

ABSTRACT
Voltage gated sodium channels are the target of a range of local anesthetic, anti-epileptic and anti-arrhythmic compounds. But, gaining a molecular level understanding of their mode of action is difficult as we only have atomic resolution structures of bacterial sodium channels not their eukaryotic counterparts. In this study we used molecular dynamics simulations to demonstrate that the binding sites of both the local anesthetic benzocaine and the anti-epileptic phenytoin to the bacterial sodium channel NavAb can be altered significantly by the introduction of point mutations. Free energy techniques were applied to show that increased aromaticity in the pore of the channel, used to emulate the aromatic residues observed in eukaryotic Nav1.2, led to changes in the location of binding and dissociation constants of each drug relative to wild type NavAb. Further, binding locations and dissociation constants obtained for both benzocaine (660 μM) and phenytoin (1 μ M) in the mutant channels were within the range expected from experimental values obtained from drug binding to eukaryotic sodium channels, indicating that these mutant NavAb may be a better model for drug binding to eukaryotic channels than the wild type.
KEYWORDS: benzocaine, drug binding, local anesthetic, NavAb, phenytoin, sodium channel
Introduction
Eukaryotic voltage-gated sodium channels (eNav) are responsible for the initiation and propagation of action potentials in excitable cells. By opening in response to small depolarising signals they allow the rapid influx of sodium ions into the cell before entering a distinct non-conductive inactivated state. Humans have 9 isoforms of eNav1 (annotated as Nav1.1 to Nav1.9)2 localized in different tissues around the body. Mutations or aberrant expression of these sodium channels lead to a series of disorders such as cardiac arrhythmia, epilepsy and chronic pain syndromes.3-7 Hence eNavs are a common target for sodium channel blockers such as anti-arrhythmics, local-anesthetics and anti-epileptics.8-11 However, in spite of being so widely applied, sodium channel blockers are not isoform specific, leading to many undesirable health effects. Improving the specificity of these drugs is imperative, but this is hard due to the sequence similarity of the isoforms and uncertainty as to where the drugs bind, how they reach this site and how they alter channel function.
While we are currently limited by the fact that no eNav structures have been obtained, there are now several structures of bacterial voltage gated sodium channels (bNav).12-17 These structures are currently being used as model systems for gaining insight into the structure-function relationship of drug binding within eNavs.18-22 However, there are several fundamental differences between bNavs and eNavs. eNav have 4 heterologous domains (annotated as DI to DIV) encoded in a single polypeptide chain. Each domain has 6 transmembrane helices, S1-S4 that form the voltage sensing domains and S5-S6 that form the central pore of the channel. In contrast, bNav are homotetramers of 4 identical polypeptide chains. Each subunit has 6 helices (S1-S6) which arrange in the same manner as that observed in eNavs. However, bNav lack certain important domains found in eNav such as the DIII-DIV linker, found to be important in eNav for fast inactivation.23 As some bNav have delayed entry into and recovery from inactivated states they cannot easily be used to model the use-dependent block observed in eNavs.24 In addition, bNav have significant sequence differences to eNav, including many residues known to be involved in the binding of the pore blocking drugs. Nevertheless, it has been shown that bNavs can be used as functional models for some aspects of eNav behavior 18,19 including tonic block. For example, Bagneris et al.19 recently demonstrated that eukaryotic channel blockers were able to reversibly bind to and inhibit a bNav with a similar potency to that observed for Nav1.2. In addition, site directed mutagenesis was used to show non-identical but overlapping binding sites for a local-anesthetic and an anti-epileptic similar to results found for rat Nav1.1.19
Ragsdale et al.25 found that the binding of cationic and hydrophobic drugs to the inner pore of eNavs formed by the 4 S6 helices is due to the presence of several highly conserved aromatic residues. For example, in Nav1.2 it was determined that mutation of F1764 and Y1771 to alanine greatly reduced the binding affinity of the drugs phenytoin, lidocaine and etidocaine.25,10 Furthermore, in Nav1.4, an equivalent residue F1579 was found to interact with lidocaine through pi-stacking.26 These 2 crucial residues from Nav1.2 are indicated in Figure 1. It should be noted that while only DIV contains an aromatic residue at the position equivalent to F1764, 3 of the 4 Nav1.2 domains have an aromatic residue at the position equivalent to Y1771. In contrast, none of the bNav for which we have structures have aromatic residues at the position equivalent to F1764, and only NavMs has an aromatic equivalent to Y1771. In this study we have tried to determine whether we can make a bNav (NavAb) a better model for local anesthetic binding in eNav by incorporating aromatic mutations in the S6 helical domain at these critical positions (residues 206 and 213 for NavAb).
Figure 1.
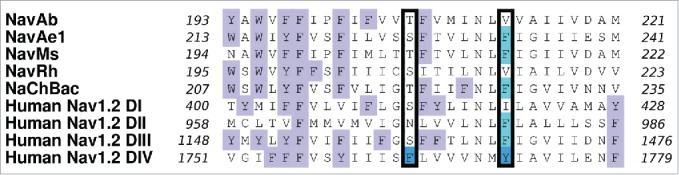
Sequence alignments of the S6 domains of NavAb (Arcobacter butzleri), NavAe1 (Alkalilimnicola ehrlichei), NavMs (Magnetococcus marinus), NavRh (Rickettsiales sp HIMB114) and NaChBac (Bacillus haldurans), as well as from domains I to IV of human Nav1.2, are shown as determined using ClustalW2. F1764 and Y1771 in Nav1.2 DIV (bright blue) are implicated in the binding of local anesthetic drugs.25 The corresponding aromatic residues in each sequence alignment are indicated with pale blue boxes and the NavAb residues, T206 and V213, are indicated in the black outlined box. It should be noted that 3 of the 4 domains in Nav1.2 have an aromatic residue at the position equivalent to 1771 in DIV. All other aromatic residues are indicated by a lilac box.
It has previously been shown using molecular dynamics (MD) simulations that small molecule blockers have preferred binding sites in the NavAb pore, and that some compounds can enter through lateral fenestrations.20-22 Here we do similar studies to examine the binding of the drugs benzocaine and phenytoin to mutant NavAb in which aromatic residues are added to the pore to better emulate eNavs. By comparing to results obtained by Martin and Corry21 in WT NavAb we are able to investigate the specific changes in drug binding induced by the additional aromatic residues in the mutant channels.
Results
In this study we mutated 2 residues within the S6 helix of NavAb, introducing a phenylalanine at position 206 (F206T) and a tyrosine at position 213 (Y213V) in order to emulate the aromatic residues found in Nav1.2. These residues were mutated in either one of the 4 subunits (the 1S simulations), to emulate Nav1.2 DIV, or in all 4 subunits (the 4S simulations), in order to increase the overall aromaticity of the pore as observed in all domains of Nav1.2 (Fig. 1). We have observed marked changes in how and where the drugs benzocaine and phenytoin bind relative to WT NavAb.21
A clustering analysis was performed on 300 ns of simulation coming from 3 independent simulations for each drug / mutant system allowing the most heavily populated drug binding positions to be determined. This is illustrated in Figure 2 where the most populated clusters are indicated in blue. In the case of benzocaine in the 1S system (Fig. 2A) it did not spend any time in the fenestrations, instead it moved progressively down into the activation gate in a position observed in previous simulations of WT NavAb.21 Conversely, in the 4S system all of the clusters occupied by benzocaine are in the vicinity of the fenestrations as the bulky tyrosines introduced at residue 213 occlude the region around the activation gate. This fenestration position is similar to what is observed for phenytoin for both the 1S and 4S systems. Due to its bulky nature, the added aromatic groups are sufficient to prevent phenytoin from moving into the activation gate during the timescale of these simulations.
Figure 2.
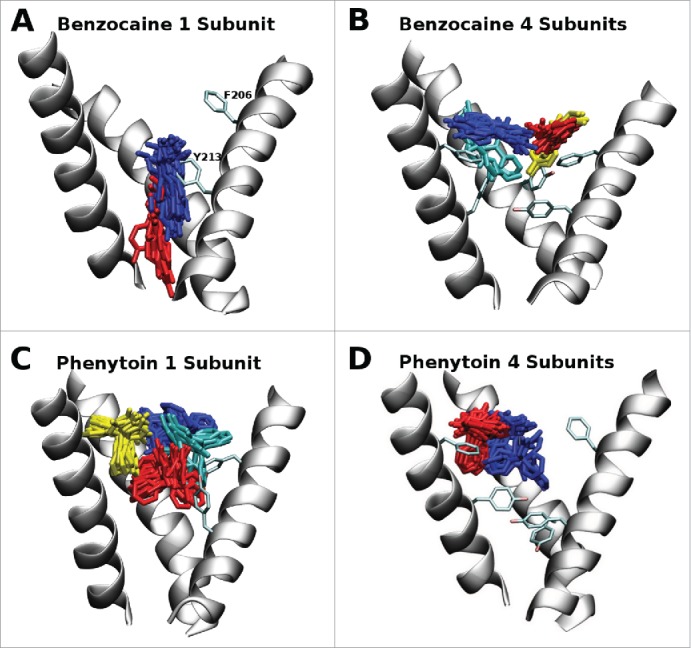
Most populated clusters of the drugs benzocaine and phenytoin obtained from 300 ns of molecular dynamics simulation where blue, red, yellow and cyan represent progressively less populated clusters. While the full pore forming domain of NavAb (residues 115–221) was used in these simulations, for clarity, only selected residues (200–221) from 3 of the 4 S6 helices are shown in this figure. A: In the 1S system benzocaine remained in the activation gate. B: In the 4S system benzocaine was not able to pass the bulky aromatic residues and remained in the vicinity of the fenestrations. The blue cluster is pi-stacking with F206. C: In the 1S system phenytoin was able to sit in the center of the cavity (red) and move and rotate in the vicinity of the fenestrations (blue, yellow and cyan clusters). D: In the 4S system phenytoin is more restricted in its movement and remains in a pocket formed by the aromatic residues and M209 (blue) or straddles F206 in the fenestration (red).
In our previous studies of WT NavAb we found that both benzocaine and phenytoin can bind at 2 distinct positions: in the activation gate and near the internal entrance of the fenestrations. Surprisingly, we found that binding at the activation gate was slightly more favorable than the fenestration site21 even though experimental mutation studies suggest the expected local anesthetic site to be close to the fenestrations. Our new results suggest that the aromatic residues found in the pore of eNavs can prevent the drugs from binding at the activation gate. The lack of aromatic residues in our simulations of WT NavAb can explain our previous results.
As the results above are determined from equilibrium simulations run over a limited timescale it is possible that the drug does not sample all of the possible positions inside the pore at the same probability as in longer simulations. To overcome this, and to quantify the relative energetics of the potential binding sites, we used metadynamics to sample all of the drug positions in the central cavity of the pore. While the same protein systems as above were used for this study (residues 115–221) in order to reduce the computational burden we restricted our analysis of drug binding positions to specific quadrants, making use of the inherent protein symmetry. For the 1S system, we ran 2 separate simulations, one for the quadrant opposite the mutation, and one for the quadrant with the mutation. The results obtained for the quadrant opposite the mutation (Fig. 3A and B) are very similar to those obtained previously for both benzocaine and phenytoin in WT NavAb.21 A different pattern is evident in the free energy surface of benzocaine when it is allowed near the mutated subunit (Fig. 3A). Specifically, binding in the fenestration becomes much more favorable, with new binding sites appearing further from the pore axis. This extended binding pocket increases the probability that benzocaine will find a favorable binding position in the fenestration and potentially remain there longer. Furthermore, while a free energy well is present at the activation gate it is slightly reduced in size compared to WT. Adding mutations on all 4 subunits completely removes the energy minima at the activation gate (Fig. 3C). Instead a network of favorable binding sites are located in the fenestrations. Detailed pictures of the most favorable binding positions are shown in Figure 5, highlighting the residues with the greatest interaction with each drug. The binding sites in the fenestrations are present in both the 1S and 4S system as benzocaine is able to pi-stack with F206 (Fig. 5D) with an average distance of 3.95 Å from the benzyl ring to the center of mass of the drug in the most populated cluster from the equilibrium simulations.
Figure 3.
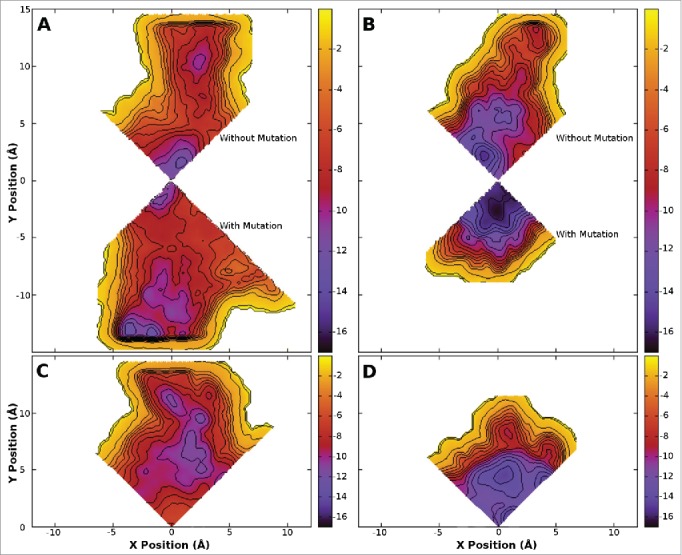
Free energy surfaces obtained from metadynamics simulation for the 1S (800 ns - A and B) and 4S systems (400 ns - C and D) in the presence of benzocaine (A and C) and phenytoin (B and D). The surfaces are integrated onto a 2D surface and viewed looking down the axis of the pore from the extracellular space. Two quadrants are shown for the 1S systems where the upper quadrant was obtained opposite to the site of the mutation and the lower quadrant was obtained at the site of the mutation. A single quadrant is shown for the 4S systems as the mutations introduced into each subunit maintain the 4 fold symmetry of the protein. Contours are shown every 1 kcal/mol. The full pore forming domain of NavAb (residues 115–221) was used in these simulations.
Figure 5.
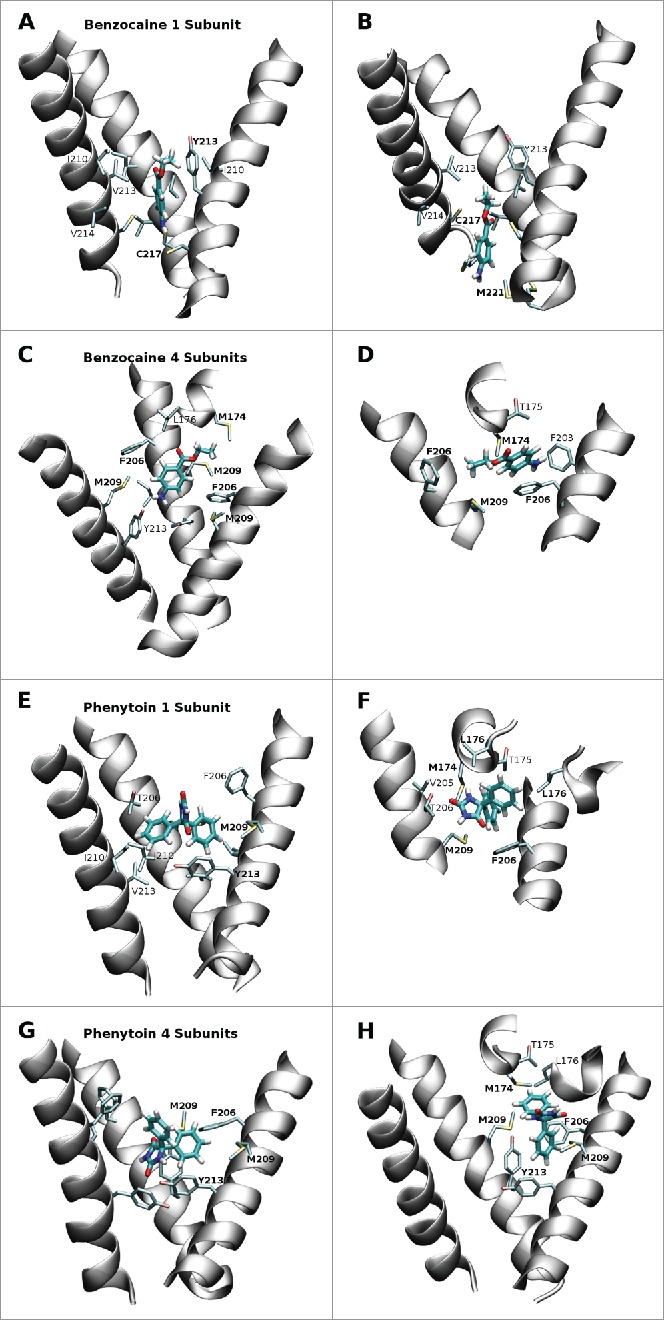
Snapshots from the equilibrium simulations representing the 2 most favored positions of benzocaine and phenytoin in each system. While the full pore forming domain of NavAb (residues 115–221) was used in these simulations, for clarity, only selected residues from the S6 helices (residues 200–221) and the region between the S5 and S6 helices (residues 170–178) are shown in this figure. A and B: Benzocaine in the 1S system in the region of the activation gate. C and D: benzocaine in the 4S system with the amine oriented toward Y213 (C) and pi-stacking with F206 (D). E and F: phenytoin in the 1S system occupying the central cavity above Y213 (E) and in the fenestration (F). G and H: phenytoin in the 4S system with (G) the amine oriented toward Y213 while the benzyl rings are surrounded by the bulky residues M209, F206 and Y213; or (H) straddling F206 and with one benzyl ring protuding into the fenestration.
The strong interaction between benzocaine and F206 is further supported by an analysis of the strength of the interaction between the drug and each protein residue presented in Figure 6. The interaction energies obtained for clusters 1 to 3 (Fig. 6B) in the 4S system all show a significant interaction with F206. M209 is also a pivotal residue in the region of the fenestrations. In clusters 1 and 2 from the 1S system all interactions occur with residues in the activation gate - of particular importance are V/Y 213, C217 and M221.
Figure 6.
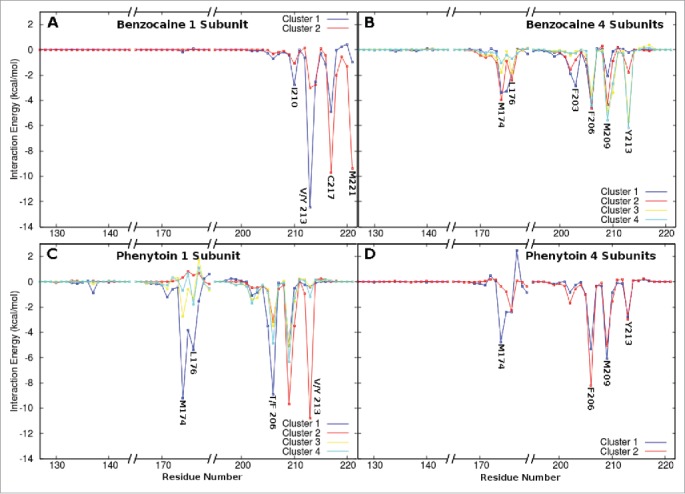
Interaction energies between each drug and the residues lining the channel lumen for each of the most populated clusters. (A) Benzocaine in the 1S system where cluster 1 and 2 are located above and in the activation gate respectively. (B) Benzocaine in the 4S system in which Clusters 1 to 4 are all located in the fenestrations but the drug is oriented in different directions. (C) Phenytoin in the 1S system in which clusters 1, 3 and 4 are all located in the fenestrations while cluster 2 is located in the activation gate. (D) Phenytoin in the 4S system. Only regions of the sequence with significant interactions are shown.
Previous simulations have shown that benzocaine faces only a small barrier to move through the closed activation gate,21 and it has been suggested that there could be a second aqueous route of drug entry into the pore via the intracellular space and the activation gate.22 Figure 4A demonstrates that, while benzocaine faces a larger energy barrier than that observed previously for the WT,21 in the 1S system benzocaine still sees only a small barrier slowing movement through the activation gate, supporting observations from the equilibrium simulations (Fig. 2). In contrast, in the 4S system (Fig. 4C) it is highly unlikely that benzocaine will leave the channel through the activation gate as there is a very large barrier present. Thus, while the aqueous drug entry route may exist in WT NavAb, it is possible that this will be closed off by the additional aromatic residues present in eNavs as seen in the 4S mutant channel.
Figure 4.
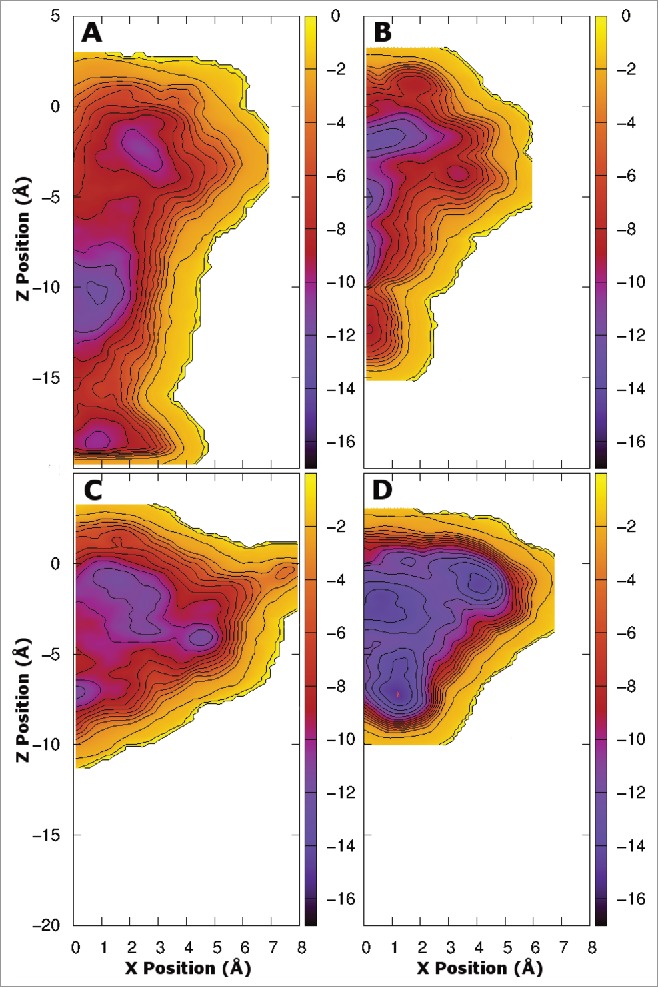
Free energy surfaces obtained from metadynamics simulation for the 1S (800 ns - A and B) and 4S systems (400 ns - C and D) in the presence of benzocaine (A and C) and phenytoin (B and D) viewed from in the plane of the membrane. Contours are shown every 1 kcal/mol.
The binding free energies of benzocaine to the protein in the 4S system were determined using free energy perturbation for the 2 major clusters. The values obtained (−4.3 and −5.1 kcal/mol respectively) and the average value (−4.7 kcal/mol) shown in Table 1 are very similar to that obtained for benzocaine in WT NavAb (−5.6 kcal/mol).21 This indicates that even though benzocaine can now pi-stack with F206 in the mutant protein this may disrupt other interactions which occur in the WT protein. For example, Boiteux et al.22 observed pi-stacking with F203 in WT NavAb and the energetic contribution from that is expected to be equivalent to the energetics observed here. Thus, although benzocaine binds in slightly different positions in the mutant channels in our simulations, there is no evidence that the binding is stronger.
Table 1.
Drug Binding Affinities. The binding free energy and dissociation constants found from the simulations of the most populated positions in both WT and 4S (T206F V213Y) NavAb are compared to experimental data for eNavs.
The location and characteristics of phenytoin binding to the channel are substantially different in both the 1S and 4S systems compared to WT NavAb as evidenced by the free energy landscapes shown in (Fig. 4B and D). The introduction of additional aromatic residues has reduced the size of the activation gate binding site, with Y213 occupying much of the space in this region. In both mutant systems (Fig. 4B and D) the energy barrier that must be overcome by phenytoin in order to leave the channel through the activation gate is much higher than that observed in the WT channel21 making this event unlikely, suggesting that there is no aqueous entry route for phenytoin in any of these proteins. In addition, the added bulk of F206 makes it harder for phenytoin to pass through the fenestrations. We thus suspect that the T206F mutation will slow the entry of phenytoin into the channel, potentially prolonging the time taken to tonically block the channel.
The difference in the binding positions of phenytoin in the WT and mutant channels is most obvious when looking at the free energy surfaces for the 4S system (Fig. 3D). Phenytoin has a less clearly defined binding site in the center of the cavity where it can interact with aromatic residues on all sides. This position is similar to the 2 main clusters found in the equilibrium simulations in which the most important interactions occur between phenytoin and M174, F206, M209 and Y213.
Free energy perturbation was applied to determine the binding free energy for phenytoin in the positions seen in the 2 major clusters found in the equilibrium simulations. Given the similarity of the 2 clusters in terms of location and specific interactions (the only difference being the interaction with M174 in the major cluster which is absent in the second cluster), it is not surprising that the binding free energies obtained (−8.1 and −8.2 kcal/mol in each case) are so similar. The values are significantly higher than those obtained for phenytoin in WT NavAb (−6.1 kcal/mol)21 indicating that introducing F206 and Y213 has led to a stronger association of phenytoin to the channel. A representative position of phenytoin in each of these clusters in illustrated in Figure 5G and H. In each case phenytoin is surrounded by F206 and Y213, and in Cluster 2 it actually appears to straddle F206. Adding these residues has effectively provided a hydrophobic pocket of ideal size to accommodate phenytoin yielding stronger binding.
Discussion
The major interactions for the high affinity S6 binding site of eNav, involving residues F1764 and Y1771 in Nav1.2,25 F1579 and Y1586 in Nav1.4, and F1759 and Y1766 in Nav1.5,27 are known to be hydrophobic. The lack of these aromatic residues within the pore of bacterial channels has been proposed as an explanation for why many drugs which bind to eNavs and share this site 28 do not show such a high affinity for bNav. Through these simulations we have demonstrated that the inclusion of aromatic residues in the pore of NavAb, to mimic what is observed in eNav, changes the locations of drug binding for both the local anesthetic benzocaine and the anti-epileptic phenytoin.
The main results are summarised in Figure 7. Previous analysis has shown at least 2 binding sites exist in the pore, one in the activation gate and one near the fenestrations 21 with additional sites possibly located further away from the pore center in the fenestrations.22 Surprisingly, the most favorable position in the WT protein was found to lie in the activation gate as indicated in Figure 7 A and D,21 in contrast to the expectation from mutagenesis studies.25,10 The presence of additional aromatic residues in NavAb to emulate those found in eukaryotic proteins, reduces the space near the activation gate and hence makes it more likely for benzocaine and phenytoin to bind near the lateral fenestrations as indicated in Figures 7C, F and F. The addition of increased aromaticity in the fenestration binding site also altered the energetic interactions of both drugs with the protein. For example, a significant interaction is now evident with F206 (Fig. 2), whereas the residue at this position only had a minor interaction with the drugs in the WT channel.21 As a consequence we find that phenytoin binds more strongly to these mutant channels than to WT NavAb, as the oddly shaped molecule can now form aromatic stacking interactions. The V213Y mutation also reduced the space in the activation gate which appears to decrease the likelihood of these drugs entering the pore via the closed activation gate.
Figure 7.
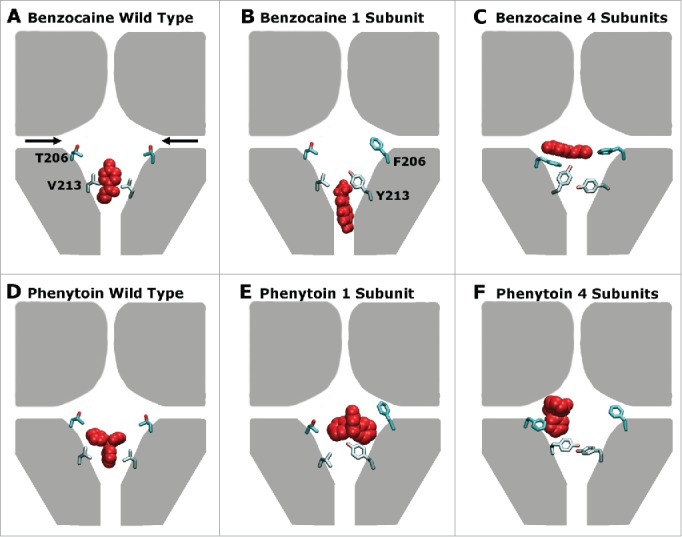
A schematic representation of the preferred drug binding positions (red) in the pore forming domain of NavAb (represented in gray) with the residues mutated in this study (206 and 213) shown explicitly for 2 of the 4 domains in each case. The fenestrations are indicated with black arrows in panel (A). Panels (A), (B) and (C) show the wild type, 1S, 4S channels in the presence of benzocaine while panels (D), (E) and (F) show the wild type, 1S, and 4S channels in the presence of phenytoin. Including increased aromaticity moves both drugs away from the activation gate site, and toward the fenestrations.
It has been shown that the local anesthetics bind in the central cavity of the channel near the internal entrance of the fenestrations 25 with non-specific hydrophobic interactions to several residues in the asymmetric binding site found in eNavs.26 Relative to simulations conducted on WT NavAb by Martin et al.21 the Kd values obtained here for benzocaine in the 4S system were significantly higher. The Kd (660 μ M) for the most populated drug binding position of benzocaine in mutant NavAb is comparable to values obtained experimentally in eNavs which range from 300–1200 μM.29,30,31 This value is close to that obtained for the bNav NachBac (650 μM), which has an aromatic residue (F227) at the position equivalent to F978, F1468 and Y1771 in DII to DIV of Nav1.218 supporting the concept that increased aromaticity in the channel pore has led to the decreased affinity observed in mutant NavAb for benzocaine.
For phenytoin, the Kd values obtained here (1.1 μM) is significantly lower than the value obtained for binding in the fenestrations of WT NavAb (30 μM) but equivalent to the value obtained in the activation gate (1 μM).21 The values obtained here are comparable to experimentally determined Kd, ranging from 4–9 μM, for phenytoin binding to eNavs.32,33,34 Phenytoin has a bulky shape with 3 planar groups protruding in different directions, which makes interaction with flat surfaces difficult. The inclusion of aromatic residues allows for favorable pi-stacking accounting for the increase in affinity at the fenestration site. The difference in interaction from WT to mutant is not as great for the linear benzocaine which can fit into hydrophobic pockets in all cases.
We believe that our simulations of the interaction of benzocaine and phenytoin with the pore domains of a bacterial sodium channel help to understand the binding modes of the compounds, however there are many issues which we cannot address with this approach. Firstly, most channel blocking compounds are known to have different affinities for the different functional states of the protein. In particular, there is a general trend for such compounds to stabilize the inactivated state.24 In our case we have used the structure from a single state, crystalised with a closed pore and an activated voltage sensor.12 Such a conformation is likely to be good for studying binding to the closed state of the pore domain as seen in tonic block, but it is difficult to infer much about binding to other protein conformations. Both benzocaine and phenytoin display binding to resting channels (tonic block),18,35 however, phenytoin is also know to block in a use dependent manner.33 That is, the rate of binding and the binding affinity are both increased after the channel has cycled through the open and inactivated states. Although we have previously examined potential entry routes for these compounds into the pore21,36 our present study does not elucidate the mechanism of use dependence. By focusing only on the pore domain, our simulations also cannot answer if, or how, drug binding alters the interaction between the pore-domain and voltage sensor. We have also compared our dissociation constants to those measured for eukaryotic channels from a range of tissue types which can contain different channel subtypes, making it hard to present a difinitive comparison. Finally, by using a simple bacterial sodium channel as a model, we are unable to determine if there are any differences in the binding of benzocaine or phenytoin to different eukaryotic sodium channel isoforms, or even between different bacterial homologues. Elucidating the subtle differences in the interaction of each isoform with channel blocking drugs will be essential if subtype selective pore blocking medication is to be developed.
Eukaryotic Nav have been extensively characterized for their interactions with specific drugs used to treat epilepsy, cardiac arrhythmia and pain disorders. We have been able to demonstrate that in our simulations, mutations in the pore of a bacterial sodium channel create a pattern of drug binding that is more consistent with data available for eukaryotic channels. In this case, Kd values obtained for binding to a mutant bNav (NavAb) are in line with experimental results obtained for both the local anesthetic benzocaine and the anti-epileptic phenytoin in eNavs, and the locations of binding are more consistent with expectations obtained from site directed mutagenesis experiments. If the bacterial channels can be made to tolerate such mutations, then our simulations suggest that such mutant bNavs may provide an even better model of drug binding to eNavs than the WT bacterial channels. The correlation of our theoretical results with experimental values obtained from eNavs further highlights the importance of bNavs in providing a simple system to model eNav behavior and warrants further experimental study.
Methods
Simulation systems
A pre-equilibrated and solvated system of closed NavAb in a lipid membrane was obtained from Martin and Corry et al.21 In this system the coordinates of closed/pre-open NavAb were obtained from the protein databank (pdb accession number 3RVY).12 The voltage sensing domains were cleaved off prior to inserting the central pore forming domain of NavAb (helices S5-S6 with residues 115–221) into a pre-equilibrated POPC membrane and solvating in a TIP3P water box with dimensions of 72 × 72 × 82 Å with 250 mM NaCl. The protein was then held fixed while the water, ions and lipid were allowed to equilibrate for 2 ns. The protein C α atoms were restrained with a force constant which reduced in 4 steps over 10 ns from 10 to 0.1 kcal/mol.
The removal of the voltage sensing domains makes it impossible to directly study how drug binding can influence the interaction between the pore and voltage sensors. However, it has been demonstrated that pore blocking drugs such as benzocaine and phenytoin interact directly with sites on the S6 helix in the pore forming domain.10,25,26 Furthermore, it has been shown that some pore blockers are still effective on bacterial sodium channels lacking voltage sensors,37 and that some blocking drugs can be seen in crystal structures channels lacking the voltage sensors.19 In addition, previous simulation studies of just the pore forming domain have been able to identify drug binding sites in line with experimental expectations.21 Thus, we expect to be able to gain valuable insights into drug binding using this simplified system and expect that the introduction of increased aromaticity will allow further alignment of the simulation studies with the results obtained for eNav's.
The coordinates from this equilibrated system were used as the starting point for the following simulation systems. In the first system (called ‘1 subunit’ or ‘1S’ hereafter) residues 206 and 213 on the first subunit of NavAb were mutated to phenylalanine and tyrosine respectively. In the second system (called ‘4 subunits’ or ‘4S’ hereafter) these residues were mutated in each of the 4 subunits. The mutated systems were subsequently energy minimised for 10,000 steps. Constant temperature (298 K) and pressure (1 atm) were maintained using Langevin dynamics and a Langevin piston and the particle mesh Ewald method was employed to compute the complete electrostatics of the system.38 All bonds to hydrogen were kept fixed allowing the use of 2 fs timesteps. Protein and lipid parameters were obtained from the CHARMM2739 and CHARMM3640 force fields respectively. Ion parameters were obtained from Joung and Cheatham.41 Parameters for benzocaine and phenytoin were previously determined by Martin et al.42
Equilibrium simulations
In order to identify likely drug binding positions, 3 repeated simulations lasting 125 ns were obtained for each protein-substrate system with the drug starting in different orientations in the central cavity of the pore in each simulation. The initial 25 ns of simulation time was subsequently excluded to allow for the protein and drug to equilibrate and the remaining 100 ns trajectories were combined leading to a total of 300 ns of simulation time for each drug in each protein system. The protein coordinates were then aligned prior to a Cluster analysis being performed on the RMSD of the drug coordinates with a cut-off of 3 Å using the quality threshold algorithm.43 This allowed the identification of the most commonly occupied drug positions in the protein. Furthermore, in order to determine the interactions between the drug and specific residues in the protein for each potential binding site, the NAMDEnergy plugin was used to analyze the frames from each cluster.
Metadynamics
Metadynamics44,45 was subsequently applied to each of the mutant systems in order to determine the energetics for the binding of benzocaine and phenytoin and to ensure the drugs sample the complete interior of the channel. Well-tempered metadynamics46 was implemented with a bias factor of 10 in the PLUMED47 package. The collective variables sampled in these simulations were the x, y and z positions of the center of mass of the drug. As restraints with a force constant of 0.2 kcal/mol were applied to the C α carbons of the S5 helix of NavAb, the Cartesian coordinates of the simulation can be utilised as a direct measure of the drugs position relative to the protein throughout the simulation. Gaussians potentials, with a width and height of 0.25 kcal/mol, were deposited every 1000 steps at each position sampled by the drug. Due to the 4-fold symmetry of NavAb, analysis was performed on single quadrants of the x-y plane of the channel and flat bottomed harmonic potentials were applied to maintain the drug in the desired region. Each quadrant represents 400 ns of metadynamics simulation. For the 1S system 2 quadrants were obtained - one in the vicinity of the mutations and the quadrant opposite the mutations. In contrast, as the 4S system has the mutations in each subunit, maintaining the symmetry of the protein, only 1 quadrant was obtained in each case. Boundaries were applied at the lipid end of the fenestrations and at the end of the activation gate in order to prevent the drug leaving the pore of the channel.
Binding free energies
The free energy difference between each drug in the solvated form and bound to the protein was determined using free energy perturbation.48-53 Two independent sets of simulations were obtained; one in which the drug appears or disappears from the protein binding site, and another in which the drug appears or disappears from a box of solvent and ions. The drug binding positions in the protein were obtained from the 2 most populated clusters of the equilibrium simulations obtained for each drug. 40 evenly spaced λ windows (0.025) were utilised with 0.5 ns of equilibration and 2 ns of ensemble averaging to ensure a high level of convergence. Soft core potentials54 were used to scale the electrostatic interactions from λ = 0.5 to 1.0 and the van der Waals interactions from λ = 0.0 to 1.0 for annihilated particles. As the drugs disappear the interactions between the protein and the drug reduce progressively - to ensure that the drug remains in an appropriate position a flat bottomed harmonic potential was applied with a radius of 2 Å defined relative to the center of mass of residues chosen from the equilibrium simulation. This restraint holds a single atom of the drug in a sphere of 2 Å radius with a force constant of 5 kcal/mol. An identical restraint was also applied to the drug in bulk water but it was restrained relative to a dummy water atom. The final free energy value obtained for the drug in bulk solution was corrected to account for the loss in translational entropy. The following equation was then applied to determine the equilibrium constant55 for the binding of each drug to the mutant proteins:
where Kb is the binding affinity, ΔG is the free energy required for the drug to enter the protein from bulk solution and bind to the protein, k is the Boltzmann constant and T is the absolute temperature. The value of Kb can then be utilised to determine the dissociation constant (Kd) using the relationship Kd = 1/Kb.
Sequence alignments
The S6 sequences of NavAb from Arcobacter butzleri, NavAe1 from Alkalilimnicola ehrlichei, NavMs from Magnetococcus marinus, NavRh from Rickettsiales sp. HIMB114, NaChBac from Bacillus haldurans and from domains I to IV of human Nav1.2 were aligned using ClustalW2.56-58 The output was presented using Aline.59
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This research was undertaken with the assistance of funding from the Australian Research Council (FT130100781) and resources provided at the NCI National Facility at the Australian National University and the Pawsey Center through the National Merit allocation Scheme supported by the Australian Government and through the ANU partner Scheme.
References
- [1].Catterall WA, Goldin AL, Waxman SG. International union of pharmacology. XLVII. nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev 2005; 57:397-409; PMID:16382098; http://dx.doi.org/ 10.1124/pr.57.4.4 [DOI] [PubMed] [Google Scholar]
- [2].Goldin AL, Barchi RL, Caldwell JH, Hofmann JH, Howe JR, Hunter JC, Kallen RG, Mandel G., Meisler MH, Netter YB, et al.. Nomenclature of voltage-gated sodium channels. Neuron 2000; 28:365-368; PMID:11144347; http://dx.doi.org/ 10.1016/S0896-6273(00)00116-1 [DOI] [PubMed] [Google Scholar]
- [3].Amd N, Liu YR, Priori SG. Sodium channel mutations and arrhythmias. Nat Rev Cardiol 6:337-348; PMID:19377496 [DOI] [PubMed] [Google Scholar]
- [4].Roden DM, George AL. The cardiac ion channels: relevance to management of arrhythmias. Annu Rev Med 1996; 47:135-148; PMID:8712768; http://dx.doi.org/ 10.1146/annurev.med.47.1.135 [DOI] [PubMed] [Google Scholar]
- [5].Waxman SG, Hains BC. Fire and phantoms after spinal cord injury: Na+ channels and central pain. Trends Neurosci 2006; 29:207-215; PMID:16494954; http://dx.doi.org/ 10.1016/j.tins.2006.02.003 [DOI] [PubMed] [Google Scholar]
- [6].Waxman SG, Dib-Hajj S, Cummins TR, Black JA. Sodium channels and pain. Proc Natl Acad Sci USA 1999; 96:7635-7639; PMID:10393872; http://dx.doi.org/ 10.1073/pnas.96.14.7635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Meisler MH, Kearney JA. Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest 2005; 115:2010-2017; PMID:16075041; http://dx.doi.org/; http://dx.doi.org/ 10.1172/JCI25466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Liu G, Yarov-Yarovoy V, Nobbs M, Clare J, Scheuer T, Catterall W. Differential interactions of lamotrigine and related drugs with transmembrane segment ivs6 of voltage-gated sodium channels. Neuropharmacology 2003; 44:413-422; PMID:12604088;http://dx.doi.org/ 10.1016/S0028-3908(02)00400-8 [DOI] [PubMed] [Google Scholar]
- [9].Hanck D, Nikitina E, McNulty M, Fozzard H, Lipkind G, Sheets M. Using lidocaine and benzocaine to link sodium channel molecular conformations to state-dependent antiarrhythmic drug affinity. Circ Res 2009; 105:492-499; PMID:19661462; http://dx.doi.org/ 10.1161/CIRCRESAHA.109.198572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of state-dependent block of voltage gated Na+ channels. Proc Natl Acad Sci U.S.A 1996; 93:9270-9275; PMID:8799190; http://dx.doi.org/ 10.1073/pnas.93.17.9270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wang G, Quan C, Wang S. Local anesthetic block of batrachotoxin-resistant muscle Na+ channels. Mol Pharmacol 1998; 54:389-396; PMID:9687581 [DOI] [PubMed] [Google Scholar]
- [12].Payandeh J, Scheuer T, Zheng N, Catterall WA. The crystal structure of a voltage-gated sodium channel. Nature 2011; 475:353-358; PMID:21743477; http://dx.doi.org/ 10.1038/nature10238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Payandeh J, Gamal El-Din TM, Scheuer T, Zheng N, Catterall WA. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 2012; 486:135-139; PMID:22678296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].McCusker EC, Bagneris C, Naylor CE, Cole AR, A'Avanzo N, Nichols CG, Wallace BA. Structure of a bacterial voltage-gated sodium channel pore reveals mechanisms of opening and closing. Nature Commun 2012; 3:1-10; http://dx.doi.org/ 10.1038/ncomms2077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhang L, Ren W, DaCaen P, Yan C, Tao X, Tang L, Wang J, Hasegawa K, Kumasaka T, He J. et al.. Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature 2012; 486:130-134; PMID:22678295;http://dx.doi.org/ 10.1038/486323e [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bagneris C, Decaen PG, Hall BA, Naylor CE, Clapham DE, Kay CW, Wallace BA. Role of the c-terminal domain in the structure and function of tetrameric sodium channels. Nat Commun 2013; 4:1-10; http://dx.doi.org/ 10.1038/ncomms3465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Shaya D, Findeisen F, Abderemane-Ali F, Arrigoni C, Wong S, Nurva S, Loussouarn G, Minor D Jr.. Structure of a prokaryotic sodium channel pore reveals essential gating elements and an outer ion binding site common to eukaryotic channels. J Mol Biol 2014; 426:467-483; PMID:24120938;http://dx.doi.org/ 10.1016/j.jmb.2013.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lee S, Goodchild SJ, Ahern CA. Local anesthetic inhibition of a bacterial sodium channel. J Gen Physiol 2012; 139:507-516; PMID:22641643; http://dx.doi.org/ 10.1085/jgp.201210779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bagneris C, Decaen PG, Naylor CE, Pryde DC, Nobelli I, Clapham DE, Wallace BA. Prokaryotic NavMs channel as a structural and functional model for eukaryotic sodium channel antagonism. P Natl Scad Sci USA 2014; 111:8428-8433; http://dx.doi.org/ 10.1073/pnas.1406855111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Raju SG, Barber AF, LeBard DN, Klein ML, Carnevale V. Exploring volatile general anesthetic binding to a closed membrane-bound bacterial voltage-gated sodium channel via computation. PLoS Comput Biol 2013; 9:e1003090; PMID:23785267; http://dx.doi.org/ 10.1371/journal.pcbi.1003090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Martin LJ, Corry B. Locating the entry route and binding sites of benzocaine and phenytoin in a bacterial voltage gated sodium channel. PLoS Comput Biol 2014; 10(7) e1003688; PMID:24992293; http://dx.doi.org/ 10.1371/journal.pcbi.1003688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Boiteux C, Vorobyov I, French RJ, French C, Yarov-Yaravoy V, Allen TW. Local anesthetic and antiepileptic drug access and binding to a bacterial voltage-gated sodium channel. P Natl Acad Sci USA 2014; 111:13057-13062; http://dx.doi.org/ 10.1073/pnas.1408710111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na+ -channel inactivation. Proc Natl Acad Sci USA 1992; 89:10910-10914; PMID:1332060; http://dx.doi.org/ 10.1073/pnas.89.22.10910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Corry B, Lee S, Ahern C. Pharmacological insights and quirks of bacterial sodium channels. Handb Exp Pharmacol 2014; 221:251-267; PMID:24737240; http://dx.doi.org/ 10.1007/978-3-642-41588-3_12 [DOI] [PubMed] [Google Scholar]
- [25].Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science 1994; 265:1724-1728; PMID:8085162; http://dx.doi.org/ 10.1126/science.8085162 [DOI] [PubMed] [Google Scholar]
- [26].Ahern CA, Eastwood AL, Dougherty DA, Horn R. Electrostatic contributions of aromatic residues in the local anesthetic receptor of voltage-gated sodium channels. Circ Res 2008; 102:86-94; PMID:17967784; http://dx.doi.org/ 10.1161/CIRCRESAHA.107.160663 [DOI] [PubMed] [Google Scholar]
- [27].McNulty MM, Edgerton GB, Shah RD, Hanck DA, Fozzard HA, Lipkind GM. Charge at the lidocaine binding site residue phe-1759 affects permeation in human cardiac voltage-gated sodium channels. J Physiol 2007; 581:741-755; PMID:17363383; http://dx.doi.org/ 10.1113/jphysiol.2007.130161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mike A, Lukacs P. The enigmatic drug binding site for sodium channel inhibitors. Curr Mol Pharmacol 2010; 3:129-144; PMID:20565383; http://dx.doi.org/ 10.2174/1874467211003030129 [DOI] [PubMed] [Google Scholar]
- [29].Wang S, Mitchell J, Moczydlowski E, Wang GK. Block of inactivation-deficient Na+ channels by local anesthetics in stably transfected mammalian cells: evidence for drug binding along the activation pathway. J Gen Physiol 2004; 124:691-701; PMID:15545401; http://dx.doi.org/ 10.1085/jgp.200409128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Meeder T, Ulbricht W. Action of benzocaine on sodium channels of frog nodes of Ranvier treated with chloramine-T. Pflugers Arch 1987; 409:265-273; PMID:2442701; http://dx.doi.org/ 10.1007/BF00583475 [DOI] [PubMed] [Google Scholar]
- [31].Neguliaev IA, Nosyreva ED. Comparative study of the action of procaine and benzocaine on normal and aconitine-modified sodium channels. Tsitologiia 1979; 21:697-702; PMID:462546 [PubMed] [Google Scholar]
- [32].Schwarz JR, Grigat G. Phenytoin and carbamazepine: potential- and frequency-dependent block of Na currents in mammalian myelinated nerve fibres. Epilepsia 1989; 30:286-294; PMID:2542011; http://dx.doi.org/ 10.1111/j.1528-1157.1989.tb05300.x [DOI] [PubMed] [Google Scholar]
- [33].Kuo CC, Bean BP. Slow binding of phenytoin to inactivated sodium channels in rat hippocampal neurons. Mol Pharmacol 1994; 46:716-725; PMID:7969051 [PubMed] [Google Scholar]
- [34].Kuo CC, Chen R, Lu L, Chen R. Carbamazepine inhibition of neuronal Na+ currents: quantitative distinction from phenytoin and possible therapeutic implications. Mol Pharmacol 1997; 51:1077-1083; PMID:9187275 [DOI] [PubMed] [Google Scholar]
- [35].Lee S, Goodchild SJ, Ahern CA. Molecular and functional determinants of local anesthetic inhibition of NaChBac. Channels 2012; 6:403-406; PMID:22992485; http://dx.doi.org/ 10.4161/chan.21807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kaczmarski JA, Corry B. Investigating the size and dynamics of voltage gated sodium channel fenestrations: a molecular dynamics study. Channels 2014; 8:264-277; PMID:24632677; http://dx.doi.org/ 10.4161/chan.28136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Shaya D, Kreir M, Robbins R, Wong S, Hammon J, Brüggemann A, Minor D Jr. Voltage-gated sodium channel (NaV) protein dissection creates a set of functional pore-only proteins. Proc Natl Acad Sci USA 2011; 108:12313-12318; PMID:21746903; http://dx.doi.org/ 10.1073/pnas.1106811108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Essmann U, Perera L, Berkowitz ML. A smooth particle mesh Ewald method. J Chem Phys 1995; 103:8577-8593; http://dx.doi.org/ 10.1063/1.470117 [DOI] [Google Scholar]
- [39].MacKerell AD Jr., Bashford D, Bellott M, Dunbrack RL Jr., Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S. et al.. All-atom empirical potential for molecular modelling and dynamics studies of proteins. J Phys Chem B 1998; 102:3586-3616; PMID:24889800; http://dx.doi.org/ 10.1021/jp973084f [DOI] [PubMed] [Google Scholar]
- [40].Klauda JB, Venable RM, Freites JA, O'Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD Jr, Pastor RW. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J Phys Chem B 2010; 114:7830-7843; PMID:20496934; http://dx.doi.org/ 10.1021/jp101759q [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Joung IS, Cheatham TE. Determination of alkali and halide monovalent ion parameters for use in explicit solvated biomolecular simulations. J Phys Chem B 2008; 112:9020-9041; PMID:18593145; http://dx.doi.org/ 10.1021/jp8001614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Martin LJ, Chao R, Corry B. Molecular dynamics simulation of the partitioning of benzocaine and phenytoin into a lipid bilayer. Biophys Chem 2013; 185:98-107; PMID:24406394; http://dx.doi.org/ 10.1016/j.bpc.2013.12.003 [DOI] [PubMed] [Google Scholar]
- [43].Heyer L, Kruglyak S, Yooseph S. Exploring expression data: Identification and analysis of coexpressed genes. Genome Res 1999; 9:1106-1115; PMID:10568750; http://dx.doi.org/ 10.1101/gr.9.11.1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Laio A, Gervasio FL. Metadynamics: a method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep Prog Phys 2008; 71:126601-126622; http://dx.doi.org/ 10.1088/0034-4885/71/12/126601 [DOI] [Google Scholar]
- [45].Laio A, Parrinello M. Escaping free-energy minima. Proc Natl Acad Sci USA 2002; 99:12562-12566; PMID:12271136; http://dx.doi.org/ 10.1073/pnas.202427399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Barducci A, Bussi G, Parinello M. Well-tempered metadynamics: A smoothly converging and tunable free-energy method. Phys Rev Lett 2008; 100:020603-020607; PMID:18232845; http://dx.doi.org/ 10.1103/PhysRevLett.100.020603 [DOI] [PubMed] [Google Scholar]
- [47].Bonomi M, Branduardi D, Bussi G, Camilloni C, Provasi D, Raiteri P, Donadio D, Marinelli F, Pietrucci F, Broglia RA, et al.. PLUMED: a portable plugin for free-energy calculations with molecular dynamics. Comput Phys Commun 2009; 180:1961-1972; http://dx.doi.org/ 10.1016/j.cpc.2009.05.011 [DOI] [Google Scholar]
- [48].Chipot C, Pearlman DA. Free energy calculations. the long and winding gilded road. Mol Sim 2002; 28:1-12; http://dx.doi.org/ 10.1080/08927020211974 [DOI] [Google Scholar]
- [49].Chipot C, Pohorille A. Calculating free energy differences using perturbation theory in free energy calculations Theory and applications in chemistry and biology 2007; Berlin, Heidelberg: Springer-Verlag. [Google Scholar]
- [50].Zwanzig RW. High-temperature equation of state by a perturbation method. i. nonpolar gases. J Chem Phys 1954; 22:1420-1426; http://dx.doi.org/ 10.1063/1.1740193 [DOI] [Google Scholar]
- [51].Pearlman DA. A comparison of alternative approaches to free energy calculations. J Phys Chem 1994; 98:1487-1493; http://dx.doi.org/ 10.1021/j100056a020 [DOI] [Google Scholar]
- [52].Simonson T, Archontis G, Karplus M. Free energy simulations come of age: Protein-Ligand recognition. Acc Chem Res 2002; 35:430-437; PMID:12069628; http://dx.doi.org/ 10.1021/ar010030m [DOI] [PubMed] [Google Scholar]
- [53].Kollman PA. Free energy calculations: Applications to chemical and biochemical phenomena. Chem Rev 1993; 93:2395-2417; http://dx.doi.org/ 10.1021/cr00023a004 [DOI] [Google Scholar]
- [54].Beutler T, Mark A, van Schaik R, Gerber P, van Gunsteren W. Avoiding singularities and numerical instabilities in free energy calcu-lations based on molecular simulations. Chem Phys Lett 1994; 222:529-539; http://dx.doi.org/ 10.1016/0009-2614(94)00397-1 [DOI] [Google Scholar]
- [55].Deng Y, Roux B. Computations of standard binding free energies with molecular dynamics simulations. J Phys Chem 2009; 113:2234-2246; http://dx.doi.org/ 10.1021/jp807701h [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Larkin M, Blackshields G, Brown N, Chenna R, McGettigan P, McWilliam H, Valentin F, Wallace I, Wilm A, Lopez R, et al.. Clustal w and clustal x version 2.0. Bioinformatics 2007; 23(21):2947-2948; PMID:17846036; http://dx.doi.org/ 10.1093/bioinformatics/btm404 [DOI] [PubMed] [Google Scholar]
- [57].Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez R. A new bioinformatics analysis framework at EMBL-EBI. Nucl Acid Res 2010; 38: W695-699; http://dx.doi.org/ 10.1093/nar/gkq313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, Cowley AP, Lopez R. Analysis tool web services from the EMBL-EBI. Nucleic acids research 2013; 41: W597-600; PMID:23671338; http://dx.doi.org/ 10.1093/nar/gkt376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Bond C, Schuttelkopf A. An extensible WYSIWYG protein sequence alignment editor for publication quality figures. Acta Cryst D 2009; 65:510-512; http://dx.doi.org/ 10.1107/S0907444909007835 [DOI] [PubMed] [Google Scholar]