

Abstract
In search for the function of local chromatin environment on pre-mRNA processing we established a new tool, which allows for the modification of chromatin using a targeted approach. Using Transcription Activator-Like Effector domains fused to histone modifying enzymes (TALE-HME), we show locally restricted alteration of histone methylation modulates the splicing of target exons. We provide evidence that a local increase in H3K9 di- and trimethylation promotes inclusion of the target alternative exon, while demethylation by JMJD2D leads to exon skipping. We further demonstrate that H3K9me3 is localized on internal exons genome-wide suggesting a general role in splicing. Consistently, targeting of the H3K9 demethylase to a weak constitutive exon reduced co-transcriptional splicing. Together our data show H3K9 methylation within the gene body is a factor influencing recognition of both constitutive and alternative exons.
The removal of introns and ligation of exons in the process of pre-mRNA splicing takes place predominantly co-transcriptionally1,2,3. In budding yeast, an RNA polymerase II (Pol II) pause site in the terminal exon ensures that splicing is completed before termination of transcription, suggesting that co-transcriptional splicing is of functional importance2,4. In fact, mounting evidence suggests transcription, chromatin and pre-mRNA splicing are functionally coupled. The first evidence implying chromatin affects alternative splicing came from genome-wide ChIP-Seq studies that correlated the presence of certain histone modifications with exon inclusion rates5,6. However, determining whether these histone marks are causative for splicing changes or simply a consequence of splicing cannot be addressed by this correlative approach. While it is widely assumed that chromatin modifications modulate alternative splicing, several studies have provided evidence that H3K4 and H3K36 methylation is splicing dependent7,8,9. These data suggest a more complex regulatory network where splicing can feed back on chromatin to label histones for the following rounds of transcription.
In order to experimentally determine the role of chromatin in alternative splicing regulation, most studies have used a global approach to perturb histone modifications genome-wide, such as small molecule inhibitors or knockdown/overexpression of histone modifying enzymes (HMEs)10,11,12,13,14,15. As this method globally affects the transcriptional program of the cell, secondary effects cannot be fully excluded. The central question remains, what is the minimal amount of chromatin change required to influence splicing and how local is this effect? In this study, we set out to determine the role of chromatin by direct, local manipulation of the chromatin environment at a selected target exon. We use Transcription-Activator-Like-Effector (TALE) domains to tether HMEs to a target exon locus, analogous to TALENs for genome engineering or TALE-TFs for transcriptional regulation16,17,18. This strategy has been successfully utilized to modify chromatin modifications at enhancers19. As proof of principle, we chose to investigate the effect of H3K9 and H3K36 methylation11,12,15,20,21, histone marks previously shown to modulate alternative splicing, on alternative splicing of the endogenous human fibronectin EDB exon, a widely studied example of a cassette exon22,23,24. We further studied the global distribution of H3K9me3 within genes and showed this histone mark plays an unanticipated role in splicing of constitutive exons.
Results
TALEs for targeted modulation of chromatin
To test whether locally restricted changes in the chromatin context can directly affect splicing of an alternative exon, we altered H3K36 and H3K9 methylation at the EDB exon in human fibronectin (FN1). Methylation of H3K9 and H3K36 were previously proposed to regulate alternative splicing11,12,15,20,21, and splicing of the EDB exon was described to be sensitive to the chromatin environment13,23,24. To specifically and locally target chromatin around the EDB exon we designed a TALE domain recognizing 23 nt of the plus strand close to the 3′ splice site (SS) of the EDB exon using the TAL Effector Targeter25 (https://tale-nt.cac.cornell.edu/node/add/single-tale). No off targets in the human genome were predicted for this binding sequence. To modulate H3K9 methylation, the TALE domain was fused to the H3K9 tri-methyltransferase Suv39H1, the catalytic domain of the di-methyltransferase G9a (EHMT2) or the H3K9me2/3 demethylase JMJD2D (KDM4D)26,27,28,29. For H3K36me3 we selected the methyltransferase ASH1L and the catalytically active domain of SETD230,31 (Fig. 1A). The HA-tag was included as a linker between the TALE domain and HME to allow detection of fusion proteins by Western blot (Supplementary Fig. S1). As a control, the TALE domain was fused to GFP and all results were normalized or compared to the TALE-GFP construct in order to exclude unspecific effects due to the binding of the TALE construct to DNA.
Figure 1. Local manipulation of histone modifications by TALE-HME affects alternative splicing of the target exon.
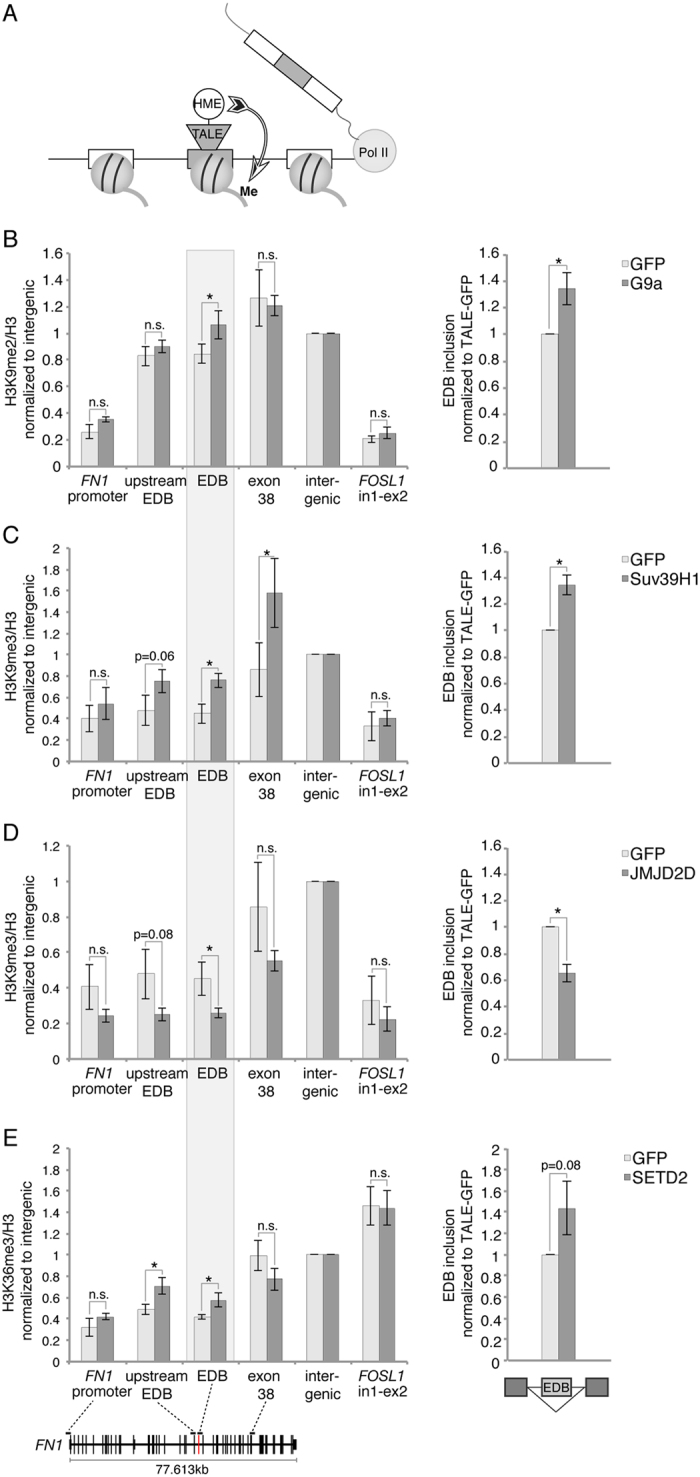
(A) Schematic representation of the approach. A TALE domain was assembled to bind the target exon and fused to HMEs. Upon binding of TALE-HME to the target exon, the local chromatin environment is modified. (B–E) Upon transient transfection with TALE-HME or TALE-GFP as a control, the effect on chromatin was monitored by ChIP (left panel) and EDB inclusion levels were assessed by RT-qPCR (right panel). ChIP signals are calculated as immunoprecipitated DNA over input signal and normalized to total H3 and an intergenic region on chromosome 10. Amplicon positions for FN1 are indicated in the gene diagram at the bottom. EDB inclusion rates are calculated as the ratio of the EDB exon to the upstream constitutive exon 24 and normalized to TALE-GFP. Mean ± SEM are shown, n = 3–4. Statistical significance of all results was analyzed by t-test and significant changes (p < 0.05) were indicated by * .
Local changes in chromatin alter EDB inclusion
TALE-HME or TALE-GFP constructs were transiently transfected into HeLa cells and the effect of TALE-HME expression on chromatin was monitored by chromatin immunoprecipitation (ChIP). Tethering of the H3K9me2 methyltransferase construct TALE-G9a to the EDB exon resulted in a significant ~20% increase in H3K9me2 at the target site, while the immediate upstream exon (1.5 kb distant from EDB), a far downstream exon (17.8 kb distant from EDB) and the promoter of FN1 were unaffected (Fig. 1B, left panel). Likewise, methylation of a non-targeted gene, FOSL1, remained unchanged. The effect of chromatin alteration on alternative splicing was assessed using RT-qPCR. Expression of TALE-G9a resulted in a ~30% increase in EDB inclusion compared to the tethering of control TALE-GFP, thus proving the principle that a local change in the chromatin environment impacts alternative splicing (Fig. 1B, right panel).
Similarly to H3K9me2, targeting the H3K9me3 methyltransferase Suv39H1 led to higher levels of H3K9me3 at the target exon (Fig. 1C). However, here methylation was also elevated at both the downstream exon and the immediate upstream exon, suggesting the possibility of H3K9me3 spreading, as previously described for heterochromatin32. Nevertheless, the promoter region of FN1 and the control gene FOSL1 were not affected indicating gene specific targeting of Suv39H1. Analogous to H3K9 dimethylation by TALE-G9a, increased H3K9 trimethylation promoted EDB inclusion. Since H3K9me2 serves as a substrate for Suv39H1, it is possible the increased H3K9 dimethylation upon TALE-G9a is converted to H3K9me3 by endogenous enzymes. However, TALE-G9a tethering did not exhibit elevated H3K9me3 levels along FN1, indicating that H3K9me2 and H3K9me3 can independently promote exon inclusion (Supplementary Fig. S1). To further test the role of H3K9me2/3 on alternative splicing, the H3K9 demethylase JMJD2D was targeted to the EDB exon subsequently leading to reduced H3K9me3 at the target site, which in turn enhanced exon skipping (Fig. 1D). These results demonstrate the local level of H3K9 methylation directly impacts splicing of the EDB exon and that a higher methylation promotes exon inclusion.
Next, we tested the effect of two H3K36me3 methyltransferases - SETD2 and ASH1L. TALE-SETD2 locally increased H3K36me3 levels at the target exon and the immediate upstream exon but the changes in alternative splicing were below statistical significance (Fig. 1E). TALE-ASH1L had no effect on the chromatin level nor on alternative splicing (Supplementary Fig. S1). However, the latest construct was poorly expressed (Supplementary Fig. S1).
Internal exons are tri-methylated at H3K9
Given the positive effect of local H3K9me3 on alternative exon inclusion (Fig. 121,33), we speculated this histone modification may help to recognize exons in general. We first determined H3K9me3 abundance around gene architecture landmarks using publicly available H3K9me3 ChIP-seq data from three different human cell lines (K562, HeLa and MCF-7) generated by the ENCODE project34. H3K9me3 was depleted around transcriptional start sites and poly(A) sites and enriched around internal exons in all tested cells (Fig. 2 and Supplementary Fig. S2). While H3K9 profiles at internal exons resemble the general distribution of nucleosomes5,6, it confirms that nucleosomes localized on internal exons carry H3K9 trimethylation and is consistent with the model that this histone modification is important for exon recognition.
Figure 2. H3K9me3 signal at internal exons.

Relative H3K9me3 abundance profiles in HeLa cells 1kb up and downstream of transcriptional start sites (TSS, upper left panel), polyadenylation sites (p(A) site, upper right panel), 3′ splice sites (lower left panel) and 5′ splice sites (lower right panel) of internal exons were plotted versus relative genomic positions 1 kb up and downstream of gene architecture hallmark of interest. Genome wide abundance profiles were derived from chromatin immunoprecipitation sequencing (ChIP-seq) experiments performed in the ENCODE project. Schematic gene architecture hallmarks are displayed above. See also Supplementary Fig. S2 for K562 and MCF-7 analysis.
H3K9me3 influences co-transcriptional splicing of constitutive exons
To experimentally test the effect of H3K9 methylation on splicing efficiency of constitutive exons we analyzed co-transcriptional splicing of the exon 3 in FOSL1. This exon is surrounded by weak splice sites and H3K9me3 might promote its inclusion similarly to the alternative EDB exon. To modulate H3K9me3 we utilized two alternative approaches - global overexpression of the methyltransferase Suv39H1 and demethylase JMJD2D and local targeting of HMEs via TALE to FOSL1 exon 3. To avoid potential artifacts connected to the cell adapting to HME overexpression, we established stable cell lines containing Suv39H1 or JMJD2D under doxycycline inducible promoter. Suv39H1 or JMJD2D were tagged with mRFP to monitor their expression. Suv39H1-mRFP or JMJD2D-mRFP induction with doxycycline led to a global increase or decrease of H3K9me3, respectively (Supplementary Fig. 3). H3K9me3 ChIP on FOSL1 exon 3 confirmed that induction of JMJD2D expression reduced H3K9me3 levels by 70%. However, overexpression of Suv39H1 had no effect on H3K9me3 levels (Fig. 3B). Since the H3K9me3 signal was already high in uninduced cells, further increase in methylation may not have been possible. To monitor the splicing efficiency of the constitutive exon 3 in FOSL1, we analyzed co-transcriptional splicing of nascent RNA to avoid potential artifacts due to high amount of spliced mRNA present in total RNA samples. We used a primer downstream of the poly(A) cleavage site for reverse transcription, thereby selecting for nascent transcripts still attached to chromatin through the transcription machinery2,35 (Fig. 3A). The ratio of pre-mRNA to mRNA revealed an accumulation of unspliced transcripts after JMJD2D overexpression and depletion of H3K9me3 (Fig. 3C). Suv39H1 overexpression had no additional effect in enhancing the co-transcriptional splicing of the constitutive exon, which was consistent with ChIP data, when we observed no increase in H3K9me3.
Figure 3. H3K9me3 promotes co-transcriptional splicing of FOSL1.

(A) Stable U2OS-TO cell lines for doxycycline inducible overexpression of JMJD2D and Suv39H1 were established to test the effect of H3K9me3 on co-transcriptional splicing of exons 3 to 4 of FOSL1. Schematic representation of the Tet-On constructs and the endogenous FOSL1 gene are shown. To select nascent transcripts a primer downstream of the poly(A) cleavage site was used for reverse transcription. qPCR primers used for detection of pre-mRNA or mRNA are indicated below the gene diagram; above are the primers for ChIP. (B) H3K9me3 ChIP on FOSL1 exon 3 showing reduced H3K9me3 levels after JMJD2D overexpression, while Suv39H1 had no additional effect. ChIP signals are calculated as immunoprecipitated DNA over input and normalized to the uninduced control cells. Mean ± SEM are shown, n = 3. Statistical significance of all results was analyzed by t-test and significant changes (p < 0.05) were indicated by*. (C) Unspliced pre-mRNA accumulates after JMJD2D overexpression and depletion of H3K9me3 levels, while Suv39H1 had no effect on co-transcriptional splicing of FOSL1 exons 3–4 as determined by RT-qPCR. Ratios of unspliced pre-mRNA to spliced mRNA are normalized to uninduced control cells. Mean ± SEM are shown, n = 3. Statistical significance of all results was analyzed by t-test and significant changes (p < 0.05) were indicated by*. (D) Co-transcriptional splicing of FOSL1 exons 3–4 was assessed after targeting H3K9 methyltransferases and demethylases by TALE-HMEs binding exon 3. A primer downstream of the poly(A) cleavage site was used for reverse transcription to select nascent transcripts. Tethering of JMJD2D, but not the catalytically inactive JMJD2D-N202M, resulted in accumulation of unspliced transcripts. In contrast, tethering of methyltransferases G9a or Suv39H1 did not further improve co-transcriptional splicing. Ratios of unspliced pre-mRNA to spliced mRNA are normalized to TALE-GFP. Mean ± SEM are shown, n = 3. Statistical significance of all results was analyzed by t-test and significant changes (p < 0.05) were indicated by*.
To further investigate the role of H3K9me3 in exon recognition, we utilized the TALE-HME system and expressed Suv39H1, G9a or JMJD2D fused with the TALE domain targeting FOSL1 exon 3 (Supplementary Fig. 3). Consistent with global overexpression, tethering of JMJD2D significantly reduced splicing efficiency, while TALE-Suv39H1 or TALE-G9a had no effect (Fig. 3D). These results demonstrate H3K9me3 has a functional role in the splicing of constitutive exons.
Discussion
In recent years there has been mounting evidence that pre-mRNA splicing is closely coupled with RNA synthesis and the chromatin environment of template DNA4,36. The original findings suggested that chromatin modifications modulate alternative splicing11,13,14,21,23,37,38 and further studies have shown that pre-mRNA splicing reciprocally feeds back on histone modifications7,8,9. Most studies have relied on global depletion/inhibition or overexpression of chromatin modifying enzymes. These experiments, though thoroughly controlled, can never exclude the possibility that the observed changes in alternative splicing are caused by downstream secondary effects. The first attempt to investigate local effects of chromatin on splicing utilized siRNA-induced chromatin methylation22. However, the molecular mechanism of how siRNAs modulate chromatin modifications in mammalian cells is still not fully understood39 and the histone marks altered by this approach can not be controlled. A recent study utilized a zinc finger protein to tether the histone methyltransferase G9a to the promoter region of a target gene. This method increased H3K9me2 at both the promoter and the downstream region within the gene body, thus leaving the question of how local chromatin at the target exon affects splicing unaddressed15. To address whether a change of local histone methylation can modulate splicing, we used TALEs as a tool to navigate HMEs to a specific and unique sequence within the targeted gene. Given the specificity of TALE binding, secondary effects due to global changes in the transcriptional output can be eliminated, thus providing a tool to directly test the influence of local histone modifications on splicing.
First we targeted the alternative exon EDB in the FN1 gene, which was previously shown to be sensitive to chromatin regulation13,23. Four out of five tested TALE-HME constructs exhibited the desired effect on chromatin. SETD2 targeting increased H3K36 trimethylation at the target exon but we did not detect any robust changes in alternative splicing. H3K36me3 was reported to impact alternative splicing by the recruitment of splicing regulatory factors11,12,20. However, this effect is apparently context dependent and H3K36me3 alone is not sufficient to change the fate of the alternative target exon.
In contrast to H3K36me3, alteration of H3K9 methylation status exhibited a clear effect on the EDB exon. Expression of TALE fused with methyltransferases G9a or Suv39H1 increased H3K9me2 and me3 respectively, which in both cases promoted inclusion of the alternative exon. Conversely, TALE targeting of histone demethylase JMJD2D reduced H3K9me3, which reduced EDB inclusion. Furthermore, we showed that H3K9me3 is present over internal exons genome-wide, suggesting this histone mark plays a general role in exon recognition. Although the genome-wide abundance profiles mirror nucleosome position as derived from MNase ChIP-Seq data5,6, our data supports the fact that these nucleosomes carry H3K9me3 and this histone modification is a functional element in splicing regulation. Indeed, overexpression or TALE targeting of histone demethylase JMJD2D to constitutively spliced exon 3 of the FOSL1 gene reduced co-transcriptional splicing of this exon. Based on the bioinformatic analysis and the JMJD2D expression data we conclude that H3K9me3 at nucleosomes within the gene body affect exon recognition. Two mechanistic models can explain the effect of H3K9me3 on splicing. First, methylated H3K9 is bound by HP1γ, which in turn can recruit the splicing regulatory protein SRSF115. Alternatively, H3K9 methylation might affect alternative splicing through kinetic coupling by slowing down Pol II elongation21,22,37. Nucleosomes positioned over exons were previously proposed to act as speed bumps for Pol II elongation and H3K9 methylation on these exon-centered nucleosomes may increase the effect on Pol II speed40. Furthermore, splicing of the EDB exon was shown to be sensitive to Pol II elongation rate13,23,24, rendering kinetic coupling the most likely mechanism by which TALE-directed H3K9 methylation changes EDB inclusion rates.
In general, our data point to H3K9 methylation as the mark important for proper splicing pattern. This supports a model in which histone modifications together with DNA sequence, transcription machinery and the spliceosome form a complex unit, acting in cooperation to produce functional mRNA.
Material and Methods
Cell culture and treatments
HeLa and U2OS-TO cells were cultured in high glucose (4.5 g/l) DMEM (Sigma) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin (Penicillin/Streptomycin, Gibco) and 10% (v/v) fetal bovine serum (FBS, Gibco) at 37 °C and 5% CO2. Expression of mRFP-JMJD2D and mRFP-Suv39H1 in U2OS-TO cells was induced with 15 or 30 μg/ml doxycycline for 20 h. FOSL1 was induced by addition of 2 μg/ml ionomycin calcium salt (LifeTechnologies) for 30 min.
Plasmids and stable cell lines
TALE-domains targeting the EDB exon in human FN1 and exon 3 in human FOSL1 were designed using TAL Effector Targeter25 (https://tale-nt.cac.cornell.edu/node/add/single-tale) and assembled into pTALEN41 (Addgene) using the Golden Gate TALEN and TAL Effector Kit 2.0 (Daniel Voytas, Addgene)17. GFP or histone modifying enzymes Suv39H1, ASH1L, JMJD2D and the catalytically active domains of G9a and SETD2 were cloned into the pTALEN41 backbone using NaeI and XbaI restriction sites, replacing the nuclease domain. The HA-tag was included as a linker between the TALE-domain and the HME. Plasmids were transiently transfected into HeLa or U2OS-TO cells using Lipofectamin LTX reagent (LifeTechnologies) according to manufacturer’s instructions and incubated for 48 h.
Suv39H1 and JMJD2D were cloned into mRFP-C3 (Clontech) with BamHI/EcoRI or BamHI/XhoI restriction sites, respectively. The resulting Suv39H1-mRFP or JMJD2D-mRFP constructs were subcloned into pcDNA4/TO (LifeTechnologies) with KpnI/BamHI or HindIII/BamHI restriction sites, respectively. Stable cell lines in U2OS-TO cells were established after transfection with Lipofectamin LTX (LifeTechnologies) and selection with 300 μg/ml Zeocin (LifeTechnologies). Single-cell-colonies were expanded, tested for doxycycline inducible expression of Suv39H1-mRFP or JMJD2D-mRFP and individual clones were selected for experiments. To generate catalytically inactive JMJD2D, we introduced the point mutation N202M, which disrupts the coordination of α-ketoglutarate within JMJD2D catalytic site42. Mutagenic primers were designed using NEBaseChanger tool (F: 5′- TACAGCATCAtgTACCTGCACCTTGG - 3′; R: 5′ - AAGGTCCATGTCCTCTGTATG - 3′) and verified by DNA sequencing.
RNA isolation, reverse transcription and qPCR
Cells were grown to 90% confluency in a 6-well tissue culture plate (~106 cells). RNA was isolated using TRIZOL reagent (LifeTechnologies), precipitated with isopropanol, resuspended in nuclease free water and treated with Turbo DNase (Ambion) according to manufacturer’s protocol. Reverse transcription was performed with SuperScript III (LifeTechnologies) using 2 μg of RNA. Total RNA was reverse transcribed with random hexamer primers, while nascent RNA from FOSL1 was selectively enriched using a primer downstream of the poly(A) cleavage site2,35. The cDNA was analyzed by quantitative PCR. A list of primers is provided in the supplemental material.
Antibodies
The following antibodies were used for ChIP: rabbit α-H3K9me3 (Abcam ab8898), rabbit α-H3K36me3 (Abcam ab9050), mouse α-H3K9me2 (Millipore 05-1249), rabbit α-H3 (Abcam ab1719) and mouse α-IgG (Sigma I5381).
For Western blot the following antibodies were used: mouse α-GAPDH (Abcam ab9484), rabbit α-mRFP (Apronex), rat α-HA (Roche), mouse α-GFP (Santa Cruz sc-9996), rabbit α-H3K9me3 (Abcam ab8898), mouse α-tubulin kindly provided by Pavel Draber (Institute of Molecular Genetics of the ASCR, Prague, Czech Republic), goat α-mouse-HRP (Jackson ImmunoResearch), goat α-rabbit-HRP (Jackson ImmunoResearch), goat α-rat-HRP (Santa Cruz sc-2006).
Protein extraction and Western Plot
Proteins were isolated from TRIZOL fractions at the same time as RNA, precipitated with isopropanol and resuspended in NEST-2 buffer (50 mM Tris-HCl pH 6.8, 20 mM EDTA, 5% (w/v) SDS). Proteins were resolved on a 10% SDS-PAGE, blotted onto a nitrocellulose membrane and detected using the antibodies indicated above and SuperSignal Pico West (Thermo Fisher Scientific).
Native Chromatin Immunoprecipitation
Native ChIP assays were performed as previously described13. Cells were grown to 90% confluency in a 6-well plate, washed twice and scraped in PBS with proteinase inhibitor cocktail (Calbiochem). Four wells were combined for one ChIP experiment and the native ChIP assay was performed as previously described13. A list of primers is provided in the supplemental material. ChIP signals were calculated as IP over Input (2(Ct(input)–Ct(IP)) and normalized to a non-transcribed intergenic control region.
Cross-linked Chromatin Immunoprecipitation
Cross-linked ChIP assays were performed as previously described13. Cells were grown to 90% confluency in a 6-well plated, crosslinked with 1% formaldehyde in PBS for 10 min and ChIP assays were performed as previously described13. ChIP signals were calculated as IP over Input (2(Ct(input)–Ct(IP)) and normalized to a non-transcribed intergenic control region.
H3K9me3 ChIP profiles
H3K9me3 ChIP-seq experiments for different cell lines were performed by the ENCODE project34 and are publicly available (https://www.encodeproject.org/experiments/). The annotated references were obtained from GENCODE (Release 19)43. Processed data were obtained in bigwig format for the human genome version 19 (hg19) of the HeLa (ENCFF000BBL), K562 (ENCFF000BYX) and MCF-7 (ENCFF000VFL) cell lines. Mean abundance profiles in a window 1 kb up- and downstream for annotated transcriptional start sites (TSSs), polyA-sites as well as 5′ SSs and 3′ SSs of internal exons were determined using Sitepro of CEAS package44.
Additional Information
How to cite this article: Bieberstein, N. I. et al. TALE-directed local modulation of H3K9 methylation shapes exon recognition. Sci. Rep. 6, 29961; doi: 10.1038/srep29961 (2016).
Supplementary Material
Acknowledgments
We thank Radek Malik, Radek Jankele, Pavel Draber and Petr Svoboda for advice on design and assembly of TALE domains and for kindly providing reagents and plasmids and Kim Kotovic for English corrections. This work was supported by the Czech Science Foundation (P305/12/G034), the institutional support (RVO68378050) and the program for postdoctoral support (L200521501 to NIB) from the Academy of Sciences of the Czech Republic, and the DAAD PostDoc Fellowship Program to NIB.
Footnotes
Author Contributions N.I.B., E.K., M.H. and D.S. designed the study. N.I.B. performed experiments using the TALE-HME constructs. M.H. established clonal cell lines for inducible expression of Suv39H1 and JMJD2D. E.K. performed experiments using the inducible cell lines. F.C.O. and P.K.T. contributed the bioinformatics analysis. M.K. prepared catalytically inactive JMJD2D N202M and Z.K. performed experiments using this mutated protein, N.I.B. and D.S. wrote the paper.
References
- Brugiolo M., Herzel L. & Neugebauer K. M. Counting on co-transcriptional splicing. F1000prime reports 5, 9, 10.12703/P5-9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo Oesterreich F., Preibisch S. & Neugebauer K. M. Global analysis of nascent RNA reveals transcriptional pausing in terminal exons. Mol. Cell 40, 571–581, 10.1016/j.molcel.2010.11.004 (2010). [DOI] [PubMed] [Google Scholar]
- Tilgner H. et al. Deep sequencing of subcellular RNA fractions shows splicing to be predominantly co-transcriptional in the human genome but inefficient for lncRNAs. Genome Res. 22, 1616–1625, 10.1101/gr.134445.111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo Oesterreich F., Bieberstein N. & Neugebauer K. M. Pause locally, splice globally. Trends in Cell Biology 21, 328–335, 10.1016/j.tcb.2011.03.002 (2011). [DOI] [PubMed] [Google Scholar]
- Schwartz S., Meshorer E. & Ast G. Chromatin organization marks exon-intron structure. Nat Struct Mol Biol 16, 990–995, 10.1038/nsmb.1659 (2009). [DOI] [PubMed] [Google Scholar]
- Tilgner H. et al. Nucleosome positioning as a determinant of exon recognition. Nat Struct Mol Biol 16, 996–1001, 10.1038/nsmb.1658 (2009). [DOI] [PubMed] [Google Scholar]
- Bieberstein N. I., Oesterreich F. C., Straube K. & Neugebauer K. M. First exon length controls active chromatin signatures and transcription. Cell reports 2, 62–68, 10.1016/j.celrep.2012.05.019 (2012). [DOI] [PubMed] [Google Scholar]
- de Almeida S. F. et al. Splicing enhances recruitment of methyltransferase HYPB/Setd2 and methylation of histone H3 Lys36. Nat Struct Mol Biol 18, 977–983, 10.1038/nsmb.2123 (2011). [DOI] [PubMed] [Google Scholar]
- Kim S., Kim H., Fong N., Erickson B. & Bentley D. L. Pre-mRNA splicing is a determinant of histone H3K36 methylation. Proceedings of the National Academy of Sciences 108, 13564–13569, 10.1073/pnas.1109475108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnilicova J. et al. The C-terminal domain of Brd2 is important for chromatin interaction and regulation of transcription and alternative splicing. Mol Biol Cell 24, 3557–3568, 10.1091/mbc.E13-06-0303 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luco R. et al. Regulation of Alternative Splicing by Histone Modifications. Science 327, 996–1000, 10.1126/science.1184208 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo R. et al. BS69/ZMYND11 reads and connects histone H3.3 lysine 36 trimethylation-decorated chromatin to regulated pre-mRNA processing. Mol. Cell 56, 298–310, 10.1016/j.molcel.2014.08.022 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnilicová J. et al. Histone deacetylase activity modulates alternative splicing. PLoS One 6, e16727, 10.1371/journal.pone.0016727 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims R. J. et al. Recognition of trimethylated histone H3 lysine 4 facilitates the recruitment of transcription postinitiation factors and pre-mRNA splicing. Mol. Cell 28, 665–676, 10.1016/j.molcel.2007.11.010 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salton M., Voss T. C. & Misteli T. Identification by high-throughput imaging of the histone methyltransferase EHMT2 as an epigenetic regulator of VEGFA alternative splicing. Nucleic Acids Res. 42, 13662–13673, 10.1093/nar/gku1226 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cermak T., Starker C. G. & Voytas D. F. Efficient design and assembly of custom TALENs using the Golden Gate platform. Methods Mol. Biol. 1239, 133–159, 10.1007/978-1-4939-1862-1_7 (2015). [DOI] [PubMed] [Google Scholar]
- Cermak T. et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 39, e82, 10.1093/nar/gkr218 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J. C. et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 29, 143–148, 10.1038/nbt.1755 (2011). [DOI] [PubMed] [Google Scholar]
- Mendenhall E. M. et al. Locus-specific editing of histone modifications at endogenous enhancers. Nature biotechnology 31, 1133–1136, 10.1038/nbt.2701 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradeepa M. M., Sutherland H. G., Ule J., Grimes G. R. & Bickmore W. A. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS genetics 8, e1002717, 10.1371/journal.pgen.1002717 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint-André V., Batsché E., Rachez C. & Muchardt C. Histone H3 lysine 9 trimethylation and HP1γ favor inclusion of alternative exons. Nat Struct Mol Biol 18, 337–344, 10.1038/nsmb.1995 (2011). [DOI] [PubMed] [Google Scholar]
- Alló M. et al. Control of alternative splicing through siRNA-mediated transcriptional gene silencing. Nat Struct Mol Biol 16, 717–724, 10.1038/nsmb.1620 (2009). [DOI] [PubMed] [Google Scholar]
- Duskova E., Hnilicova J. & Stanek D. CRE promoter sites modulate alternative splicing via p300-mediated histone acetylation. Rna Biol 11, 865–874, 10.4161/Rna.29441 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagani F., Stuani C., Zuccato E., Kornblihtt A. R. & Baralle F. E. Promoter architecture modulates CFTR exon 9 skipping. The Journal of biological chemistry 278, 1511–1517, 10.1074/jbc.M209676200 (2003). [DOI] [PubMed] [Google Scholar]
- Doyle E. L. et al. TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res. 40, W117–122, 10.1093/nar/gks608 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M., Sugimoto K., Fukushima T. & Shinkai Y. Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. The Journal of biological chemistry 276, 25309–25317, 10.1074/jbc.M101914200 (2001). [DOI] [PubMed] [Google Scholar]
- Wu H. et al. Structural biology of human H3K9 methyltransferases. PLoS One 5, e8570, 10.1371/journal.pone.0008570 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea S. et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406, 593–599, 10.1038/35020506 (2000). [DOI] [PubMed] [Google Scholar]
- Whetstine J. R. et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 125, 467–481, 10.1016/j.cell.2006.03.028 (2006). [DOI] [PubMed] [Google Scholar]
- Sun X. J. et al. Identification and characterization of a novel human histone H3 lysine 36-specific methyltransferase. The Journal of biological chemistry 280, 35261–35271, 10.1074/jbc.M504012200 (2005). [DOI] [PubMed] [Google Scholar]
- An S., Yeo K. J., Jeon Y. H. & Song J. J. Crystal structure of the human histone methyltransferase ASH1L catalytic domain and its implications for the regulatory mechanism. The Journal of biological chemistry 286, 8369–8374, 10.1074/jbc.M110.203380 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbert P. B. & Henikoff S. Spreading of silent chromatin: inaction at a distance. Nature reviews. Genetics 7, 793–803, 10.1038/nrg1920 (2006). [DOI] [PubMed] [Google Scholar]
- Schor I. E., Fiszbein A., Petrillo E. & Kornblihtt A. R. Intragenic epigenetic changes modulate NCAM alternative splicing in neuronal differentiation. EMBO J. 32, 2264–2274 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74, 10.1038/nature11247 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya-Jones A. & Black D. L. Co-transcriptional splicing of constitutive and alternative exons. RNA 15, 1896–1908, rna.1714509/10.1261/rna.1714509 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnilicova J. & Stanek D. Where splicing joins chromatin. Nucleus 2, 182–188 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schor I. E., Rascovan N., Pelisch F., Allo M. & Kornblihtt A. R. Neuronal cell depolarization induces intragenic chromatin modifications affecting NCAM alternative splicing. Proc. Natl. Acad. Sci. USA 106, 4325–4330 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogues G., Kadener S., Cramer P., Bentley D. & Kornblihtt A. R. Transcriptional activators differ in their abilities to control alternative splicing. The Journal of biological chemistry 277, 43110–43114, 10.1074/jbc.M208418200 (2002). [DOI] [PubMed] [Google Scholar]
- Malik R. & Svoboda P. In Toxicology and Epigenetics (ed Saura C. Sahu) Ch. 15, (John Wiley & Sons, Ltd, 2012). [Google Scholar]
- Kwak H., Fuda N. J., Core L. J. & Lis J. T. Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science 339, 950–953, 10.1126/science.1229386 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana N. E. et al. A transcription activator-like effector toolbox for genome engineering. Nat Protoc 7, 171–192, 10.1038/nprot.2011.431 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoabi M. et al. RNA-dependent chromatin localization of KDM4D lysine demethylase promotes H3K9me3 demethylation. Nucleic Acids Res 42, 13026–13038, 10.1093/nar/gku1021 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774, 10.1101/gr.135350.111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H., Liu T., Manrai A. K. & Liu X. S. CEAS: cis-regulatory element annotation system. Bioinformatics 25, 2605–2606, 10.1093/bioinformatics/btp479 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.