

Abstract
Objectives
Carbapenemases are the most important mechanism responsible for carbapenem resistance in Enterobacteriaceae. Among carbapenemases, OXA-48 presents unique challenges as it is resistant to β-lactam inhibitors. Here, we test the capacity of the compound LN-1-255, a 6-alkylidene-2′-substituted penicillanic acid sulfone, to inhibit the activity of the carbapenemase OXA-48.
Methods
The OXA-48 gene was cloned and expressed in Klebsiella pneumoniae and Escherichia coli in order to obtain MICs in the presence of inhibitors (clavulanic acid, tazobactam and sulbactam) and LN-1-255. OXA-48 was purified and steady-state kinetics was performed with LN-1-255 and tazobactam. The covalent binding mode of LN-1-255 with OXA-48 was studied by docking assays.
Results
Both OXA-48-producing clinical and transformant strains displayed increased susceptibility to carbapenem antibiotics in the presence of 4 mg/L LN-1-255 (2–32-fold increased susceptibility) and 16 mg/L LN-1-255 (4–64-fold increased susceptibility). Kinetic assays demonstrated that LN-1-255 is able to inhibit OXA-48 with an acylation efficiency (k2/K) of 10 ± 1 × 104 M−1 s−1 and a slow deacylation rate (koff) of 7 ± 1 × 10−4 s−1. IC50 was 3 nM for LN-1-255 and 1.5 μM for tazobactam. Lastly, kcat/kinact was 500-fold lower for LN-1-255 than for tazobactam.
Conclusions
In these studies, carbapenem antibiotics used in combination with LN-1-255 are effective against the carbapenemase OXA-48, an important emerging mechanism of antibiotic resistance. This provides an incentive for further investigations to maximize the efficacy of penicillin sulfone inhibition of class D plasmid-carried Enterobacteriaceae carbapenemases.
Introduction
Antimicrobial resistance rates of bacterial pathogens have steadily increased in recent years and are now considered a world health crisis by major international agencies.1 Enterobacteriaceae are a bacterial family in which significantly increased rates of resistance have been observed. The main resistance mechanism—as for Gram-negative bacteria in general—is β-lactamase-mediated hydrolysis of antimicrobial compounds.2 The prevalence and plasmid-mediated dissemination of β-lactamase genes are important clinical challenges. Until the first decade of the 21st century, the more clinically relevant β-lactamases were the ESBLs and the chromosomal and plasmid-mediated cephalosporinases (AmpCs).3 Although carbapenem antibiotics remained active against β-lactamase-expressing bacteria, in recent years there has been an increase in the number of β-lactamases able to hydrolyse carbapenems. Of these enzymes, denoted ‘carbapenemases’, a growing group of OXA enzymes—the class D β-lactamases—is especially problematic.4,5 These carbapenemases are mostly observed in Acinetobacter baumannii,5,6 but the frequency of OXA-48 expression is increasing in Enterobacteriaceae,7 largely in Klebsiella pneumoniae, and in Escherichia coli, Enterobacter cloacae, Citrobacter freundii and Providencia rettgeri.8–13
Currently, an important epidemiological change is occurring, a rapid increase in the number of isolates of carbapenemase-producing Enterobacteriaceae, mainly K. pneumoniae.14 Thus, numerous hospital outbreaks of OXA-48-carrying Enterobacteriaceae have been reported worldwide8,10,15 and the rising numbers of clinical isolates reported show OXA-48 as the most frequently detected carbapenemase in France, Belgium and Malta.12,16 The rapid increase of such carbapenem-resistant isolates is facilitated by two main methods of dissemination: (i) horizontal transfer of plasmids encoding OXA-48; and (ii) vertical dissemination of successful clones.17,18 The blaOXA-48-like genes are encoded in transposon Tn1999, carried on IncL/M-type plasmids which are broad-host-range, self-conjugative and possess a high conjugation rate.19 Along with KPC- and NDM-type carbapenemases, OXA-48 represents a major obstacle in the preservation of carbapenem efficacy against serious bacterial infections in Enterobacteriaceae.
Currently, few novel options for treatment of such MDR bacterial strains exist. One of the more promising avenues is the combination of β-lactam antibiotics with β-lactamase inhibitors, but currently commercial inhibitors are not effective against class D carbapenemases. Unlike class A enzymes, class D carbapenemases are not inhibited by β-lactamase inhibitors such as clavulanic acid, sulbactam or tazobactam.2 Bacteria harbouring and expressing these class D β-lactamases are resistant to most antibiotics with few exceptions (e.g. tigecycline and colistin), although resistance rates to these antimicrobials are also increasing.20–22 Considering that carbapenems are the main drug of choice to treat multiresistant hospital-acquired infections, the development of efficient inhibitors of OXA-48 enzymes is urgently needed in order to maintain the efficacy of β-lactam antibiotics.8,23 Avibactam has generated great interest among the medical community and has very recently been approved by the FDA for treating complicated infections caused by antibiotic-resistant pathogens.24 Avibactam demonstrates activity against OXA-48 but not against the class D carbapenemases of A. baumannii, such as OXA-23 or OXA-24/40, which are mainly responsible for carbapenem resistance in this MDR pathogen.25,26 Buynak27 has designed several β-lactamase inhibitors, of which the compound LN-1-255 represents a promising candidate in the quest for new OXA β-lactamase inhibitors. The design and synthesis of this 6-alkylidene-2′-substituted penicillin sulfone was previously reported.28 LN-1-255 demonstrated efficacy to inhibit the class D carbapenemase OXA-24/40 of A. baumannii (implicating the hydrophobic barrier established through an arrangement of the Tyr-112 and Met-223 amino acids). This barrier is not present in the OXA-48 enzyme, similarly to OXA-10, another class D β-lactamase that lacks carbapenem hydrolytic activity.29,30 LN-1-255 is also able to inhibit class A β-lactamases, such as SHV-1 and SHV-2.31 However, the potential of LN-1-255 as an inhibitor of Enterobacteriaceae-derived class D carbapenemases has not yet been evaluated. Here, we analyse the ability of LN-1-255 to inhibit OXA-48 carbapenemase, similarly to what happens with the OXA-24/40 carbapenemase. Also, we study the synergy of this inhibitor with carbapenems against clinical isolates of K. pneumoniae and transformants of E. coli and K. pneumoniae.
Materials and methods
Bacterial strains, culture medium and plasmids
For microbiological studies, the blaOXA-48 gene was amplified from a clinical strain of K. pneumoniae derived from an outbreak in our hospital (A Coruña Hospital, Spain), which began in 2013 and continues until today, affecting >160 patients. Epidemiological studies using PFGE and MLST demonstrate that the outbreak resulted from the dissemination of a single clone of serotype ST-15. All carbapenem-resistant strains involved in the outbreak harbour the same conjugative plasmid, which carries the OXA-48 and CTX-M-15 genes integrated in the composite transposon Tn1999.2.32 The strains utilized to transform the genetic constructs to study the MICs were obtained during previous studies and have porin deficits: E. coli J53 ΔompC/F lacks porins OmpC and OmpF33 and K. pneumoniae ΔompK35/36 lacks porins OmpK35 and OmpK36.34 These strains were chosen in order to increase the genetic background of resistance upon transformation. All strains were cultured in LB medium at 37°C and stored at −80°C until analysis in LB medium containing 15% glycerol. When necessary, the LB medium was supplemented with ampicillin and/or kanamycin (Sigma–Genosys, UK).
Susceptibility testing of antibiotics and inhibitors
OXA-48 was amplified from genomic DNA using the Expand High Fidelity PCR System (Roche, Basel, Switzerland) and a primer pair containing recognition sites for the restriction enzymes BamHI (5′-AAAGGATCCATGCGTGTATTAGCCTTAT-3′) and EcoRI (5′-AAAGAATTCCTAGGGAATAATTTTTTCCTGTTT-3′). The amplified DNA fragment was then digested with BamHI and EcoRI (Fermentas, Glen Burnie, MA, USA) and ligated to a BamHI/EcoRI-digested pBGS18 plasmid.35 This plasmid construct (pBGS18-OXA-48) was used to transform the strain E. coli DH5α for subcloning and then transformed into E. coli J53 ΔompC/F and K. pneumoniae ΔompK35/36.
Antibiotic susceptibility profiles were determined by microdilution following CLSI criteria.36 Susceptibility to imipenem, meropenem and ertapenem (Sigma–Genosys, UK), in combination with the inhibitor LN-1-255 at fixed concentrations of 4 or 16 mg/L, was determined. For comparison, the inhibitor tazobactam was used at the same concentrations, as well as sulbactam and clavulanic acid (Sigma–Genosys, UK) at 4 mg/L. Etest of carbapenems (bioMérieux, Marcy-l’Étoile, France) in conjunction with inhibitors was performed on Mueller–Hinton II agar plates to confirm MICs. The MICs reported are the mean of three independent replicates. LN-1-255 was prepared in the laboratories of Southern Methodist University, Dallas, TX, USA, as described previously.27 The chemical structures of LN-1-255 and tazobactam are shown in Figure 1.
Figure 1.
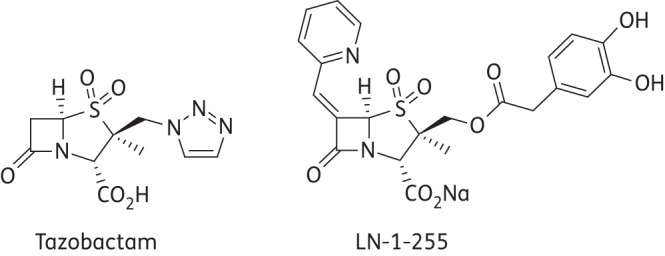
β-Lactamase inhibitors used in this study.
Synergy studies were also performed using the chequerboard method using both K. pneumoniae strains. Synergy of imipenem (range 0.12–256 mg/L) in the absence and presence of LN-1-255 at 16 mg/L was combined with tigecycline, colistin and amikacin (range 0.12–256 mg/L), important treatment options against MDR pathogens. The fractional inhibitory concentration index (FICI) was calculated and interpreted as follows: FICI ≤0.5, synergy; and FICI >0.5–4, no interaction.37
Purification of OXA-48
For kinetic studies, blaOXA-48 was directionally cloned into the p-GEX-6p-1 plasmid for expression and purification (GE Healthcare, Little Chalfont, UK). As above, this gene was amplified from genomic DNA using the Expand High Fidelity PCR System (Roche, Basel, Switzerland) with a primer pair containing recognition sites for the restriction enzymes BamHI (5′-AAAGGATCCAAGGAATGGCAAGAAAACAAA-3′) and EcoRI (5′-AAAGAATTCCTAGGGAATAATTTTTTCCTGTTT-3′). The amplified DNA fragment was then digested with BamHI and EcoRI and ligated to BamHI/EcoRI-digested p-GEX-6p-1. The recombinant plasmid was electroporated into a protease-deficient strain, E. coli BL21 (DE3), to generate the fusion protein glutathione S-transferase (GST)/OXA-48. This protein was purified to homogeneity using the GST Gene Fusion System (Amersham Pharmacia Biotech, Europe) in conjunction with the manufacturer's instructions. The purification was confirmed using SDS–PAGE gels as a band of ∼29 kDa (>95% purity) and later identification with a MALDI-TOF/TOF spectrometer (Bruker Daltonics, Billerica, MA, USA).
Kinetic experiments
Kinetic constants of OXA-48 β-lactamase were determined by continuous assays at room temperature (25°C) using a Nicolet Evolution 300 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Each experiment was performed in triplicate in 50 mM sodium phosphate with 20 mM sodium bicarbonate38 and using 0.2 and 1.0 cm pathlength cuvettes. Measurements of hydrolysis were performed with nitrocefin (Oxoid, Hampshire, UK) at 590 nM, imipenem and ertapenem, both at 299 nM. The extinction coefficients used were 15 900 M−1 cm−1 for nitrocefin, 9970 M−1 cm−1 for imipenem and 10 930 M−1 cm−1 for ertapenem. Measurements of inhibition were performed in the presence of LN-1-255 and tazobactam, using nitrocefin at 200 μM as the reporter substrate. The kinetic assays were previously described30,39,40 and are briefly explained below. Vmax, kcat and Km were determined by initial velocity kinetic analysis. The data were fitted to the Michaelis–Menten equation (Equation 1) using non-linear least-squares regression analysis:
| (1) |
The inhibitor concentration resulting in 50% reduction of nitrocefin hydrolysis after 10 min of pre-incubation of the enzyme and inhibitor at 25°C was denoted as the IC50, as previously described.30
The apparent Km for tazobactam and LN-1-255 was obtained as a competitive inhibition constant (Ki app) in the presence of nitrocefin.40 Inverse initial velocities (1/v0) were plotted against the inhibitor concentration ([I]) to obtain linear plots. Initial velocity (v0) was determined by Equation (2):
| (2) |
Ki app was determined by dividing the value of the y-intercept of the linear plot by the slope and corrected for nitrocefin affinity using Equation (3):
| (3) |
The inhibitor complex inactivation rate (kinact) in the presence of nitrocefin was measured and KI determined as previously described.41 The kobs values were determined using non-linear least-squares fitting of the data, employing Origin 7.5® software and Equation (4):
| (4) |
Here, A is absorbance, v0 (expressed in variation of absorbance per unit time) is initial velocity, vf is the final velocity and t is time. Each kobs was plotted against [I] and fitting performed to determine kinact and KI using Equation (5). The KI value was corrected using Equation (3).
| (5) |
The k2/K (observed) was determined as the slope of the line obtained by plotting [I] against kobs. This value was corrected for nitrocefin affinity as in Equation (6):
| (6) |
The koff for LN-1-255 was performed using a jump dilution method followed by continuous assays as previously described.42 Briefly, 1 μM enzyme was incubated with 10 μM LN-1-255 for 5 min at 37°C, the reaction solution was diluted 40 000-fold in assay buffer and hydrolysis of 200 μM nitrocefin was measured in 500 μL of reaction solution. Reaction solutions without OXA-48 or containing OXA-48 without LN-1-255 were used as controls.
The Kd (dissociation constant) was determined by Equation (7):
| (7) |
The partition ratio (kcat/kinact) or turnover number (tn) is defined as the ratio of inhibitor/enzyme (I : E) necessary for >90% inhibition of nitrocefin hydrolysis.30 The tn values were determined after 5 h of incubation with increasing concentrations of inhibitor and 10 nM enzyme, varying the molar ratios of I : E. Longer incubation times could not be used due to enzyme instability and loss of activity. Incubations were done in a final reaction volume of 500 μL and 200 μM nitrocefin was added immediately before measurement, in order to determine the residual enzyme activity during 60 s.
Docking studies
The covalent binding mode of LN-1-255 with OXA-48 from K. pneumoniae was studied using GOLD 5.243 and the enzyme geometries found in the crystal structure of the OXA-48/avibactam adduct (PDB 4S2K,44 2.1 Å). Water molecules and ligands were removed from the crystal structure. Ligand geometry was minimized using AM1 Hamiltonian as implemented in the program Gaussian 0945 and used as MOL2 files. The ligand was docked in 25 independent genetic algorithm (GA) runs and for each of these, a maximum number of 100 000 GA operations were performed on a single population of 50 individuals. Covalent docking was applied between the catalytic Ser-81 (oxygen atom side chain) and the carboxylate group (oxygen atom) of the indolizine obtained after covalent modification of OXA-24/40 from A. baumannii with LN-1-255.26 Operator weights for crossover, mutation and migration in the entry box were used as default parameters (95, 95 and 10, respectively), as well as hydrogen bonding (4.0 Å) and van der Waals (2.5 Å) parameters. The position of avibactam in the crystal structure was used to define the active site and the radius was set to 7 Å. The ‘flip ring corners’ flag was switched on, while all other flags were off. The GOLD scoring function was used to rank the ligands in order of fit.
Results
Antimicrobial susceptibility assays
To determine the synergy of LN-1-255 with different carbapenems against OXA-48, in vitro susceptibility to imipenem/LN-1-255, meropenem/LN-1-255 and ertapenem/LN-1-255 was compared with susceptibility to these carbapenems in combination with the β-lactamase inhibitors clavulanic acid, sulbactam and tazobactam. Results are shown in Table 1. Type strains without any resistance mechanism were not presented because they showed very low carbapenem MIC values when OXA-48 was cloned into them (data not shown), thus the used transformants were porin deficient. As discussed above, carbapenem MICs were determined using 4 mg/L of all inhibitors and 16 mg/L LN-1-255 and tazobactam. Clavulanic acid and sulbactam could not be tested at 16 mg/L for K. pneumoniae strains or at 4 mg/L for E. coli strains, as the MICs of these two inhibitors for K. pneumoniae strains were 32–256 mg/L and for E. coli strains were 16 mg/L sulbactam and 16–128 mg/L clavulanic acid. This intrinsic susceptibility to sulbactam and clavulanic acid prevents clear observation of OXA-48 inhibition.
Table 1.
Carbapenem MIC values (mg/L) for the clinical and transformed bacterial strains used
| β-Lactam + inhibitor |
K. pneumoniae clinical strain (OXA-48) |
K. pneumoniae ΔompK35/36 |
K. pneumoniae ΔompK35/36 (OXA-48) |
E. coli J53 ΔompC/F |
E. coli J53 ΔompC/F (OXA-48) |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| 4 mg/L | 16 mg/L | 4 mg/L | 16 mg/L | 4 mg/L | 16 mg/L | 4 mg/L | 16 mg/L | 4 mg/L | 16 mg/L | |
| Imipenem | 64 | 64 | 0.5 | 0.5 | 128 | 128 | 0.25 | 0.25 | 1 | 1 |
| Imipenem + TZB | 64 | 32 | 0.5 | 0.25 | 128 | 128 | 0.25 | 0.125 | 1 | 0.5 |
| Imipenem + SUL | 64 | NA | 0.5 | NA | 128 | NA | NA | NA | NA | NA |
| Imipenem + CLA | 64 | NA | 0.5 | NA | 128 | NA | NA | NA | NA | NA |
| Imipenem + LN | 16 | 4 | 0.5 | 0.25 | 32 | 16 | 0.25 | 0.25 | 0.5 | 0.125 |
| Meropenem | 16 | 16 | 0.25 | 0.25 | 64 | 64 | 0.03 | 0.03 | 2 | 2 |
| Meropenem + TZB | 16 | 8 | 0.25 | 0.25 | 32 | 32 | 0.03 | 0.015 | 1 | 1 |
| Meropenem + SUL | 16 | NA | 0.25 | NA | 32 | NA | NA | NA | NA | NA |
| Meropenem + CLA | 16 | NA | 0.25 | NA | 32 | NA | NA | NA | NA | NA |
| Meropenem + LN | 8 | 4 | 0.25 | 0.25 | 16 | 8 | 0.03 | 0.03 | 0.25 | 0.06 |
| Ertapenem | 128 | 128 | 2 | 2 | 256 | 256 | 0.015 | 0.015 | 0.5 | 0.5 |
| Ertapenem + TZB | 128 | 32 | 2 | 1 | 256 | 128 | 0.015 | 0.007 | 0.5 | 0.25 |
| Ertapenem + SUL | 128 | NA | 2 | NA | 256 | NA | NA | NA | NA | NA |
| Ertapenem + CLA | 128 | NA | 2 | NA | 256 | NA | NA | NA | NA | NA |
| Ertapenem + LN | 64 | 16 | 2 | 1 | 64 | 64 | 0.015 | 0.015 | 0.015 | 0.007 |
TZB, tazobactam; SUL, sulbactam; CLA, clavulanic acid; LN, LN-1-255.
Data represent the means of three independent experiments.
The four inhibitors were tested at 4 mg/L using K. pneumoniae clinical and transformed strains. Tazobactam and LN-1-255 were tested up to 4 mg/L using transformed E. coli; and tazobactam and LN-1-255 were tested up to 16 mg/L using K. pneumoniae clinical and transformed strains.
NA, some combinations of carbapenems and inhibitors could not be tested due to intrinsic strain susceptibility. MICs for transformed E. coli were 16 mg/L sulbactam and 16–128 mg/L clavulanic acid and for K. pneumoniae strains were 32–256 mg/L (both inhibitors).
Neither sulbactam nor clavulanic acid at 4 mg/L were able to inhibit OXA-48 activity at any dilution in the performed assays. At 4 mg/L, tazobactam was able to inhibit OXA-48 activity in only one dilution and otherwise did not reduce MICs compared with carbapenems alone. At 4 mg/L, LN-1-255 was able to reduce carbapenem MICs 2–32-fold. These differences were amplified when 16 mg/L LN-1-255 or tazobactam was used.
The clinical K. pneumoniae isolate displayed an imipenem MIC of 64 mg/L, which was decreased 16-fold to 4 mg/L in the presence of LN-1-255 and 2-fold to 32 mg/L in the presence of tazobactam. Similarly, transformed K. pneumoniae ΔompK35/36 displayed an imipenem MIC of 128 mg/L, which was decreased 8-fold to 16 mg/L in the presence of 16 mg/L LN-1-255, but 0-fold in the presence of 16 mg/L tazobactam.
More pronounced differences were observed in transformed E. coli ΔompC/F, with 8-, 32- and 64-fold reductions in imipenem, meropenem and ertapenem MICs in the presence of LN-1-255, compared with a 2-fold MIC decrease in the presence of tazobactam. The remaining data are shown in Table 1. Using transformed E. coli DH5α, the effect of 4 mg/L LN-1-255 on the carbapenem MIC was also more significant than that of 4 mg/L tazobactam. However, at inhibitor concentrations of 16 mg/L, this transformed strain was highly susceptible to the antimicrobial effect of the inhibitors, precluding quantitative analysis of effect sizes (data not shown).
By the chequerboard method, the combination of imipenem/LN-1-255 or imipenem alone was tested against tigecycline, colistin and amikacin. The presence of LN-1-255 decreased the MICs of tigecycline and amikacin by one dilution for the K. pneumoniae clinical strain and also the MIC of amikacin by one dilution for the ΔompK35/36 mutant. However, the FICI was >0.5, so no synergy was observed with these antimicrobials (data not shown).
Kinetics and inhibition studies
Kinetic properties of substrate hydrolysis by OXA-48 are shown in Table 2. The Km for nitrocefin was 65.7 μM, which allowed us to use nitrocefin at 200 μM as a reporter substrate in the studies of inhibition kinetics. Similar Km values were observed for both carbapenems tested (imipenem and ertapenem, 60.3 and 123.7 μM, respectively), but pronounced differences were observed in hydrolysis velocity (kcat) for carbapenems. When converted to catalytic efficiency, this value was 10-fold higher for imipenem than ertapenem (0.025 and 0.002 μM−1 s−1, respectively) while the catalytic efficiency of nitrocefin (4.8 μM−1 s−1) hydrolysis was 192-fold higher than that of imipenem.
Table 2.
Carbapenem and nitrocefin hydrolysis kinetics of OXA-48
| Substrate | Km (μM) | kcat (s−1) | kcat/Km (μM−1 s−1) |
|---|---|---|---|
| Nitrocefin | 65.7 ± 18.9 | 314.7 ± 18.9 | 4.79 |
| Imipenem | 60.3 ± 12.4 | 1.5 ± 0.1 | 0.025 |
| Ertapenem | 123.7 ± 36.2 | 0.3 ± 0.02 | 0.002 |
Data represent the means of three independent experiments.
The OXA-48 inhibitory activities of LN-1-255 and tazobactam were determined and are summarized in Table 3. Using nitrocefin as the indicator substrate, LN-1-255 showed inhibitory activity at much lower concentrations than tazobactam. Thus, the Ki app—analogous to the Km for inhibitors—showed a higher affinity between the carbapenemase and LN-1-255 than between the carbapenemase and tazobactam (Ki app = 0.17 and 30 μM, respectively).
Table 3.
OXA-48 inhibition kinetics of LN-1-255 and tazobactam
| Substrate | IC50 (μM) | Ki app (μM) | KI (μM) | kinact (s−1) | kinact/KI (M−1 s−1) | k2/K (M−1 s−1) | koff (s−1) | t1/2,off (min) | Kd (nM) | tn |
|---|---|---|---|---|---|---|---|---|---|---|
| LN-1-255 | 0.003 ± 0.0003 | 0.17 ± 0.01 | NA | NA | NA | (10 ± 1) × 104 | (7 ± 1) × 10−4 | 16.5 | 7 | 2 |
| Tazobactam | 1.5 ± 0.5 | 30 ± 3 | 14 ± 2 | 0.038 ± 0.004 | 3000 ± 500 | NA | NA | NA | NA | 1000 |
Data represent the means of three independent experiments. NA, not adequate.
In order to determine kinact values and inactivation efficiencies (kinact/KI) of LN-1-255 and tazobactam, progress curves were fitted to Equation (3) to obtain kobs values (Figure 2). These kobs values were plotted against concentrations of both inhibitors and indicated faster acylation at lower concentrations of LN-1-255 than of tazobactam. However, in the case of LN-1-255, this determination could not be completed due to an inability to achieve the maximum rate. Instead, k2/K was determined as an approximation of the inactivation efficiency (10 × 104 M−1 s−1). The kinact/KI of tazobactam was 3000 M−1 s−1, showing an OXA-48 inactivation efficiency >33-fold lower than that of LN-1-255. Similarly, the IC50 of LN-1-255 (0.003 μM) was much lower (∼500-fold) than that of tazobactam (1.5 μM), indicating a higher relative efficacy of LN-1-255. Separate experiments (koff) indicated that the off rate of LN-1-255 was 7 × 10−4 s−1, representing a slow return of carbapenemase activity, with a half-life (t1/2) of 16.5 min (Figure 3). In the case of tazobactam, an irreversible inhibitor, it was not possible to determine the off rate.46
Figure 2.
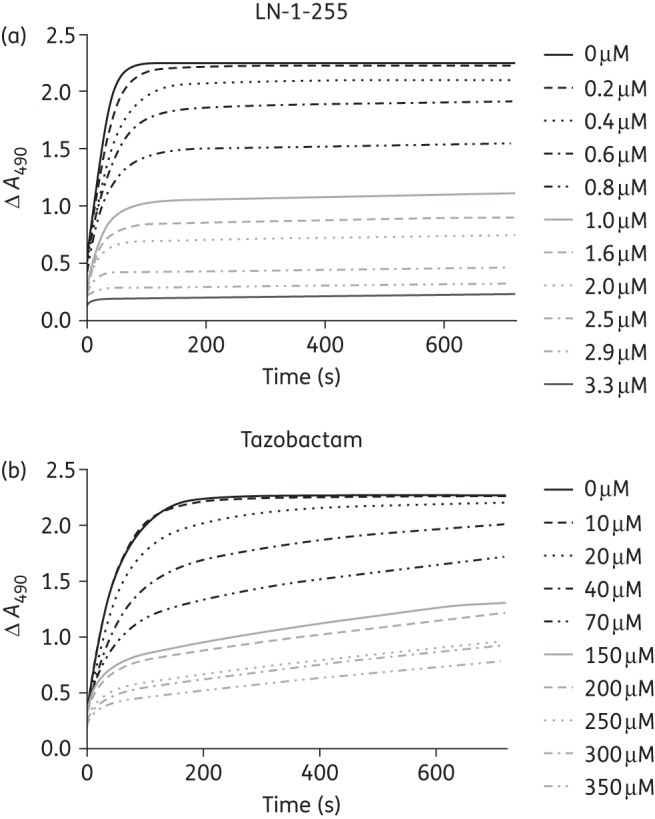
Progress curves of 200 µM nitrocefin hydrolysis with increasing inhibitor concentrations (0.2 µM–3.3 µM LN-1-255 and 10 µM–350 µM tazobactam) and a fixed concentration of OXA-48 (8.6 nM).
Figure 3.
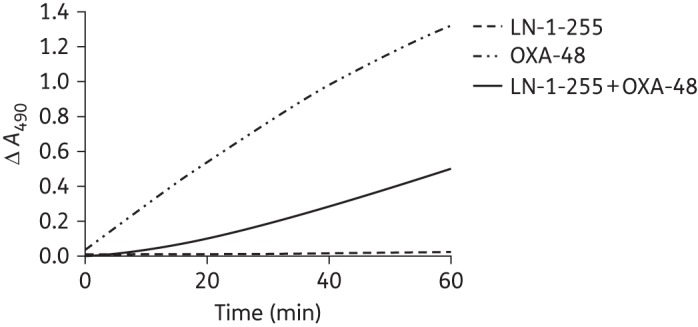
Progress curves for determination of off rates (koff) for OXA-48/LN-1-255 and controls (OXA-48 alone and LN-1-255 alone) with 200 µM nitrocefin. OXA-48 was incubated with LN-1-255 to allow formation of the acyl-enzyme adduct and then diluted to permit enzyme reactivation.
Lastly, in determining the partition ratio (tn) of LN-1-255 and tazobactam, the data showed a relative I : E significantly lower for LN-1-255 (tn = 2) than for tazobactam (tn = 1000), indicating 90% inhibition of OXA-48 at a much lower concentration (∼500-fold) of LN-1-255 than of tazobactam (Figure 4).
Figure 4.
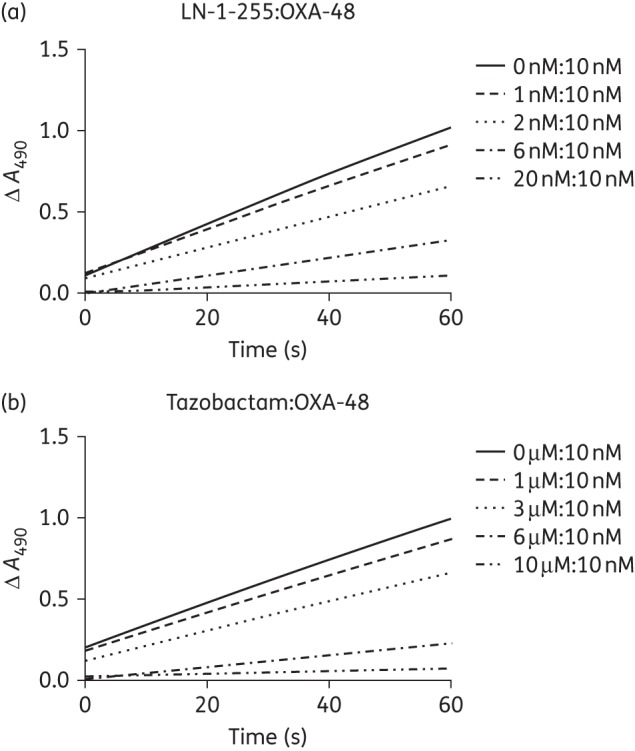
Progress curves for determination of tn. OXA-48 at 10 nM was incubated with increasing concentrations of LN-1-255 (a) or tazobactam (b).
Docking studies
Similar to a previously reported OXA-48/avibactam adduct (PDB 4S2K,38 2.1 Å; Figure 5a and c), our docking studies showed that the indolizine adduct obtained after covalent modification of the catalytic Ser-70 by LN-1-255 would also interact with residues Arg-250, Lys-208, Tyr-211 and Thr-209 (Figure 5b and d). Specifically, the carboxylate group of the modified ligand would be anchored in the active site by electrostatic interactions with the guanidinium group of Arg-250 and the ϵ-amino group of Lys-208 and hydrogen-bonding interaction with the hydroxyl group of Thr-209. In addition, the sulfinate group would establish an electrostatic interaction with the guanidinium group of Arg-250 and also the carboxylate group of the ester linkage would interact by hydrogen bonding with the main chain amide of Tyr-211. In contrast, our molecular docking studies predict that the catechol side chain would be more flexible. One of the phenol groups of the catechol moiety would interact by hydrogen bonding with the ϵ-amino group of Lys-208. This catechol moiety would partially occupy the large pocket close to the active site.
Figure 5.

Docking of avibactam and LN-1-255 into the active site of OXA-48. Carbapenemase OXA-48 from K. pneumoniae covalently modified by avibactam and LN-1-255. (a and c) Crystal structure of OXA-48/avibactam adduct (PDB 4S2K,44 2.1 Å). (b and d) Inactivation of OXA-48 by LN-1-255 obtained by molecular docking studies. Hydrogen bonding and electrostatic interactions between the ligand and OXA-48 are shown as dashed blue lines. Relevant side chain residues are shown and labelled. Figure created using the PyMOL Molecular Graphics System.53 This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
Discussion
Carbapenem-hydrolysing class D β-lactamases can confer resistance to carbapenems, mostly in A. baumannii or Enterobacteriaceae with OXA-24/40 and OXA-48 as respective examples of such enzymes, limiting therapeutic options for infection with these pathogens.20,21 The increasing number of clinical isolates and outbreaks of strains carrying class D carbapenemases underscores the need for new β-lactamase inhibitors that can restore carbapenem efficacy. The penicillin sulfone inhibitor LN-1-255 is a novel inhibitor of β-lactamases.28 Initially, LN-1-255 was shown to be effective in inhibiting class A β-lactamases31 and, subsequently, class D enzymes.29,30 Similarly to OXA-24/40, carbapenem MICs in the presence of LN-1-255 were more significantly reduced when 16 mg/L rather than 4 mg/L LN-1-255 was used.30
We have thus observed inhibition of OXA-48 carbapenemases, both in a clinical isolate of K. pneumoniae and in porin-deficient transformed K. pneumoniae and E. coli strains. Depending on which carbapenem was used, we obtained 2–64-fold reductions in carbapenem MICs. However, in most cases, use of the inhibitors (clavulanic acid, sulbactam and tazobactam) did not alter carbapenem MICs. These results are consistent with those obtained in similar studies on OXA-24/40 inhibition, where carbapenem MICs decreased 8–32-fold in the presence of LN-1-255, but only up to 2-fold in the presence of tazobactam.30 In this study, the porin deficit in K. pneumoniae slightly decreased the efficacy of LN-1-255 at 16 mg/L when combined with imipenem and ertapenem compared with the clinical strain (16-fold for imipenem and 8-fold for ertapenem); however, it increased the efficacy when combined with meropenem (MIC decreased 8-fold in the presence of 1-1-255 in the ΔompK35/36 mutant and just 4-fold in the clinical strain. Thus, it seems that LN-1-255 is able to pass through the outer membrane, despite the porin deficit. In fact, this inhibitor was designed with a catecholic functionality that resembles a natural bacterial siderophore, enabling it to utilize the iron uptake system to traverse the outer membrane.28
The carbapenem hydrolysis kinetics of OXA-48 (Km and kcat) are largely concordant with those previously published by Poirel et al.47,48 LN-1-255 was highly effective at inhibiting OXA-48 β-lactamase. LN-1-255 demonstrated a 33-fold higher inhibition efficiency than tazobactam, with k2/K = 10 × 104 M−1 s−1 and kinact/KI = 3000 M−1 s−1, respectively. The inhibition due to LN-1-255 is characterized by high binding affinity, with Ki = 0.17 μM and IC50 = 0.003 μM, which are 180- and 500-fold lower, respectively, than those yielded using tazobactam. The off rate of LN-1-255 was moderately slow, with koff = 7 × 10−4 s−1, yielding an enzyme reactivation half-life of 16.5 min. Lastly, a partition rate (tn) of 2 for LN-1-255 compares very favourably with that for tazobactam (>1000) required for 90% inactivation of OXA-48. These data likely indicate that LN-1-255 molecules are more slowly hydrolysed by OXA-48 than tazobactam molecules.
Regarding the A. baumannii OXA-24/40 carbapenemase, LN-1-255-mediated inhibition, which was previously tested by our group, proved to be similar to the binding affinity of LN-1-255 for OXA-48, with Ki app = 0.7 and 0.17 μM, respectively. LN-1-255 inhibition efficacies were also similar between these two enzymes, with k2/K = 10 × 104 M−1 s−1 for OXA-48 and kinact/KI = 21 × 104 M−1 s−1 for OXA-24/40.30 The crystal structure of both carbapenemases has been previously determined, OXA-48 by Docquier et al.49 and OXA-24/40 by our group.50 These two β-lactamases were the first class D carbapenemase crystal structures published and may be considered as two different carbapenemase models: although they share a common evolutionary origin, they eventually acquired their carbapenemase activity through distinct evolutionary pathways. Significant differences in the size and shape of their active sites are evident. OXA-24/40 presents a ‘hydrophobic barrier’ (consisting of Tyr-112 and Met-223) covering the active site, which is implicated in correct orientation of small substrates such as the carbapenems. However, this is not a conserved structure in the OXA carbapenemases and OXA-48 lacks this tunnel-like structure. By contrast, OXA-48 displays more homology with OXA-10, which is not a carbapenemase but is highly active in hydrolysing oxacillin. This ability is shared by OXA-48 but not OXA-24/40.30,49 Despite these important differences, the compound LN-1-255 was able to inhibit both enzymes (OXA-24/40 and OXA-48) in the nM range. Later, De Luca et al.51 showed that the small β5-β6 loop of OXA-48, close to the active site, is implicated in the ability of OXA β-lactamase to hydrolyse carbapenems by facilitating movement of the deacylating water molecule towards the acylated catalytic Ser-70 residue. The relevance of this loop has been confirmed and it is present in OXA-23, OXA-24/40 and OXA-48 carbapenemases; meanwhile, it presents significant structural differences in OXA-10 β-lactamase.49,51 This suggests that LN-1-255 interacts with this loop to prevent carbapenem access to the active site of the carbapenemases. However, further structural and biochemical work is necessary to identify the precise inhibition mechanism.
When comparing our results with those obtained by Ehmann et al.52 studying avibactam and OXA-48, a new β-lactamase inhibitor, we observed a faster on rate with LN-1-255 (10 × 104 M−1 s−1 compared with 1.4 × 103 M−1 s−1 of avibactam, 71-fold lower). However, avibactam displayed a slower koff (1.2 × 10−5 s−1) compared with the off rate of LN-1-255 (7 × 10−4 s−1), resulting in a 60-fold higher avibactam half-life.52 Thus, the half-life of LN-1-255 of 16.5 min could be a significant drawback if used in the treatment of infections. How these kinetic data correlate to the clinical impact of these inhibitors is an issue that should be studied in experimental models of infection in the near future.
Figure 5 represents avibactam and LN-1-255 at the active binding site of OXA-48; the docking of LN-1-255 on the active site supports the inhibitory role of this compound. LN-1-255 is anchored to OXA-48, similarly to the structure of avibactam and OXA-48 published by King and Strynadka.44 Furthermore, although avibactam is able to inhibit OXA-48, it poorly inhibits OXA-24/40 of A. baumannii, in contrast to LN-1-255.26 Structurally, it has been suggested that the inhibition efficiency of LN-1-255 is mainly due to: (i) the presence of the catechol moiety that facilitates entry through the outer membrane via the cellular iron uptake pathway; and (ii) the incorporation of a (2-pyridyl)methylene group at C6 in the sulbactam core that results in the formation of an heterocyclic (indolizine) ester derivative that is resistant to hydrolysis.26 Despite the high stability of the heterocyclic esters, LN-1-255 proved to have a higher koff (7 × 10−4 s−1) against OXA-48 from K. pneumoniae than avibactam (1.2 × 10−5 s−1). It has been suggested that avibactam does not decompose via a hydrolytic mechanism of the carbamoyl linkage with the catalytic serine of the carbapenemase. Instead, it seems to occur via regeneration of the diazobicyclooctane moiety. Both acyl-enzyme intermediates are electronically stabilized towards attack of water through resonance interactions of the acyl-enzyme carbonyl group with the lone pair of electrons on an adjacent nitrogen atom. Instead of being an ester linkage, the avibactam acyl-enzyme is a urethane, while that of LN-1-255 is a vinylogous urethane. It is likely that the more sterically bulky environment surrounding the avibactam acyl-enzyme linkage interferes with the approach of the hydrolytic water, while the more two-dimensional vinylogous urethane allows this approach to a limited extent. Also, the more bulky nature of the avibactam moiety in the site may preclude conformational movement of the inhibitor fragment to allow approach of nucleophiles to the carbonyl, thus limiting the approaching nucleophiles to the already well-positioned sulfamoyl nitrogen and regenerating the inhibitor itself. LN-1-255, by contrast, likely has more conformational freedom, potentially leading to movement that disrupts the planarity needed for resonance stabilization of the acyl-enzyme carbonyl group.
We consider that, in addition, there are some differences in the binding mode of both modified ligands that could also contribute to the lower stability of the OXA-48/LN-1-255 adduct than the corresponding OXA-48/avibactam. Comparison of the binding mode of the modified avibactam shown in the crystal structure of the OXA-48/avibactam adduct (PDB 4S2K)44 and the predicted binding mode of the corresponding OXA-48/LN-1-255 adduct obtained by molecular docking studies revealed that both adducts interact with mainly the same active site residues, specifically, Arg-250, Lys-208, Tyr-211 and Thr-209. However, as illustrated in Figure 5, there are some differences in the binding contacts of the groups that most contribute to anchor the ligand in the active site, the sulphate of avibactam and sulfinate/carboxylate of LN-1-255 groups that might explain their different behaviour. While the sulphate group in the avibactam adduct is deeply anchored in the active site through a salt bridge with Arg-250 and hydrogen bonding with the Thr-209 side chain (Figure 5c), the sulfinate group in the modified LN-1-255 only interacts by electrostatic interaction with Arg-250 (Figure 5d). We consider that the latter is a consequence of the lack of the hydrophobic barrier present in OXA-24/40, which involves mainly residues Tyr-112 and Met-223.30 In this case, the carboxylate group of the modified LN-1-255 interacts by hydrogen bonding with the phenol group of Tyr-112. As a consequence, the sulfinate group can be located pointing inwards. As in the crystal structure of the OXA-48/avibactam adduct, the latter group would interact by a salt bridge with the guanidinium group of Arg-261 and hydrogen bonding with the Ser-128 side chain. These binding differences might account for the overall difference in stability of the two adducts as experimentally observed (on and off rates).
Due to structural and mechanistic differences among class D carbapenemases,49 this area of drug discovery requires further studies to identify additional inhibitors as possible therapeutic options. The results presented here provide stimulus for further in vitro and in vivo investigations to maximize the efficacy of penicillin sulfone inhibition of class D carbapenemases in Enterobacteriaceae.
Funding
This work was supported by the Spanish National Plans for Scientific Research, Development and Technological Innovation 2013-16 and funded by the ISCIII-General Subdirection of Assessment and Promotion of the Research-European Regional Development Fund (ERDF) ‘A way of making Europe’: PI12/00552 to G. B. and PI14/00059 to M. P. and A. B. Also, research reported in this publication was supported in part by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (USA) under Award Numbers R01AI100560, R01AI063517 and R01AI072219 to R. A. B. This study was supported in part by funds and/or facilities provided by the Cleveland Department of Veterans Affairs (USA), Award Number 1I01BX001974 to R. A. B. from the Biomedical Laboratory Research & Development Service of the VA Office of Research and Development and the Geriatric Research Education and Clinical Center VISN 10 (USA) to R. A. B. This study was also supported by the Spanish Ministry of Economy and Competiveness (SAF2013-42899-R), Xunta de Galicia (Spain) (GRC2013-041) and the European Regional Development Fund (ERDF) to C. G.-B, and supported by National Institutes of Health (USA) to J. D. B. (1R15AI109624). J. V. A. was financially supported by the Sara Borrell Programme ISCIII-FEDER (CD13/00373). J. V. H. and A. B. were financially supported by the Miguel Servet Programme ISCIII-FEDER (CP13/00226).
Transparency declarations
None to declare.
Disclaimer
The content is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health or the Department of Veterans Affairs.
Acknowledgements
We thank Dr Sebastian Albetí for the kind gift of the strain K. pneumoniae ΔompK35/36.
References
- 1.The White House. National Strategy to Combat Antibiotic-Resistant Bacteria. 2014. http://www.cdc.gov/drugresistance/federal-engagement-in-ar/national-strategy/index.html.
- 2.Bush K, Jacoby GA. Updated functional classification of β-lactamases. Antimicrob Agents Chemother 2010; 54: 969–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oteo J, Perez-Vazquez M, Campos J. Extended-spectrum β-lactamase producing Escherichia coli: changing epidemiology and clinical impact. Curr Opin Infect Dis 2010; 23: 320–6. [DOI] [PubMed] [Google Scholar]
- 4.Walsh TR. Emerging carbapenemases: a global perspective. Int J Antimicrob Agents 2010; 36 Suppl 3: S8–14. [DOI] [PubMed] [Google Scholar]
- 5.Poirel L, Naas T, Nordmann P. Diversity, epidemiology, and genetics of class D β-lactamases. Antimicrob Agents Chemother 2010; 54: 24–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonnin RA, Nordmann P, Poirel L. Screening and deciphering antibiotic resistance in Acinetobacter baumannii: a state of the art. Expert Rev Anti Infect Ther 2013; 11: 571–83. [DOI] [PubMed] [Google Scholar]
- 7.Poirel L, Potron A, Nordmann P. OXA-48-like carbapenemases: the phantom menace. J Antimicrob Chemother 2012; 67: 1597–606. [DOI] [PubMed] [Google Scholar]
- 8.Nordmann P, Poirel L. The difficult-to-control spread of carbapenemase producers among Enterobacteriaceae worldwide. Clin Microbiol Infect 2014; 20: 821–30. [DOI] [PubMed] [Google Scholar]
- 9.Arana DM, Saez D, Garcia-Hierro P et al. Concurrent interspecies and clonal dissemination of OXA-48 carbapenemase. Clin Microbiol Infect 2015; 21: 148.e1-4. [DOI] [PubMed] [Google Scholar]
- 10.Branas P, Villa J, Viedma E et al. Molecular epidemiology of carbapenemase-producing Klebsiella pneumoniae in a hospital in Madrid: successful establishment of an OXA-48 ST11 clone. Int J Antimicrob Agents 2015; 46: 111–6. [DOI] [PubMed] [Google Scholar]
- 11.Grundmann H, Livermore DM, Giske CG et al. Carbapenem-non-susceptible Enterobacteriaceae in Europe: conclusions from a meeting of national experts. Euro Surveill 2010; 15: pii=19711. [DOI] [PubMed] [Google Scholar]
- 12.Dortet L, Cuzon G, Nordmann P. Dissemination of carbapenemase-producing Enterobacteriaceae in France, 2012. J Antimicrob Chemother 2014; 69: 623–7. [DOI] [PubMed] [Google Scholar]
- 13.Nordmann P, Naas T, Poirel L. Global spread of carbapenemase-producing Enterobacteriaceae. Emerg Infect Dis 2011; 17: 1791–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oteo J, Miró E, Pérez-Vázquez M et al. Evolution of carbapenemase-producing Enterobacteriaceae at the global and national level: what should be expected in the future? Enferm Infecc Microbiol Clin 2014; 32 Suppl 4: 17–23. [DOI] [PubMed] [Google Scholar]
- 15.Canton R, Akova M, Carmeli Y et al. Rapid evolution and spread of carbapenemases among Enterobacteriaceae in Europe. Clin Microbiol Infect 2012; 18: 413–31. [DOI] [PubMed] [Google Scholar]
- 16.Glasner C, Albiger B, Buist G et al. Carbapenemase-producing Enterobacteriaceae in Europe: a survey among national experts from 39 countries, February 2013. Euro Surveill 2013; 18: pii=20525. [DOI] [PubMed] [Google Scholar]
- 17.Pitart C, Sole M, Roca I et al. First outbreak of a plasmid-mediated carbapenem-hydrolyzing OXA-48 β-lactamase in Klebsiella pneumoniae in Spain. Antimicrob Agents Chemother 2011; 55: 4398–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Potron A, Kalpoe J, Poirel L et al. European dissemination of a single OXA-48-producing Klebsiella pneumoniae clone. Clin Microbiol Infect 2011; 17: E24–6. [DOI] [PubMed] [Google Scholar]
- 19.Poirel L, Bonnin RA, Nordmann P. Genetic features of the widespread plasmid coding for the carbapenemase OXA-48. Antimicrob Agents Chemother 2012; 56: 559–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Falagas ME, Karageorgopoulos DE, Nordmann P. Therapeutic options for infections with Enterobacteriaceae producing carbapenem-hydrolyzing enzymes. Future Microbiol 2011; 6: 653–66. [DOI] [PubMed] [Google Scholar]
- 21.Bradford PA, Kazmierczak KM, Biedenbach DJ et al. Colistin-resistant Enterobacteriaceae: correlation of β-lactamase production and colistin resistance among isolates from a global surveillance program. Antimicrob Agents Chemother 2015; 60: 1385–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu YY, Wang Y, Walsh TR et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect Dis 2015; 16: 161–8. [DOI] [PubMed] [Google Scholar]
- 23.Perez-Llarena FJ, Bou G. β-Lactamase inhibitors: the story so far. Curr Med Chem 2009; 16: 3740–65. [DOI] [PubMed] [Google Scholar]
- 24.Zhanel GG, Lawson CD, Adam H et al. Ceftazidime-avibactam: a novel cephalosporin/β-lactamase inhibitor combination. Drugs 2013; 73: 159–77. [DOI] [PubMed] [Google Scholar]
- 25.Drawz SM, Papp-Wallace KM, Bonomo RA. New β-lactamase inhibitors: a therapeutic renaissance in an MDR world. Antimicrob Agents Chemother 2014; 58: 1835–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lahiri SD, Mangani S, Jahic H et al. Molecular basis of selective inhibition and slow reversibility of avibactam against class D carbapenemases: a structure-guided study of OXA-24 and OXA-48. ACS Chem Biol 2015; 10: 591–600. [DOI] [PubMed] [Google Scholar]
- 27.Buynak JD. The discovery and development of modified penicillin- and cephalosporin-derived β-lactamase inhibitors. Curr Med Chem 2004; 11: 1951–64. [DOI] [PubMed] [Google Scholar]
- 28.Buynak JD, Rao AS, Doppalapudi VR et al. The synthesis and evaluation of 6-alkylidene-2′β-substituted penam sulfones as β-lactamase inhibitors. Bioorg Med Chem Lett 1999; 9: 1997–2002. [DOI] [PubMed] [Google Scholar]
- 29.Drawz SM, Bethel CR, Doppalapudi VR et al. Penicillin sulfone inhibitors of class D β-lactamases. Antimicrob Agents Chemother 2010; 54: 1414–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bou G, Santillana E, Sheri A et al. Design, synthesis, and crystal structures of 6-alkylidene-2′-substituted penicillanic acid sulfones as potent inhibitors of Acinetobacter baumannii OXA-24 carbapenemase. J Am Chem Soc 2010; 132: 13320–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pattanaik P, Bethel CR, Hujer AM et al. Strategic design of an effective β-lactamase inhibitor: LN-1-255, a 6-alkylidene-2′-substituted penicillin sulfone. J Biol Chem 2009; 284: 945–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barba MJ, Fernandez A, Vindel A et al. Brotes simultáneos producidos por una cepa de Klebsiella pneumoniae productora de CTX-M-15 y OXA-48 y por otra cepa diferente productora de CTX-M-15 en el Complejo Hospitalario Universitario A Coruña (CHUAC). In: Abstracts of the Eighteenth Spanish Meeting of Clinical Microbiology and Infectious Diseases, Valencia, 2014 Abstract 51, p. 42. Spanish Society of Clinical Microbiology and Infectious Diseases, Madrid, Spain. [Google Scholar]
- 33.Beceiro A, Maharjan S, Gaulton T et al. False extended-spectrum β-lactamase phenotype in clinical isolates of Escherichia coli associated with increased expression of OXA-1 or TEM-1 penicillinases and loss of porins. J Antimicrob Chemother 2011; 66: 2006–10. [DOI] [PubMed] [Google Scholar]
- 34.Garcia-Sureda L, Juan C, Domenech-Sanchez A et al. Role of Klebsiella pneumoniae LamB porin in antimicrobial resistance. Antimicrob Agents Chemother 2011; 55: 1803–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beceiro A, Perez-Llarena FJ, Perez A et al. Molecular characterization of the gene encoding a new AmpC β-lactamase in Acinetobacter baylyi. J Antimicrob Chemother 2007; 59: 996–1000. [DOI] [PubMed] [Google Scholar]
- 36.Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically–Tenth Edition: Approved Standard M07-A10. CLSI, Wayne, PA, USA, 2015. [Google Scholar]
- 37.Odds FC. Synergy, antagonism, and what the chequerboard puts between them. J Antimicrob Chemother 2003; 52: 1. [DOI] [PubMed] [Google Scholar]
- 38.Golemi D, Maveyraud L, Vakulenko S et al. Critical involvement of a carbamylated lysine in catalytic function of class D β-lactamases. Proc Natl Acad Sci USA 2001; 98: 14280–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Papp-Wallace KM, Winkler ML, Taracila MA et al. Variants of β-lactamase KPC-2 that are resistant to inhibition by avibactam. Antimicrob Agents Chemother 2015; 59: 3710–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Papp-Wallace KM, Mallo S, Bethel CR et al. A kinetic analysis of the inhibition of FOX-4 β-lactamase, a plasmid-mediated AmpC cephalosporinase, by monocyclic β-lactams and carbapenems. J Antimicrob Chemother 2014; 69: 682–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bethel CR, Taracila M, Shyr T et al. Exploring the inhibition of CTX-M-9 by β-lactamase inhibitors and carbapenems. Antimicrob Agents Chemother 2011; 55: 3465–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ehmann DE, Jahic H, Ross PL et al. Avibactam is a covalent, reversible, non-β-lactam β-lactamase inhibitor. Proc Natl Acad Sci USA 2012; 109: 11663–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cambridge Crystallographic Data Centre. http://www.ccdc.cam.ac.uk/products/life_sciencies/gold/.
- 44.King DT, Strynadka NCJ. The X-ray crystal structure is available from the Protein Data Bank (PDB: 4S2K). OXA-48 in complex with avibactam at pH 7.5. http://www.rcsb.org/pdb/explore/explore.do?structureId=4S2K.
- 45.Frisch GWT, Schlegel HB, Scuseria GE et al. Gaussian 09, Revision A.2. Wallingford, CT: Gaussian, 2009. [Google Scholar]
- 46.Bush K, Macalintal C, Rasmussen BA et al. Kinetic interactions of tazobactam with β-lactamases from all major structural classes. Antimicrob Agents Chemother 1993; 37: 851–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Poirel L, Castanheira M, Carrer A et al. OXA-163, an OXA-48-related class D β-lactamase with extended activity toward expanded-spectrum cephalosporins. Antimicrob Agents Chemother 2011; 55: 2546–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Poirel L, Heritier C, Tolun V et al. Emergence of oxacillinase-mediated resistance to imipenem in Klebsiella pneumoniae. Antimicrob Agents Chemother 2004; 48: 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Docquier JD, Calderone V, De Luca F et al. Crystal structure of the OXA-48 β-lactamase reveals mechanistic diversity among class D carbapenemases. Chem Biol 2009; 16: 540–7. [DOI] [PubMed] [Google Scholar]
- 50.Santillana E, Beceiro A, Bou G et al. Crystal structure of the carbapenemase OXA-24 reveals insights into the mechanism of carbapenem hydrolysis. Proc Natl Acad Sci USA 2007; 104: 5354–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Luca F, Benvenuti M, Carboni F et al. Evolution to carbapenem-hydrolyzing activity in noncarbapenemase class D β-lactamase OXA-10 by rational protein design. Proc Natl Acad Sci USA 2011; 108: 18424–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ehmann DE, Jahic H, Ross PL et al. Kinetics of avibactam inhibition against class A, C, and D β-lactamases. J Biol Chem 2013; 288: 27960–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.DeLano WL. The PyMOL Molecular Graphics System. Palo Alto, CA: DeLano Scientific, 2008. http://www.pymol.org/. [Google Scholar]