

Abstract
Background
Voriconazole is a first-line agent for the prevention and treatment of a number of invasive fungal diseases. Relatively little is known about the relationship between drug exposure and the prevention of invasive fungal infections.
Patients and methods
A pharmacokinetic–pharmacodynamic substudy was performed as part of the BMT CTN 0101 trial, which was a randomized clinical trial comparing voriconazole with fluconazole for the prevention of invasive fungal infections in HSCT recipients. A previously described population pharmacokinetic model was used to calculate the maximum a posteriori Bayesian estimates for 187 patients. Drug exposure in each patient was quantified in terms of the average AUC and average trough concentrations. The relationship between drug exposure and the probability of breakthrough infection was investigated using logistic regression. AUC and trough concentrations in patients with and without breakthrough infection were compared.
Results
Pharmacokinetic data from each patient were readily described using the maximum a posteriori Bayesian estimates. There were only five patients that had a breakthrough infection while receiving voriconazole in the first 100 days post-HSCT. For these patients, there was no statistically significant relationship between the average AUC or average trough concentration and the probability of breakthrough infection [OR (95% CI) 1.026 (0.956–1.102) and 1.108 (0.475–2.581), respectively]. P value for these estimates was 0.474 and 0.813, respectively.
Conclusions
Given the very small number of proven/probable infections, it was difficult to identify any differences in drug exposure in HSCT recipients with and without breakthrough fungal infections.
Introduction
Voriconazole is a second-generation triazole antifungal agent with activity against a range of medically important opportunistic fungal pathogens.1 Voriconazole is a first-line agent for the treatment of invasive aspergillosis,2 which is largely based on the results of a large randomized clinical trial comparing voriconazole with amphotericin B deoxycholate.3 The efficacy of voriconazole has also been established for other invasive fungal diseases (IFDs) such as those caused by Candida spp.,4 Fusarium spp.5 and Scedosporium spp.6 Voriconazole is also the agent of choice for certain specific clinical conditions such as osteoarticular aspergillosis7 and cerebral aspergillosis.8 More recently, the safety and efficacy of voriconazole for the prevention of IFDs in patients receiving HSCT has been demonstrated in several clinical trials.9,10
Voriconazole is a challenging agent for routine use in clinical settings—it is complicated by highly variable pharmacokinetics (PK) (e.g. 100-fold variability in systemic drug exposure), classical non-linear (Michaelis–Menten) elimination, a multitude of drug–drug interactions and an increasingly well understood toxicity profile.1 Many experimental and clinical PK/pharmacodynamic (PD) studies consistently suggest there are important drug exposure–response and toxicity relationships that underpin clinical efficacy and safety.11–14 An in-depth understanding of these relationships is required for the safe and effective use of voriconazole.11 Therapeutic drug monitoring (TDM) is increasingly considered a standard-of-care.15,16 While arguments for the use of TDM are robust for established invasive disease, the relevance of routine use of voriconazole TDM for prophylaxis remains much less clear.
Here, we present the results of a PK/PD analysis performed as a substudy of the larger Phase III clinical trial in which the efficacy of fluconazole and voriconazole for patients with HSCT was prospectively compared.9 In that study, patients received standard regimens of both fluconazole and voriconazole. Serum concentrations of voriconazole (and fluconazole) were determined using a sparse sampling strategy. The maximum a posteriori (MAP) Bayesian estimates of PK of voriconazole for patients in the prophylaxis study were estimated using a previously described population PK model fitted to data that were obtained from both healthy volunteers and from patients enrolled in earlier clinical trials.3 Such an approach provided tractable estimates of drug exposure in patients receiving voriconazole as prophylaxis for invasive fungal infections, and was the first critical step in investigating any potential link between drug exposure and prevention of invasive fungal infections.
Patients and methods
Patients
The demographic and clinical details of the patients used in this study who were originally enrolled in a study by Wingard et al.9 are summarized in Table 1. In the original randomized control trial that compared voriconazole with fluconazole, a total of 305 patients received voriconazole. Of these 305 patients, PK data were only available for 198 patients. There were 11 patients who were <12 years of age that were excluded from the PK analysis and the population PK model because the PK in younger children is known to be markedly different from adolescents and adults.17,18 Therefore, the final number of patients available for PK/PD analysis from the Wingard et al.9 study was 187.
Table 1.
Demographics of patients in the prophylaxis study
| Age (years), median (range) | 44.23 (12.80–65.75) |
| Female, n (%) | 84 (44.9) |
| Disease, n (%) | |
| AML | 78 (41.7) |
| ALL | 34 (18.2) |
| CML | 30 (16.0) |
| myelodysplasia | 30 (16.0) |
| biphenotypic leukaemia | 2 (0.01) |
| lymphoma | 13 (8.49) |
| Proven, probable or presumptive fungal infection, n (%) | 10 (6.95) |
Study design
The Wingard et al.9 study was a Phase III randomized double-blind, multicentre prospective trial that compared the efficacy of fluconazole with voriconazole for the prevention of IFDs in allogeneic transplant recipients. Patients were randomized 1:1 to receive fluconazole or voriconazole. Voriconazole was administered intravenously over 2 h or orally 200 mg every 12 h. A loading regimen was not used. The drug was administered orally whenever possible. In circumstances when that was not possible (e.g. severe mucositis) voriconazole was administered intravenously. The voriconazole dosage was not altered according to renal function. Voriconazole was administered at least 1 h before or 1 h after food. Voriconazole was continued from days 0 until 100 post-transplantation after which patients were observed until day +360.
PK sampling
There were two sampling days, day +14 and day +28, with day 0 being the day of transplantation. On each sampling day, there were two sampling windows that encompassed early and late periods in the dosing interval. Sampling times were distributed throughout the dosing interval to inform the time course of drug concentrations via a population modelling approach. This choice supported estimation of PK parameters via population modelling. In this study, 350 samples were available for analysis. Of these, 213 (60.9%) were sampled between time 0 and 6 h post-dose, 97 (27.7%) were sampled between 6 and 12 h post-dose, 36 (10.3%) were sampled between 12 and 18 h post-dose and 4 were taken >18 h post-dose.
Measurement of voriconazole
Voriconazole concentrations in plasma were measured using HPLC with a Shimadzu Prominence (Shimadzu, Milton Keynes, Bucks, UK). Thirty μL of extracted sample was injected on to a Kinetex 2.6 μm C18 100A 75 × 4.6 mm (Phenomonex, Macclesfield, Cheshire, UK). A standard curve encompassing 0.0625–8 mg/L was constructed in plasma using stock solutions of voriconazole 1000 mg/L dissolved in methanol (Fisher Scientific, Loughborough, Leics, UK). The internal standard was 1 μg/L diazepam (Sigma-Aldrich, Dorset, UK). A gradient method was used: initial concentrations were 80% 0.1% trifluoroacetic acid in water, and 20% 0.1% trifluoroacetic acid in acetonitrile (Fisher Scientific) changing to 35% and 65%, respectively over 10 min. The overall run time was 13 min and flow rate was 1 mL/min. Voriconazole and the internal standard were detected using ultraviolet light at 254 nm; they eluted after 5.9 and 4.7 min, respectively. The coefficient of variation (CV%) was <9% over the concentration range 0.0625–8 mg/L. The limit of detection was 0.0625 mg/L. The intra- and inter-day variation was <9%.
Population PK modelling
A previously published population PK model17 was used as the Bayesian prior in the process of estimating drug exposure for the patients enrolled in the Wingard et al. study.9 This approach was required because the PK data were sparse and not suitable to fit a larger model without pre-existing information. The MAP Bayesian estimates for each patient in the prophylaxis study were estimated using the non-parametric adaptive grid algorithm, which is now embedded in the Pmetrics software package for R.19 The structural model was the same as previously used to model the PK of voriconazole.17 The three ordinary differential equations were:
Where: X(1), X(2) and X(3) represent the amount of voriconazole (in milligrams) in the gut, central compartment and peripheral compartment, respectively; Ka, first-order rate constant connecting the gut with the central compartment; Vmax, maximum rate of elimination of voriconazole (mg/h); Km (mg/L), concentration of voriconazole in the central compartment at which clearance is half-maximal; Kcp (h−1) and Kpc (h−1), first-order inter-compartmental rate constants; and B(1), instantaneous oral administration of voriconazole. Two further parameters were incorporated into this model, i.e. (i) lag function, and (ii) estimate of bioavailability. The majority information related to these parameters was contained in the Bayesian prior, which was made possible by the concomitant oral and intravenous input in the earlier studies. The estimate for time lag was primarily enabled by data from the healthy volunteer studies where there was intensive sampling immediately after oral drug administration. The estimate for oral bioavailability was enabled by both the healthy volunteer studies and Phase III clinical trial3 where there was switching between the intravenous and oral dosing.
Equations 1, 2 and 3 describe the rate of change of the amount of voriconazole in the gut, central compartment and peripheral compartment, respectively. The data were weighted by the inverse of the estimated assay variance. The fit of the model to the data was assessed using the log-likelihood value, mean weighted error (a measure of bias) and bias-adjusted mean weighted squared error (a measure of precision). A visual inspection and the coefficient of determination of a linear regression of the observed versus predicted values were also performed. Bayesian estimates for the parameters for the individual volunteers and patients were obtained.
Any switch from oral to intravenous therapy that had occurred because of significant mucositis was not collected on the clinical report forms. A retrospective search of all patients' medical records was infeasible. To circumvent this problem, all drug inputs were modelled as if they had been administered into the gut, regardless of whether voriconazole had been administered orally or intravenously for all or (more likely) part of the treatment period. Thus, there may be some impact on estimates for Ka and oral bioavailability, which may potentially deviate from the ‘true’ values for these parameters.
Monitoring for invasive fungal infections
Patients were screened twice weekly for galactomannan to day +60, and then weekly until day +100. A positive galactomannan test or other clinical features of IFD triggered a mandatory work-up for the presence of an IFD (e.g. CT scans, bronchoscopy).
Study definitions of IFD
The EORTC/MSG criteria were used to classify patients as having proven, probable, possible or having no infection.20 An additional classification of ‘presumptive’ fungal infection was used and defined in the original study protocol. Presumptive fungal infection was defined as at least one host criterion for IFD plus one clinical criterion, plus a bronchoscopic evaluation that did not reveal a bacterial or other infectious aetiology, but did not have a specific mycological finding. The clinical criteria for lower respiratory tract infection included the presence of radiological features that tend to be more specific for an IFD (i.e. halo sign, wedge-shaped infiltrate or an air crescent sign), or if a non-specific pulmonary infiltrate was present then that needed to be accompanied by clinical features such as pleural pain, pleural rub or haemoptysis.
PK and PD modelling
In the original study, the clinical trial Data Review Committee classified the clinical outcome of each patient at day +180 as either having or not having an IFD. For the purposes of these analyses, only patients experiencing an IFD while receiving voriconazole were considered (i.e. patients with an IFD occurring after day +100 were excluded, which occurred in 5 of the 10 patients with breakthrough infection).
The relationship between both the average model-predicted trough concentration (a clinically tractable measure of drug exposure) and the average model-predicted AUC0–12 (a more precise, but less readily clinically accessible measure of drug exposure) was determined from estimates for the Bayesian posterior PK values for each patient. Thus, the estimate of drug exposure for each patient was made using the population PK model rather than from the raw data. The latter was infeasible because of the relative paucity of samples (n = 4 per patient) and the absence of a sampling strategy to obtain Cmin or an estimate of AUC directly from the patient's PK data (i.e. the intention was always to use the population PK model to determine drug exposure in individual patients rather than use the raw data, and the study was designed accordingly). Using the ‘makeAUC’ function in Pmetrics, which estimates the AUC using the trapezoidal rule, the AUC0–12 for each subject was precisely determined from concentrations predicted every 12 min from the mean individual, Bayesian posterior parameter values.
The relationship between the AUC0–12 and trough concentration was explored. This was done because the AUC is the likely PD index that best links drug exposure with the observed effect, and is therefore the most robust way to resolve drug exposure–effect relationships. The validity of the AUC as the relevant PD index has been demonstrated for Candida albicans, but not Aspergillus spp.21 However, in routine clinical settings the AUC may be difficult to estimate, and the trough concentration is frequently used as a surrogate. To obtain an estimate of overall drug exposure, we determined the model-predicted average AUC0–12 and average Cmin for each patient. This approach circumvented the problem of non-linear PK where some patients with saturated clearance pathways may progressively accumulate drug leading to uncertainty as to which of AUC or Cmin is best linked with clinical outcome.
Two approaches were used in an attempt to link drug exposure with clinical outcome. In the first, logistic regression was used to examine any potential relationship between the average AUC0–12 with the probability of an IFD being diagnosed. This analysis was performed in SYSTAT version 11. To further investigate any possible relationship between drug exposure and clinical outcome, the average AUC0–12 and Cmin were compared in patients with and without IFD (i.e. failure and success of prophylaxis, respectively). Potential differences in measures of drug exposure were explored using the Mann–Whitney U-test.
Results
Patient demographics
A total of 187 patients >12 years old had undergone allogeneic transplantation, received voriconazole and had PK samples available for analysis. The patient demographics of the 187 patients are summarized in Table 1. There were 10 of 187 (5.3%) of proven, probable or presumptive IFDs up to day +180 of the study. Possible IFDs were not included as part of these analyses. Of the 10 patients, 5 had a breakthrough infection after voriconazole was stopped at day +100 and were excluded from this analysis. The time to breakthrough infection for the 10 patients is shown in Figure 1. The microbiological diagnoses of the five patients with infection while receiving voriconazole therapy up until day +100 post-HSCT were: (i) disseminated Rhizopus infection (n = 1); (ii) Candida glabrata bloodstream infection (no in vitro susceptibility testing performed); (iii) a patient with positive serum galactomannan and pulmonary infiltrates; (iv) C. glabrata isolated from bronchoalveolar lavage fluid (n = 1); and (v) growth of Beauveria spp. from bronchoalveolar lavage fluid. Cases 4 and 5 were classified as presumptive IFD because no clinically relevant pathogen was identified, but the host and clinical criteria were met for IFD and the patient underwent bronchoscopic evaluation to exclude bacteria and other clinically relevant pathogens.
Figure 1.
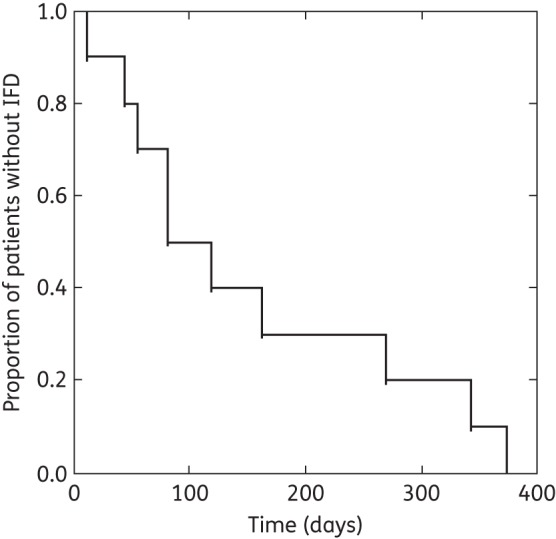
Kaplan–Meier curve showing the time to invasive fungal infection in the 10 patients with breakthrough infection. Voriconazole was used up to day +100 post-transplantation.
Population PK modelling
The fit of the population PK model to the data from the 187 patients receiving voriconazole was acceptable (Figure 2). The mean and median parameter values from each patient were both assessed in terms of their ability to account for the observed data. Both gave comparable results, but the means performed slightly better (same coefficient of determination, but slightly better slope for the linear regression of the observed–predicted values). The linear regression of the observed–predicted values was given by the following: observed = −0.027 + 1.10 × predicted; r2 = 0.77. Thus, the individual parameter estimates were used to calculate the drug exposure experienced by each patient. The distribution of the mean parameter estimates and measures of central tendency for the population of 187 patients are shown in Figure 3.
Figure 2.
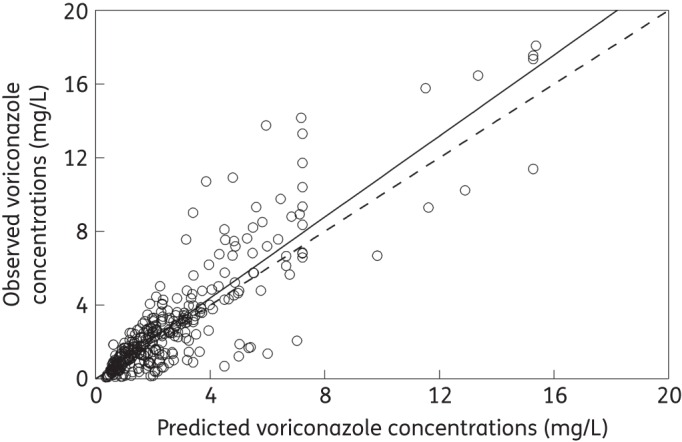
Individually observed versus predicted values for 187 patients receiving 200 mg of voriconazole every 12 h after the fit of the population model to each patient's serum concentration data and using a previous population PK model as the Bayesian prior. Observed = −0.027 + 1.10 × predicted; r2 = 0.77. The broken line is the line of identity.
Figure 3.

Density of the parameter estimates based on data from 187 patients after the MAP Bayesian analysis. Ka (h−1) is the first order rate constant that connects the gut to the central compartment, lag (h) is the delay in drug absorption from the gut, Km (mg/L) is the Michaelis constant and is the voriconazole plasma drug concentration at which enzyme activity is half-maximal, Vmax (mg/h) is the maximal rate of enzyme activity and volume (L) is the volume of the central compartment.
Relationship between drug exposure and efficacy of prophylaxis
The absence of any relationship between drug exposure and prevention of IFDs was confirmed using two logistic regression models that are summarized in Table 2. P values for average Cmin and average AUC0–12 were 0.813 and 0.474, respectively (Table 2). The ORs for both models were close to 1 for both measures of drug exposure, and the 95% CI crossed 1 in both cases.
Table 2.
Logistic regression models comparing drug exposure quantified as the average AUC0–12 and Cmin with the probability of a breakthrough fungal infection
| Parameter | Estimate | P |
|---|---|---|
| Model 1: average AUC0–12 versus outcome | ||
| constant | 3.021 | <0.001 |
| AUC0–12 | 0.026 | 0.474 |
| OR (95% CI) | 1.026 (0.956–1.102) | |
| Model 2: average Cmin versus outcome | ||
| constant | 3.413 | <0.001 |
| Cmin | 0.102 | 0.813 |
| OR (95% CI) | 1.108 (0.475–2.581) | |
There was not a statistically significant relationship between drug exposure quantified either in terms of the model-predicted average AUC0–12 or average Cmin and the ultimate clinical outcome as defined by the presence of an IFD. The mean and median ± SD AUC0–12 in patients with breakthrough IFD was 15.91 and 15.98 ± 11.37 mg·h/L, respectively. These values were close to the estimates for patients without IFD (21.62 and 15.25 ± 17.96 mg·h/L, respectively). Similarly, the mean and median ± SD trough concentrations in patients with breakthrough IFD were 1.39 and 1.04 ± 1.02 mg/L, respectively, while very similar values for patients without IFD were 1.51 and 1.08 ± 1.26 mg/L, respectively. As expected based on prior studies, between patient variability in PK and resultant drug exposure was high. There were no statistically significant differences between the model-predicted average AUC0–12 and average Cmin when assessed using the Mann–Whitney U-test with a P value of 0.475 and 0.945, respectively (Figure 4).
Figure 4.

Box and whisker plots of drug exposure [trough concentration and AUC0–12 in (a) and (b), respectively] between patients that failed therapy (‘failure’) on the left of each figure and patients for whom voriconazole therapy prevented invasive fungal infection up to day +100 (‘success’) on the right of each figure. There were no statistically significant differences in drug exposure in patients with and without breakthrough infection.
Discussion
Voriconazole is extensively used for the treatment of established IFDs in a range of clinical contexts.3,4,7 The clinical efficacy of voriconazole has been established for the prevention of IFDs in profoundly immunocompromised patients with prolonged neutropenia and following HSCT.9,10 TDM is increasingly advocated as a standard-of-care for patients with established infection.15 The evidence for routine use of the TDM for voriconazole when used for the prevention of IFDs in at-risk patients is less clear.
In this study, there was no observed relationship between drug exposure and the probability of breakthrough invasive fungal infections. Perhaps this should not be surprising given the relatively small numbers of patients with breakthrough fungal infection. A power calculation suggests that a much larger number of patients are required to achieve any confidence about potential differences in drug exposure in those with and without breakthrough infection. For example, even if a large true difference in drug exposure (e.g. average trough concentration of 1.5 versus 0.75 mg/L for therapeutic success versus failure, respectively) is present then ∼45 patients with breakthrough infection on voriconazole are required to establish statistically significant differences between these groups. Since only a small proportion of patients receiving voriconazole have a breakthrough infection this would require ∼1700 HSCT patients and 3400 HSCT patients for a two-armed comparative study. Such numbers are clearly not tenable in a clinical trial that requires several blood samples to be obtained per patient.9
There are surprisingly few patients that have had exposure–response relationships studied while receiving voriconazole for prophylaxis. The vast majority of data that supports TDM of voriconazole comes from patients with established disease. A retrospective study by Trifilio et al.22 describes 10 of 71 patients receiving voriconazole with breakthrough infection caused by Candida spp. (n = 6) and Mucorales (n = 4). Patients with Candida infection had lower voriconazole concentrations than other patients. There were no cases caused by Aspergillus spp. in that study. The absence of any observed relationship between voriconazole drug exposure and breakthrough IFD in the current study is reminiscent of the large randomized prophylaxis studies involving posaconazole23,24 in which there is no relationship between serum drug exposure (quantified in terms of average concentrations and Cmin) and the probability of breakthrough infections.25 In the analysis of Jang et al.25 a composite endpoint was used for the logistic regression modelling that comprised proven/probable IFDs, the need to administer >5 days of empirical treatment with a systemic antifungal agent other than posaconazole, all-cause mortality, discontinuation of study drug and loss to follow-up. Despite the use of a composite endpoint and the absence of any relationship when proven/probable IFDs were used as an outcome measure, TDM is often advocated as an adjunct to the use of posaconazole for prophylaxis.15
The negative findings in this study raise several interesting points. First, it is well established that voriconazole is an effective agent to treat established IFDs, such as those caused by Candida spp. and Aspergillus spp.1 Conceivably, there are a number of idiosyncrasies that could have an impact of the efficacy of an agent when used for prophylaxis versus when it is used to treat established disease (e.g. time to generate protective concentrations at the initial site of infection, partitioning into relevant tissue and cellular subcompartments that are relevant in early phases of fungal infection). The clinical trial of Wingard et al.9 suggests that the use of voriconazole is at least non-inferior to a standard-of-care (fluconazole), although its absolute benefit in terms of preventing IFDs cannot be quantified because voriconazole has not been (and never will be) compared with placebo. If an assumption is made that voriconazole prevents at least a proportion of IFDs there must be exposure–response relationships that are operational at least at some level for the patient groups of interest. Nevertheless, these relationships are not immediately apparent in this study despite our sample size of 187 patients included in the population PK/PD analysis. The potential reason(s) for this include(s) one or more of the following: (i) dosage of 200 mg of voriconazole every 12 h (intravenous or oral) results in drug exposure that induces near-maximal antifungal activity and is completely effective for prophylaxis (there are too few concentration-dependent therapeutic failures to enable any cut-off value to be observed); (ii) the number of IFDs diagnosed with an adequate degree of certainty is too small to detect a relationship between drug exposure and clinical outcome (i.e. the study is under-powered) (related to this point is the fact that PK were only available in a subset of voriconazole patients, which compromised the ability of the study to detect any differences and may have inadvertently introduced an element of bias); (iii) undetected and anticipated protocol deviations may have obscured relevant exposure–response relationships in this often critically ill patient population (for example, patients may have stopped taking voriconazole immediately prior to the time their infection broke through late in the study meaning there was a mismatch between drug exposure measured early in the post-transplantation period and the measured clinical outcome); and (iv) PK data were too imprecise to enable a robust estimate of the ‘true’ AUC and the ‘true’ trough concentration that was present in individual patients. In this situation, the sparse patient data are dominated by the parameter estimates from the Bayesian prior. Nevertheless, the use of non-parametric population PK modelling with Bayesian priors is among the most powerful data analytical methods to estimate individual drug exposures in patients with sparse PK sampling. Each of these explanations may have contributed to the absence of any relationship between voriconazole drug exposure and clinical outcome.
The absence of any demonstrable drug exposure signal in this study does not help address the question as to how (if at all) TDM should be used to manage patients after HSCT who are receiving voriconazole for prophylaxis. With all the caveats that have been mentioned, the current study does not provide evidence that TDM should be routinely used for all patients. There may, however, still be subpopulations that still benefit from TDM (e.g. patients with significant mucositis), although this study does not enable those subgroups to be better defined. Furthermore, there is no indication whether the targets for TDM (e.g. trough concentration 2 mg/L) that have been defined in the context of established clinical disease are also appropriate for prophylaxis. Until further information is available, the use of TDM for voriconazole when used for prophylaxis remains a matter of clinical judgement.
Funding
This study was supported by internal funding from the University of Liverpool. W. W. H. is supported by a Clinician Scientist Award from the National Institutes of Health Research (NIHR) in the UK. T. J. W. is a Scholar of the Henry Schueler Foundation, an Investigator of Pediatric Infectious Diseases of the Sharp Family Foundation and a Scholar of the Save our Sick Kids Foundation. M. N. N. is supported by National Institutes of Health R01 HD070886 and R01 GM068968.
Transparency declarations
W. W. H. has acted as a consultant to and/or received research grant support from Pfizer Inc., Astellas Pharma, Gilead Sciences, F2G and Pulmocide. T. J. W. has received research grants for experimental and clinical antimicrobial pharmacotherapeutics from Novartis, Merck, Pfizer and Astellas, and has served as a consultant to Astellas, ContraFect, Drais, iCo, Novartis, Pfizer, Methylgene, SigmaTau and Trius. All other authors: none to declare.
References
- 1.Denning DW, Hope WW. Therapy for fungal diseases: opportunities and priorities. Trends Microbiol 2010; 18: 195–204. [DOI] [PubMed] [Google Scholar]
- 2.Stevens DA, Kan VL, Judson MA et al. . Practice guidelines for diseases caused by Aspergillus. Infectious Diseases Society of America. Clin Infect Dis 2000; 30: 696–709. [DOI] [PubMed] [Google Scholar]
- 3.Herbrecht R, Denning DW, Patterson TF et al. . Voriconazole versus amphotericin B for primary therapy of invasive aspergillosis. N Engl J Med 2002; 347: 408–15. [DOI] [PubMed] [Google Scholar]
- 4.Kullberg BJ, Sobel JD, Ruhnke M et al. . Voriconazole versus a regimen of amphotericin B followed by fluconazole for candidaemia in non-neutropenic patients: a randomised non-inferiority trial. Lancet 2005; 366: 1435–42. [DOI] [PubMed] [Google Scholar]
- 5.Lortholary O, Obenga G, Biswas P et al. . International retrospective analysis of 73 cases of invasive fusariosis treated with voriconazole. Antimicrob Agents Chemother 2010; 54: 4446–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Troke P, Aguirrebengoa K, Arteaga C et al. . Treatment of scedosporiosis with voriconazole: clinical experience with 107 patients. Antimicrob Agents Chemother 2008; 52: 1743–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mouas H, Lutsar I, Dupont B et al. . Voriconazole for invasive bone aspergillosis: a worldwide experience of 20 cases. Clin Infect Dis 2005; 40: 1141–7. [DOI] [PubMed] [Google Scholar]
- 8.Schwartz S, Ruhnke M, Ribaud P et al. . Improved outcome in central nervous system aspergillosis, using voriconazole treatment. Blood 2005; 106: 2641–5. [DOI] [PubMed] [Google Scholar]
- 9.Wingard JR, Carter SL, Walsh TJ et al. . Randomized, double-blind trial of fluconazole versus voriconazole for prevention of invasive fungal infection after allogeneic hematopoietic cell transplantation. Blood 2010; 116: 5111–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marks DI, Pagliuca A, Kibbler CC et al. . Voriconazole versus itraconazole for antifungal prophylaxis following allogeneic haematopoietic stem-cell transplantation. Br J Haematol 2011; 155: 318–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Troke PF, Hockey HP, Hope WW. Observational study of the clinical efficacy of voriconazole and its relationship to plasma concentrations in patients. Antimicrob Agents Chemother 2011; 55: 4782–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeans AR, Howard SJ, Al-Nakeeb Z et al. . Pharmacodynamics of voriconazole in a dynamic in vitro model of invasive pulmonary aspergillosis: implications for in vitro susceptibility breakpoints. J Infect Dis 2012; 206: 442–52. [DOI] [PubMed] [Google Scholar]
- 13.Pascual A, Calandra T, Bolay S et al. . Voriconazole therapeutic drug monitoring in patients with invasive mycoses improves efficacy and safety outcomes. Clin Infect Dis 2008; 46: 201–11. [DOI] [PubMed] [Google Scholar]
- 14.Smith J, Safdar N, Knasinski V et al. . Voriconazole therapeutic drug monitoring. Antimicrob Agents Chemother 2006; 50: 1570–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ashbee HR, Barnes RA, Johnson EM et al. . Therapeutic drug monitoring (TDM) of antifungal agents: guidelines from the British Society for Medical Mycology. J Antimicrob Chemother 2014; 69: 1162–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andes D, Pascual A, Marchetti O. Antifungal therapeutic drug monitoring: established and emerging indications. Antimicrob Agents Chemother 2009; 53: 24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hope WW. Population pharmacokinetics of voriconazole in adults. Antimicrob Agents Chemother 2012; 56: 526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neely M, Margol A, Fu X et al. . Achieving target voriconazole concentrations more accurately in children and adolescents. Antimicrob Agents Chemother 2015; 59: 3090–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neely MN, van Guilder MG, Yamada WM et al. . Accurate detection of outliers and subpopulations with Pmetrics, a nonparametric and parametric pharmacometric modeling and simulation package for R. Ther Drug Monit 2012; 34: 467–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Pauw B, Walsh TJ, Donnelly JP et al. . Revised definitions of invasive fungal disease from the European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group and the National Institute of Allergy and Infectious Diseases Mycoses Study Group (EORTC/MSG). Clin Infect Dis 2008; 46: 1813–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andes D, Marchillo K, Stamstad T et al. . In vivo pharmacokinetics and pharmacodynamics of a new triazole, voriconazole, in a murine candidiasis model. Antimicrob Agents Chemother 2003; 47: 3165–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trifilio S, Singhal S, Williams S et al. . Breakthrough fungal infections after allogeneic hematopoietic stem cell transplantation in patients on prophylactic voriconazole. Bone Marrow Transplant 2007; 40: 451–6. [DOI] [PubMed] [Google Scholar]
- 23.Ullmann AJ, Lipton JH, Vesole DH et al. . Posaconazole or fluconazole for prophylaxis in severe graft-versus-host disease. N Engl J Med 2007; 356: 335–47. [DOI] [PubMed] [Google Scholar]
- 24.Cornely OA, Maertens J, Winston DJ et al. . Posaconazole vs. fluconazole or itraconazole prophylaxis in patients with neutropenia. N Engl J Med 2007; 356: 348–59. [DOI] [PubMed] [Google Scholar]
- 25.Jang SH, Colangelo PM, Gobburu JVS. Exposure-response of posaconazole used for prophylaxis against invasive fungal infections: evaluating the need to adjust doses based on drug concentrations in plasma. Clin Pharmacol Ther 2010; 88: 115–9. [DOI] [PubMed] [Google Scholar]