPSC-, and non-human primate iPSC-derived dopaminergic neurons. Stem Cells. 2013;31:1548–1562. doi: 10.1002/stem.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Asterias Biotherapeutics (initiated in 2010 by Geron Corp.). Safety study of GRNOPC1 in spinal cord injury. Available at http://clinicaltrials.gov/ct2/show/NCT01217008?term=geron&rank=9. Accessed July 1, 2015.
- 6.Ocata Therapeutics (initiated in 2011 by Advanced Cell Technology). Sub-retinal transplantation of hESC derived RPE (MA09-hRPE) cells in patients with Stargardt’s macular dystrophy. Available at http://clinicaltrials.gov/ct2/show/study/NCT01345006?term=NCT01345006&rank=1. Accessed July 1, 2015.
- 7.Ocata Therapeutics (initiated in 2011 by Advanced Cell Technology). Safety and tolerability of sub-retinal transplantation of hESC derived RPE (MA09-hRPE) cells in patients with advanced age related macular degeneration (Dry AMD). Available at http://clinicaltrials.gov/ct2/show/NCT01344993?term=rpe+advanced+cell+technology&rank=1. Accessed July 1, 2015.
- 8.Dawson L, Bateman-House AS, Mueller Agnew D, et al. Safety issues in cell-based intervention trials. Fertil Steril. 2003;80:1077–1085. doi: 10.1016/s0015-0282(03)02218-0. [DOI] [PubMed] [Google Scholar]
- 9.Kimmelman J, Baylis F, Glass KC. Stem cell trials: Lessons from gene transfer research. Hastings Cent Rep. 2006;36:23–26. doi: 10.1353/hcr.2006.0012. [DOI] [PubMed] [Google Scholar]
- 10.Master Z, McLeod M, Mendez I. Benefits, risks and ethical considerations in translation of stem cell research to clinical applications in Parkinson’s disease. J Med Ethics. 2007;33:169–173. doi: 10.1136/jme.2005.013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mathews DJH, Sugarman J, Bok H, et al. Cell-based interventions for neurologic conditions: Ethical challenges for early human trials. Neurology. 2008;71:288–293. doi: 10.1212/01.wnl.0000316436.13659.80. [DOI] [PubMed] [Google Scholar]
- 12.Hess PG. Risk of tumorigenesis in first-in-human trials of embryonic stem cell neural derivatives: Ethics in the face of long-term uncertainty. Account Res. 2009;16:175–198. doi: 10.1080/08989620903065145. [DOI] [PubMed] [Google Scholar]
- 13.Bretzner F, Gilbert F, Baylis F, et al. Target populations for first-in-human embryonic stem cell research in spinal cord injury. Cell Stem Cell. 2011;8:468–475. doi: 10.1016/j.stem.2011.04.012. [DOI] [PubMed] [Google Scholar]
- 14.Solbakk JH, Zoloth L. The tragedy of translation: The case of “first use” in human embryonic stem cell research. Cell Stem Cell. 2011;8:479–481. doi: 10.1016/j.stem.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 15.Dunnett SB, Rosser AE. Challenges for taking primary and stem cells into clinical neurotransplantation trials for neurodegenerative disease. Neurobiol Dis. 2014;61:79–89. doi: 10.1016/j.nbd.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 16.Hey SP, Kimmelman J. The risk-escalation model: A principled design strategy for early-phase trials. Kennedy Inst Ethics J. 2014;24:121–139. doi: 10.1353/ken.2014.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hurst SA, Mauron A, Momjian S, et al. Ethical criteria for human trials of stem-cell-derived dopaminergic neurons in Parkinson’s disease. AJOB Neurosci. 2015;6:52–60. [Google Scholar]
- 18.Kimmelman J. Gene Transfer and the Ethics of First-in-Human Research. Lost in Translation. New York: Cambridge University Press; 2010. [Google Scholar]
- 19.Tabar V, Studer L. Pluripotent stem cells in regenerative medicine: challenges and recent progress. Nat Rev Genet. 2014;15:82–92. doi: 10.1038/nrg3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Impurities in new drug substances, Q3A(R2) (1994). 2006. Available at http://www.ich.org/home.html. Accessed July 1, 2015.
- 21.International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Specifications: Test procedures and acceptance criteria for new drug substances and new drug products: chemical substances, Q6A (1997). 1999. Available at http://www.ich.org/home.html. Accessed July 1, 2015.
- 22.International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Stability testing of new drug substances and products, Q1A(R2) (1992). 2003. Available at http://www.ich.org/home.html. Accessed July 1, 2015.
- 23.International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Preclinical safety evaluation of biotechnology-derived pharmaceuticals, S6 (1996). 1997. Available at http://www.ich.org/home.html. Accessed July 1, 2015.
- 24.International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Note for guidance specifications: Test procedures and acceptance criteria for biotechnological/biological products, Q6B (1998). 1999. Available at http://www.ich.org/home.html. Accessed July 1, 2015.
- 25.International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Quality of biotechnological products: Stability testing of biotechnological/biological products, Q5C. 1995. Available at http://www.ich.org/home.html. Accessed July 1, 2015.
- 26.Food and Drug Administration. Biological products: General (CFR, Title 21, Part 600). Available at https://http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm?CFRPart=600. Accessed July1, 2015.
- 27.Food and Drug Administration. General biological products standards (CFR, Title 21, Part 610). Available at http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=610. Accessed July 1, 2015.
- 28.European Medicines Agency. Guideline on human cell-based medicinal products. 2008. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003894.pdf. Accessed July 1, 2015.
- 29.Food and Drug Administration. Human cells, tissues, and cellular and tissue-based products (CFR, Title 21, Part 1271). Available at http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=1271&showFR=1. Accessed July 1, 2015.
- 30.Food and Drug Administration. Center for Biologics Evaluation and Research (CBER), Guidance for reviewers. Instruction and template for chemistry, manufacturing, and control (CMC) reviewers of human somatic cell therapy investigational new drug applications (INDs). 2003. Available at http://www.fda.gov/OHRMS/DOCKETS/98fr/03d0349gdl.pdf. Accessed July 1, 2015.
- 31.Food and Drug Administration. Proposed approach to regulation of cellular and tissue-based products. 1997. Available at http://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/Tissue/UCM062601.pdf. Accessed July1, 2015. [DOI] [PubMed]
- 32.European Medicines Agency. Reflection paper on stem cell-based medicinal products, 2011. Available at http://www.cirm.ca.gov/files/PDFs/RMC/EMA_Final_Reflection_Paper.pdf. Accessed July 1, 2015.
- 33.International Society for Stem Cell Research. Guidelines for the clinical translation of stem cells. 2008. Available at http://www.isscr.org/home/publications/ClinTransGuide. Accessed July 1, 2015.
- 34.National Institutes of Health. 10. Assessing human stem cell safety. In: Stem Cell Information. Bethesda, MD: National Institutes of Health, U.S. Department of Health and Human Services. 2009. Available at http://stemcells.nih.gov/info/scireport/pages/chapter10.aspx. Accessed July 1, 2015.
- 35.Food and Drug Administration. CTGTAC Meeting # 45, Cellular therapies derived from human embryonic stem cells – Considerations for pre-clinical safety testing and patient monitoring (April 10, 2008). Available at http://www.fda.gov/ohrms/dockets/ac/08/briefing/2008-0471B1_1.pdf. Accessed July 1, 2015.
- 36.International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Good manufacturing practice guide for active pharmaceutical ingredients, Q7. 2000. Available at http://www.ich.org/home.html. Accessed July 1, 2015.
- 37.Food and Drug Administration. Guidance for industry. CGMP for Phase 1 investigational drugs, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER), Office of Regulatory Affairs (ORA), July 2008. Available at http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm070273.pdf. Accessed July 1, 2015.
- 38.Catalano J. The International Conference on Harmonization (ICH) and its relevance to cell therapy. ISCT 6th Annual Somatic Cell Therapy Symposium 2006. Available at http://www.bcg-usa.com/regulatory/docs/FDA_Presentations_Publications/SP9D06.pdf. Accessed July 1, 2015.
- 39.Deal G. Stem cell therapy regulations. The US vs the EU. Regulatory Rapporteur. 2009;6:4–6. [Google Scholar]
- 40.Halme DG, Kessler DA. FDA regulation of stem-cell-based therapies. N Engl J Med. 2006;355:1730–1735. doi: 10.1056/NEJMhpr063086. [DOI] [PubMed] [Google Scholar]
- 41.Fink DW., Jr FDA regulation of stem cell-based products. Science. 2009;324:1662–1663. doi: 10.1126/science.1173712. [DOI] [PubMed] [Google Scholar]
- Regulation (EC) No. 1394/2007 of the European Parliament and of the Council on Advanced Therapy Medicinal Products and amending Directive 2001/83/EC and Regulation (EC) No 726/2004. Official Journal of the European Union L 324/121.
- 43.Preynat-Seauve O, Burkhard PR, Villard J, et al. Pluripotent stem cells as new drugs? The example of Parkinson’s disease. Int J Pharm. 2009;381:113–121. doi: 10.1016/j.ijpharm.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 44.Bongso A, Fong C-Y, Gauthaman K. Taking stem cells to the clinic: Major challenges. J Cell Biochem. 2008;105:1352–1360. doi: 10.1002/jcb.21957. [DOI] [PubMed] [Google Scholar]
- 45.Salmikangas P, Flory E, Reinhardt J, et al. Regulatory requirements for clinical trial and marketing authorisation application for cell-based medicinal products. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz. 2010;53:24–29. doi: 10.1007/s00103-009-0991-5. [DOI] [PubMed] [Google Scholar]
- 46.Kriks S, Shim J-W, Piao J, et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature. 2011;480:547–551. doi: 10.1038/nature10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Unger C, Skottman H, Blomberg P, et al. Good manufacturing practice and clinical-grade human embryonic stem cell lines. Hum Mol Genet. 2008;17(R1):R48–R53. doi: 10.1093/hmg/ddn079. [DOI] [PubMed] [Google Scholar]
- 48.Lindvall O, Kokaia Z. Stem cells in human neurodegenerative disorders--time for clinical translation? J Clin Invest. 2010;120:29–40. doi: 10.1172/JCI40543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steinbeck JA, Studer L. Moving stem cells to the clinic: Potential and limitations for brain repair. Neuron. 2015;86:187–206. doi: 10.1016/j.neuron.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barker RA. Developing stem cell therapies for Parkinson’s disease: Waiting until the time is right. Cell Stem Cell. 2014;15:539–542. doi: 10.1016/j.stem.2014.09.016. [DOI] [PubMed] [Google Scholar]
- 51.Potts LF, Wu H, Singh A, et al. Modeling Parkinson’s disease in monkeys for translational studies, a critical analysis. Exp Neurol. 2014;256:133–143. doi: 10.1016/j.expneurol.2013.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]