

Summary
The initiation of type 1 diabetes (T1D) requires a break in peripheral tolerance. New insights into neoepitope formation indicate that post‐translational modification of islet autoantigens, for example via deamidation, may be an important component of disease initiation or exacerbation. Indeed, deamidation of islet autoantigens increases their binding affinity to the T1D highest‐risk human leucocyte antigen (HLA) haplotypes HLA‐DR3/DQ2 and ‐DR4/DQ8, increasing the chance that T cells reactive to deamidated autoantigens can be activated upon T cell receptor ligation. Here we investigated human pancreatic islets and inflammatory and tolerogenic human dendritic cells (DC and tolDC) as potential sources of deamidated islet autoantigens and examined whether deamidation is altered in an inflammatory environment. Islets, DC and tolDC contained tissue transglutaminase, the key enzyme responsible for peptide deamidation, and enzyme activity increased following an inflammatory insult. Islets treated with inflammatory cytokines were found to contain deamidated insulin C‐peptide. DC, heterozygous for the T1D highest‐risk DQ2/8, pulsed with native islet autoantigens could present naturally processed deamidated neoepitopes. HLA‐DQ2 or ‐DQ8 homozygous DC did not present deamidated islet peptides. This study identifies both human islets and DC as sources of deamidated islet autoantigens and implicates inflammatory activation of tissue transglutaminase as a potential mechanism for islet and DC deamidation.
Keywords: DC, HLA, islets, post‐translational modification, type 1 diabetes
Introduction
Type 1 diabetes (T1D) is an autoimmune disease involving the progressive destruction of the pancreatic islets of Langerhans by autoreactive CD8 and CD4 T cells 1, 2. Currently, it is unknown what triggers the initial break in peripheral tolerance that precipitates the destructive immune response, but once triggered, insulitis appears to play a key role in islet destruction 3. Recent insights indicate that post‐translational modification (PTM) of beta cell antigens play an important role in T1D pathogenesis 4. Conversion of glutamine into negatively charged glutamate by a process called deamidation is an important modification shared with coeliac disease pathogenesis. This conversion is catalysed by the enzyme tissue transglutaminase (tTG) 5. Glutamine deamidation can create far more potent binders for certain human leucocyte antigen (HLA) class II molecules. Indeed, we have shown recently that deamidation of peptides derived from islet autoantigens increases their binding affinity, relative to native peptides, to T1D predisposing HLA‐DQ molecules 6. Intriguingly, the HLA‐DQ molecule that binds deamidated islet peptides with the highest affinity also confers the highest risk for developing T1D; namely, the HLA‐DQ8trans dimer 7. Individuals heterozygous for HLA‐DQ2/8, presenting autoantigen‐derived peptides via HLA‐DQ8trans, have a more than fourfold greater risk for developing T1D than individuals who are homozygous for DQ2 or DQ8, which in themselves confer an approximately 10‐times greater risk for T1D development 8. Supporting the importance of HLA‐DQ8 in the development of T1D is a recent study identifying HLA‐DQ8cis and ‐DQ8trans restricted CD4 T cells within human pancreatic islets 9.
Currently, the cells capable of deamidating islet autoantigens, as well as the environment contributing to generating deamidated islet autoantigens, are unknown, but there is evidence that both myeloid cells and beta cells express tTG 10, 11. Here, we investigated human dendritic cells (DC) and tolerogenic (tol)DC, as professional antigen‐presenting cells capable of priming naive T cells 12, 13, 14, as well as human pancreatic islets under resting and inflammatory conditions. We aimed to identify which cells can deamidate islet autoantigens and how this could be occurring. This study indicates that both human islets and DC can deamidate islet autoantigens. Furthermore, we observed that inflammation is a trigger for increased tTG activity in islets and DC. If DC bear the capacity to target autoreactive T cells specific for both native and deamidated islet epitopes it could add significant value to current peptide immunotherapy strategies 15, 16 by increasing vaccine efficacy and simplifying vaccine design.
Materials and methods
Tissue transglutaminase activity in human islets and DC
Islets prepared for flow cytometry were treated for 48 h with 2 mM biotin–cadaverin (Biotium, Hayward, CA, USA) plus or minus interleukin (IL)−1β (2 ng/ml) and interferon (IFN)‐γ (1000 IU/ml). After 48 h the medium was replaced with Iscove's modified Dulbecco's medium (IMDM) for 1 h to remove un‐cross‐linked biotin–cadaverin. DC and tolerogenic DC were generated and the phenotype confirmed as described previously 17. Immature DC and tolDC were cultured for 24 h at 37°C, 5% CO2 with 2 mM biotin–cadaverin with or without cytokine maturation cocktail [1600 IU/ml IL‐1β, 500 IU/ml IL‐6, 335 IU/ml tumour necrosis factor (TNF)‐α and 2 μg/ml prostaglandin E2 (PGE2) in RPMI containing penicillin/streptomycin, glutamax and 10% fetal calf serum (FCS) (cRPMI)]. After 24 h the medium was replaced with fresh cRPMI for 1 h to remove un‐cross‐linked biotin–cadaverin. Following biotin–cadaverin washout, islets and DC were fixed in 1% paraformaldehyde, permiabilized in saponin buffer [0·1% saponin, 1% bovine serum albumin (BSA) in phosphate‐buffered saline (PBS)] and stained with mouse anti‐human tTG antibody (clone TG100; Thermo Scientific, Fremont, CA, USA). Cells were washed in saponin buffer then stained with an anti‐mouse secondary conjugated to phycoerythrin (PE) and streptavidin‐fluorescein isothiocyanate (FITC) to determine the level of intracellular cross‐linked biotin–cadaverin. Fluorescence was measured by flow cytometry on an LSR‐II or FacsCalibur (BD Bioscience, Breda, the Netherlands) and analysed with FlowJo software (TreeStar, Ashland, OR, USA).
Proteins and peptides
Recombinant proteins [pre‐proinsulin (PPI)], islet antigen 2 (IA‐2) and glutamic acid decarboxylase (GAD65) containing a histidine tag at the N‐terminus were produced using Gateway cloning technology (Invitrogen, Carlsbad, CA, USA) 18. Proteins were produced in Escherichia coli and affinity‐purified using anti‐His antibody (Invitrogen, Carlsbad, CA, USA). The size and purity of recombinant proteins were analysed by Western blotting. Endotoxin contents were below the detection threshold, as tested using the limulus amoebocyte lysate (LAL) assay (Cambrex, East Rutherford, NJ, USA). All proteins were tested in lymphocyte stimulation assays in order to exclude antigen non‐specific T cell stimulation. IA‐2 peptides (296‐311 and 412‐424) were synthesized according to standard Fmoc chemistry. The integrity of the peptides was checked using ultra performance liquid chromatography‐tandem mass spectrometry (UPLC‐MS) and matrix‐assisted laser desorption ionization time‐of‐flight (MALDI‐TOF) MS.
Proteome analysis of human islets
Human islets were isolated from brain‐dead organ donors, as described previously 19. Briefly, islets were isolated in Good Manufacturing Practice (GMP) facilities at the Leiden University Medical Centre. Islet purity was between 75 and 95% as assessed by 1 mmol/l dithizone (Sigma‐Aldrich, Schnelldorf, Germany) staining. Families gave informed written consent for the use of donor tissue. Islets were treated for 24 h with supernatant from GAD65‐specific T helper cells, cultured for 3 days with or without antigen stimulation 20. Islets were lysed and frozen at −80°C for proteome analysis. Proteins from lysed human islets were digested using the filter‐aided sample preparation (FASP) method 21. Briefly, protein was loaded on a 30‐kDa filter and sodium dodecyl sulphate (SDS) was removed by washing with 8 M urea. Proteins were alkylated, washed to remove excess iodoacetamide and digested overnight using endoLysC and trypsin. Tryptic peptides were desalted on C18 SepPak and fractionated by strong cation exchange (SCX) on an Agilent 1100 system equipped with an SCX column. The gradient started with 100% solvent A (70/30/0·1 water/acetonitrile/formic acid) and reached 100% solvent B (65/35/0.1 250 mM KCl/acetonitrile/formic acid), followed by 100% solvent C (65/35/0.1 500 mM KCl/acetonitrile/formic acid). Finally, the eluent was in 100% solvent A. Fifteen fractions were collected, lyophilized and reconstituted in 30 μl 95/3/0·1 (water/acetonitrile/formic acid). Fractions were analysed by online nano‐high‐performance liquid chromatography (HPLC) MS (HPLC MS with a system consisting of an Agilent 1100 gradient HPLC system; Agilent, Waldbronn, Germany) and an LTQ‐FT ultra‐mass spectrometer (Thermo, Bremen, Germany). Fractions (5 μl) were injected onto a Reprosil‐Pur C18‐AQ column and eluted via a nano‐HPLC column. The gradient was run from 0 to 30% solvent D (10/90/0·1 water/acetonitrile/formic acid). Full scan mass spectra were acquired in the Fourier transform (FT)‐MS with a resolution of 25 000 at a target value of 5 × 106. The five most intense ions were selected and fragmented in the linear ion trap using collision‐induced dissociation at a target value of 10 000. For MS2 spectral matching, Mascot 2·2·04 (Matrix Science) was used, with two parts per million (ppm) precursor and 0·5 Da fragment accuracy. Variable modifications included N‐terminal protein acetylation and methionine oxidation. Carbamidomethylation of cysteine was selected as a fixed modification. False discovery rate was set to 1%.
Generation of human DC for HLA‐peptide elutions
Isolation and generation of DC from homozygous HLA‐DQ2, homozygous ‐DQ8 or heterozygous ‐DQ2/8 healthy blood donors was performed as described previously 22. Peripheral blood mononuclear cells (PBMC) isolated from each buffy coat were cultured separately and pulsed with PPI, IA‐2 and GAD65 and iDC were matured with lipopolysaccharide (LPS) and IFN‐γ in the continuous presence of the three islet autoantigens. After 30 h, pulsed mDC were harvested, washed and lysed. Lysates were high‐speed centrifuged to remove nuclei and insoluble material. For each HLA elution a total of 40 × 106 pulsed mDCs were obtained from three donors per HLA‐DQ genotype that were pooled after pulsing with islet autoantigen prior to the HLA‐peptide elutions.
Peptide elution and isolation from affinity‐purified HLA‐DQ
Affinity purification of HLA‐DQ molecules from mDC and subsequent peptide elutions were performed as follows. Our existing peptide elution protocol 23 was optimized for low DC numbers. All HLA‐DQ isolation and washing steps were performed using a 100 µl pipette tip. DC lysate was precleared by running it through a 100 µl pipette tip containing a small filter and packed with 100 µl Sepharose beads. The precleared lysate was collected and HLA‐DQ molecules were affinity‐isolated using SPV‐L3 coupled to Sepharose beads and packed in a 100 µl pipette tip. Columns were washed with four bed volumes lysis buffer, low salt buffer, high salt buffer, no salt buffer and low Tris buffer. HLA–peptide complexes were eluted with 10% acetic acid. HLA‐DQ eluates (containing both peptides and HLA) were fractionated with an HPLC system. The material was eluted using a gradient of 0–50% acetonitrile supplemented with 0·1% trifluoroacetic acid. Eluted peptides were subsequently analysed as described previously 23.
Cell free HLA‐DQ/peptide binding assays
Binding of islet autoantigen‐derived peptides was tested for all four HLA‐DQ ‘T1D risk’ molecules. As a source of HLA‐DQ, Epstein–Barr virus lymphoblastoid B cell (EBV‐BLCL) cells lentiviral‐transduced to express a single HLA‐DQ molecule were used 23. Peptide‐binding affinities were calculated using GraphPad software version 6; the concentration of islet peptide required for half‐maximal inhibition of binding of the reporter peptide indicate the half maximal effective concentration (EC50) value. Although binding of the islet‐peptides to different HLA‐DQ molecules cannot be compared accurately (due to amino acid sequences of indicator peptides differing between the HLA‐DQ assays and different HLA‐DQ molecules having different properties), a ≥ 10‐fold difference in EC50 value was considered significant.
Results
Islets and DC express tTG, which is activated upon inflammatory insult
Pancreatic islets are the primary source of autoantigens responsible for the development of T1D. Therefore, we tested whether human islets had the potential to generate deamidated autoantigens by assessing tTG expression and tTG activity in whole islet preparations. We observed that islets express tTG, and incubation with inflammatory cytokines increased tTG activity over basal levels (Fig. 1a). Islet autoantigen uptake and presentation by high‐risk HLA‐DQ molecules expressed on the surface of professional antigen‐presenting cells, namely DC, is required to initiate an autoimmune response against pancreatic beta cells. Therefore, we tested tTG expression and activity in DC to determine whether they could also be a source of deamidated autoantigens. DC expressed tTG and there was a trend towards increased tTG activity in cytokine‐matured DC (Fig. 1b). TolDC expressed higher levels of tTG than DC, and tTG activity in tolDC was comparable to both immature and mature DC (Fig. 1c). Together, these data demonstrate that human pancreatic islets, DC and tolDC all have the potential to deamidate intracellular proteins. Furthermore, intracellular tTG activity in these cells increases in response to inflammatory cytokines.
Figure 1.
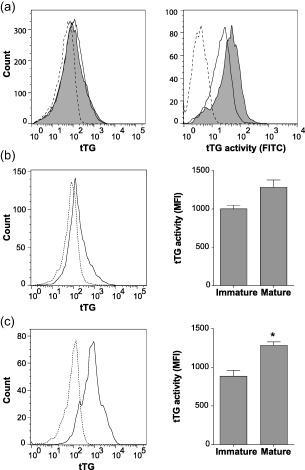
Expression and activity of human islet and dendritic cell tissue transglutaminase. (a) Islets treated with (grey‐filled) or without (solid line) inflammatory cytokines [interleukin (IL)‐1β 2 ng/ml and interferon (IFN)‐γ 1000 IU/ml] for 48 h, stained with anti‐tissue transglutaminase (tTG) antibody and phycoerythrin (PE)‐conjugated secondary antibody (left panel). Biotin–cadaverin (2 mM) was added during cytokine treatment and intracellular cross‐linked biotin–cadaverin was stained with streptavidin‐fluorescein isothiocyanate (FITC) as a measure of tTG activity (right panel). Dotted lines represent the background staining controls. Immature dendritic cell (DC) (b) and tolerogenic dendritic cell (tolDC) (c) tTG levels as measured with anti‐tTG antibody (solid line, left panels). tTG activity in immature and mature DC and tolDC as measured by biotin–cadaverin cross‐linking (right panels). Data represent mean ± standard error of the mean (n = 3) *P < 0·05 unpaired t‐test.
Inflammatory cytokine‐treated islets contain deamidated insulin C‐peptide
An increase in islet tTG activity during insulitis could result potentially in increased deamidation of islet autoantigens. To investigate the effect of T cell‐mediated islet inflammation on islet autoantigen deamidation, islets were treated with supernatant from our resting or activated GAD65‐specific CD4 T cell clone cultured in the absence or presence of its cognate GAD65 peptide; activation with peptide resulted in increased cytokine production (Supporting information, Fig. S1). Proteomic analysis of proinsulin peptides showed that islets treated with supernatant from the resting CD4 T cell clone contained only the native form of proinsulin C‐peptide. In comparison, treatment with supernatant obtained from the activated CD4 T cell clone resulted in deamidation of a glutamine at amino acid positions 62 and 65 of proinsulin C‐peptide (Table 1). Native C‐peptide was also recovered from islets conditioned in an inflammatory milieu.
Table 1.
Proteome analysis of human islets treated with supernatant from resting (control) or activated (conditioned) glutamic acid decarboxylase (GAD65)‐specific CD4 T cell clones was performed using mass spectrometry
| Control islets | |
|---|---|
| Sequence | PPI residues |
| EAEDLQVGQVELGGGPGAGSLQPLALEGSLQK | 57–88 |
| REAEDLQVGQVELGGGPGAGSLQPLALEGSLQKR | 56–89 |
| Conditioned islets | |
|---|---|
| Sequence | PPI residues |
| EAEDLQVGQVELGGGPGAGSLQPLALEGSLQK | 57–88 |
| EAEDLEVGQVELGGGPGAGSLQPLALEGSLQK | |
| EAEDLQVGEVELGGGPGAGSLQPLALEGSLQK | |
| REAEDLQVGQVELGGGPGAGSLQPLALEGSLQKR | 56–89 |
| REAEDLEVGQVELGGGPGAGSLQPLALEGSLQKR | |
| REAEDLQVGEVELGGGPGAGSLQPLALEGSLQKR | |
Residues shown in bold type within the pre‐proinsulin peptide (PPI) sequences represent the residues identified as being native (Q) or deamidated (E). In human islets conditioned with activated supernatant both native and deamidated C‐peptides were identified.
DC can process native islet autoantigens and present deamidated peptides
Having confirmed pancreatic islets as a source of deamidated C‐peptide, we then asked whether DC can also deamidate islet autoantigens. Previously, we have shown that heterozygous HLA‐DQ2/8 DC can process and present a larger variety of native islet autoantigen‐derived peptides than homozygous DQ2 or DQ8 DC 24. Interestingly, when pulsed with the native form of the islet autoantigen IA‐2, only HLA‐DQ2/8 DC presented deamidated peptides (Fig. 2a). Two length variants were found between amino acid position 296–311 of IA‐2 and one at position 412–424. Deamidated peptides were not observed in peptide elutions of HLA‐DQ2 or ‐DQ8 homozygous DC. Deamidation of the glutamine residues into glutamate was confirmed using synthetic peptides and recombinant tTG (not shown). To determine the binding affinity of the native versus deamidated forms of the peptides presented on HLA‐DQ2/8 DC, peptide binding to all four T1D‐susceptible HLA‐DQ molecules was confirmed in cell‐free peptide‐binding assays. The deamidated IA‐2 peptide at position 296–311 bound more strongly to DQ8trans and DQ8cis (EC50 1·0 ± 0·1 and 14·1 ± 1·9 µM, respectively) than the corresponding native peptide (EC50 16·0 ± 2·1 and 40·0 ± 10·8 µM, respectively) and neither bound to the DQ2cis/trans molecules (Fig. 2b). The deamidated peptide at position 412–424 bound solely to DQ8trans and a higher binding affinity was observed compared to the native peptide (EC50 1·7 ± 0·1 versus 37·0 ± 0·8 µM) (Fig. 2b). These results demonstrate that heterozygous HLA‐DQ2/8 DC processes native IA‐2 protein and presents deamidated IA‐2 peptides on the T1D highest‐risk HLA‐DQ8trans molecule. These deamidated peptides are stronger binders than their native non‐deamidated counterparts.
Figure 2.
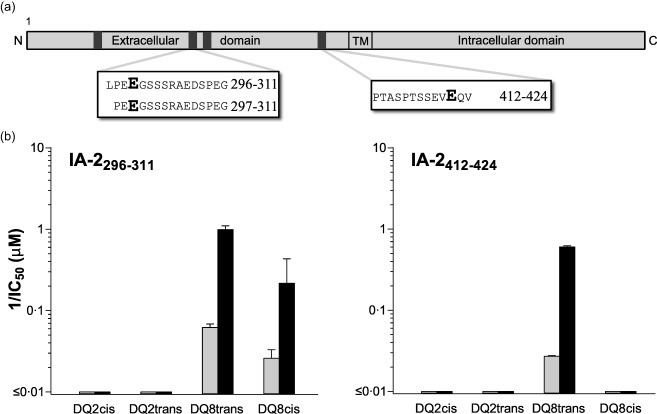
Islet antigen‐2 (IA‐2) peptides eluted from highest‐risk human leucocyte antigen (HLA)‐DQ2/8 expressing dendritic cells selectively bind HLA‐DQ8trans. (a) HLA‐DQ2 or HLA‐DQ8 homozygous and HLA‐DQ2/8 heterozygous dendritic cells (DC) from three individual donors per genotype were pulsed for 6 h with islet autoantigens pre‐proinsulin (PPI), glutamic acid decarboxylase (GAD65) and IA‐2, after which the cells were matured for 24 h with lipopolysaccharide (LPS) and interferon (IFN)‐γ in the continuous presence of the islet autoantigens. After cell lysis, donors were pooled, then HLA‐DQ was purified using SPV‐L3 (pan‐DQ antibody) and peptides were acid eluted and analysed by mass spectrometry. Peptides, covering two distinct core‐regions of IA‐2, were eluted. (b) Binding of the native (grey bars) and deamidated (black bars) forms of the eluted IA‐2 peptides was tested in competitive peptide‐binding assays for binding to HLA‐DQ2cis, ‐DQ2trans, ‐DQ8trans and ‐DQ8cis. Data represent mean ± standard error of the mean (n = 3). Shown on the x‐axes is 1/EC50, thereby illustrating that larger bars represent better binding.
Discussion
Post‐translational modification (PTM) of islet autoantigens has emerged as a strong candidate for the generation of neoepitopes as targets of autoreactive T cells in T1D pathogenesis and human islets can contribute actively to their own demise 25. Here we demonstrate this principle during an inflammatory insult with the generation of islet neoepitopes which coincides with activation of tissue transglutaminase (tTG). In addition, professional antigen‐presenting cells, namely DC, also express tTG, which is activated when DC encounter an inflammatory milieu. Upon inflammation, these DC present deamidated peptides derived from IA‐2 by the T1D highest‐risk HLA‐DQ8trans molecule. Therefore, not only inflammatory cytokine‐treated islets but also DC can generate modified islet epitopes.
T1D is a heterogeneous autoimmune disease requiring the co‐operation of several genetic and environmental factors to cause a break in immune tolerance and initiate disease 8, 26. Here we focused on a new paradigm in T1D research that may explain, in part, the break in peripheral tolerance that precipitates disease: PTM of islet autoantigens. T cells reactive to modified autoantigens are more likely to escape central tolerance, but should still undergo peripheral tolerance 27, 28. Inappropriate inflammation may be an important factor in this break in tolerance, as tTG activity and apoptosis have been reported to occur during islet isolation and inflammatory signalling induces beta cell apoptosis 29, 30, indicating that in our study the inflammatory milieu may be activating tTG by inducing beta cell apoptosis. In addition, tTG co‐localizes with insulin‐containing granules in human beta cells 10, indicating the potential for an increase in tTG activity to result in deamidation of insulin. In DC, levels of cell surface tTG increase during maturation, implying that tTG could be incorporated into endosomes during antigen uptake, thereby bringing tTG into direct contact with antigens during processing 11. In our hands, DC increased tTG activity during inflammatory maturation. Therefore, the consequences of pancreatic inflammation on the production and presentation of modified islet autoantigens could be threefold: islets deamidate proinsulin‐derived peptides after inflammatory insult, DC deamidate native islet autoantigens and once DC become activated during inflammation, they present deamidated epitopes. Conversely, we observed tTG activity in both DC and tolDC in the absence of inflammation; this could be a mechanism for inducing peripheral tolerance to modified autoantigens, whereby native islet autoantigens taken up by immature DC are deamidated and presented in a tolerogenic fashion. tTG is also present in other cells of the myeloid lineage, indicating that infiltrating monocytes and tissue resident macrophages could also contribute to the production of modified islet proteins and immune activation or tolerance induction, depending on the inflammatory context 11.
Our results indicate that DC generate deamidated peptides derived from the islet autoantigen IA‐2 and present these peptides on HLA‐DQ2/8 molecules. It is conceivable that our identified modified IA‐2 peptides represent the tip of the iceberg of the total modified islet peptide ligandome, as we did not retrieve modified peptides from GAD65 or PPI. The identified peptides in this study might represent a bias regarding more favourable binding characteristics (e.g. highest on‐rate and/or slowest off‐rate) and peptides having unfavourable properties are more challenging to identify due to technical limitations. None the less, the proof of PTM in both islets and DC is unambiguous. Deamidation and presentation of islet autoantigens by DC could increase the suppressive effect of peptide immunotherapy in T1D by targeting T cells specific for both native and deamidated epitopes. Deamidation by endogenous DC would eliminate the need for including deamidated peptides in the vaccine, making design much simpler. Peptides delivered with ex‐vivo‐generated tolDC, as those used currently in a T1D tolDC vaccine strategy could be deamidated within the tolDC to induce tolerance and generate regulatory T cells specific for both native and modified autoantigens.
The genetic susceptibility associated with T1D, especially with regard to the HLA‐DQ2 and DQ8 genotypes, indicates that T cells reactive against modified autoantigens may escape peripheral tolerance through increased binding affinity of the modified epitope presented by HLA‐DQ2/8. Indeed, we have demonstrated previously that deamidated peptides bind with higher affinity to T1D high‐risk HLA‐DQ molecules than their native counterparts 6; T1D patient T cell responses to deamidated proinsulin epitope InsB30‐C13 (TRREAEDLQVGQVELG: the two glutamines in bold type can be deamidated) differed in both quantity and quality, with more patients responding and producing IFN‐γ rather than IL‐10. We identified proinsulin C‐peptide in our islet proteome analysis, and the same glutamines as in the InsB30‐C13 epitope were deamidated exclusively in C‐peptide from islets treated with activated CD4 T cell clone supernatant, thereby linking inflammation to modified autoantigen production in islets. tTG deamidates glutamines following a particular enzyme substrate algorithm defined by the amino acids immediately following the glutamine, which permit or prevent glutamine deamidation 31. We have demonstrated previously that tTG is only able to deamidate the glutamines at positions 62 and 65 of C‐peptide 6. Deamidation of these two glutamines within C‐peptide is also demonstrated in this report, providing support for our hypothesis that tTG is responsible for deamidation in this instance.
This study indicates that activation of tTG may be the mechanistic link between inflammation and antigen deamidation in human islets and DC. While direct proof of tTG being responsible for the deamidation of autoantigens by islets and DC is difficult to provide, we propose that this is the most likely cause of PTM in this instance. First, tTG is the only deamidating enzyme known to be expressed and active in islets. Secondly, the enzyme substrate recognition motif necessary for tTG to deamidate proinsulin and IA‐2 is a perfect fit for the observed deamidation events in these two islet proteins. It proved impossible to design experiments to block tTG with a specific blocker of tTG activity (Z‐DON‐Val‐Pro‐Leu‐OMe), as the conditions required for this (e.g. the DMSO concentration required to keep the inhibitor soluble) precluded the use of this inhibitor with islets and DC. No alternative cell permeable tTG inhibitors have been reported. Alternative non‐enzymatic deamidation mechanisms (e.g. spontaneous chemical deamidation) that might explain our findings are extremely rare, and require distinct amino acid sequences as motifs 32, 33, leaving deamidation by tTG as the most plausible explanation for our observed findings.
We demonstrated that during inflammation both human islets and DC can deamidate islet autoantigens, identifying two crucial cellular sources of modified islet epitopes and the permissive environment that may lead to a break in peripheral tolerance and development of autoimmunity in T1D. These results could have implications for the design of peptide immunotherapy and may aid in simplifying the vaccine development process by removing the need to include deamidated peptides. Additionally, deamidation of immunotherapeutic peptides after delivery could be contributing to vaccine efficacy. As such, immune monitoring should include readouts that measure the immunosuppressive effects of both native and deamidated islet epitopes to investigate the impact of post‐delivery peptide deamidation on vaccine efficacy. Furthermore, these results suggest that an inflammatory insult within the pancreas may contribute to the initiation or development of an immune response against modified islet autoantigens. Therefore, if we can improve our ability to identify insulitis during the prediabetes stage, targeting deamidation could be developed as a therapeutic strategy to delay or prevent disease onset.
Disclosure
No potential disclosures relative to this paper are reported.
Author contributions
R. J. M. and M. L. designed the study, performed experiments, analysed data and wrote the manuscript. A. H. performed experiments. A. Z and E. J. K. supplied human islets and designed experiments. A. H. R. and P. A. V. designed and performed experiments. B. O. R. supervised the study and contributed to discussion and writing the paper. B. O. R. is the guarantor of this work and, as such, had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Cytokine levels in resting and activated CD4 T cell supernatants. Supernatant from resting (white bars) and activated (black bars) glutamic acid decarboxylase (GAD65)‐restricted T helper cells was measured by Luminex cytokine array.
Acknowledgements
The authors would like to thank Dr Jan Wouter Drijfhout for producing native and deamidated peptides, Dr Tatjana Nikolic for providing DC and tolDC and Jos Pool for providing recombinant proteins. B. O. R. is a member of the Danish Diabetes Academy. This work was supported by the Juvenile Diabetes Research Foundation (JDRF 2‐SRA‐2014‐295), the European Union's 7th Framework Programme (FP7/2007‐2013) under grant agreement no. 241447 (NAIMIT), by a VICI grant of the Netherlands organization for scientific research (VICI, 918.86.611) and an Expert Center Grant from the Dutch Diabetes Research Foundation.
References
- 1. Coppieters KT, Dotta F, Amirian N et al Demonstration of islet‐autoreactive CD8 T cells in insulitic lesions from recent onset and long‐term type 1 diabetes patients. J Exp Med 2012; 209:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Roep BO, Peakman M. Surrogate end points in the design of immunotherapy trials: emerging lessons from type 1 diabetes. Nat Rev Immunol 2010; 10:145–52. [DOI] [PubMed] [Google Scholar]
- 3. Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol 2009; 155:173–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van Lummel M, Zaldumbide A, Roep BO. Changing faces, unmasking the beta‐cell: post‐translational modification of antigens in type 1 diabetes. Curr Opin Endocrinol Diabetes Obes 2013; 20:299–306. [DOI] [PubMed] [Google Scholar]
- 5. van de Wal Y, Kooy Y, van Veelen P et al Selective deamidation by tissue transglutaminase strongly enhances gliadin‐specific T cell reactivity. J Immunol 1998; 161:1585–8. [PubMed] [Google Scholar]
- 6. van Lummel M, Duinkerken G, van Veelen PA et al Posttranslational modification of HLA‐DQ binding islet autoantigens in type 1 diabetes. Diabetes 2014; 63:237–47. [DOI] [PubMed] [Google Scholar]
- 7. van Lummel M, van Veelen PA, Zaldumbide A et al Type 1 diabetes‐associated HLA‐DQ8 transdimer accommodates a unique peptide repertoire. J Biol Chem 2012; 287:9514–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Koeleman BP, Lie BA, Undlien DE et al Genotype effects and epistasis in type 1 diabetes and HLA‐DQ trans dimer associations with disease. Genes Immun 2004; 5:381–8. [DOI] [PubMed] [Google Scholar]
- 9. Pathiraja V, Kuehlich JP, Campbell PD et al Proinsulin‐specific, HLA‐DQ8, and HLA‐DQ8‐transdimer‐restricted CD4+ T cells infiltrate islets in type 1 diabetes. Diabetes 2015; 64:172–82. [DOI] [PubMed] [Google Scholar]
- 10. Russo L, Marsella C, Nardo G et al Transglutaminase 2 transamidation activity during first‐phase insulin secretion: natural substrates in INS‐1E. Acta Diabetol 2013; 50:61–72. [DOI] [PubMed] [Google Scholar]
- 11. Hodrea J, Demeny MA, Majai G, Sarang Z, Korponay‐Szabo IR, Fesus L. Transglutaminase 2 is expressed and active on the surface of human monocyte‐derived dendritic cells and macrophages. Immunol Lett 2010; 130:74–81. [DOI] [PubMed] [Google Scholar]
- 12. Ganguly D, Haak S, Sisirak V, Reizis B. The role of dendritic cells in autoimmunity. Nat Rev Immunol 2013; 13:566–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mackern‐Oberti JP, Llanos C, Vega F et al Role of dendritic cells in the initiation, progress and modulation of systemic autoimmune diseases. Autoimmun Rev 2015; 14:127–39. [DOI] [PubMed] [Google Scholar]
- 14. Price JD, Tarbell KV. The role of dendritic cell subsets and innate immunity in the pathogenesis of type 1 diabetes and other autoimmune diseases. Front Immunol 2015; 6:288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gibson VB, Nikolic T, Pearce VQ, Demengeot J, Roep BO, Peakman M. Proinsulin multi‐peptide immunotherapy induces antigen‐specific regulatory T cells and limits autoimmunity in a humanized model. Clin Exp Immunol 2015; 182:251–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kleijwegt FS, Jansen DT, Teeler J et al Tolerogenic dendritic cells impede priming of naive CD8(+) T cells and deplete memory CD8(+) T cells. Eur J Immunol 2013; 43:85–92. [DOI] [PubMed] [Google Scholar]
- 17. Ferreira GB, Kleijwegt FS, Waelkens E et al Differential protein pathways in 1,25‐dihydroxyvitamin d(3) and dexamethasone modulated tolerogenic human dendritic cells. J Proteome Res 2012; 11:941–71. [DOI] [PubMed] [Google Scholar]
- 18. Franken KL, Hiemstra HS, van Meijgaarden KE et al Purification of his‐tagged proteins by immobilized chelate affinity chromatography: the benefits from the use of organic solvent. Protein Expr Purif 2000; 18:95–9. [DOI] [PubMed] [Google Scholar]
- 19. Zaldumbide A, Alkemade G, Carlotti F et al Genetically engineered human islets protected from CD8‐mediated autoimmune destruction in vivo . Mol Ther 2013; 21:1592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van der Torren CR, Zaldumbide A, Roelen DL et al Innate and adaptive immunity to human beta cell lines: implications for beta cell therapy. Diabetologia 2016; 59:170–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods 2009; 6:359–62. [DOI] [PubMed] [Google Scholar]
- 22. Unger WW, Laban S, Kleijwegt FS, van der Slik AR, Roep BO. Induction of Treg by monocyte‐derived DC modulated by vitamin D3 or dexamethasone: differential role for PD‐L1. Eur J Immunol 2009; 39:3147–59. [DOI] [PubMed] [Google Scholar]
- 23. van Lummel M, van Veelen PA, Zaldumbide A et al Type 1 diabetes associated HLA‐DQ8‐trans dimer accommodates a unique peptide repertoire. J Biol Chem 2012; 287:9514–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van Lummel M, van Veelen PA, de Ru AH et al Discovery of a selective islet peptidome presented by the highest‐risk HLA‐DQ8trans molecule. Diabetes 2016; 65:732–41. [DOI] [PubMed] [Google Scholar]
- 25. Roep BO, Kleijwegt FS, van Halteren AG et al Islet inflammation and CXCL10 in recent‐onset type 1 diabetes. Clin Exp Immunol 2010; 159:338–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nepom GT, Kwok WW. Molecular basis for HLA‐DQ associations with IDDM. Diabetes 1998; 47:1177–84. [DOI] [PubMed] [Google Scholar]
- 27. Fuhlbrigge R, Yip L. Self‐antigen expression in the peripheral immune system: roles in self‐tolerance and type 1 diabetes pathogenesis. Curr Diab Rep 2014; 14:525. [DOI] [PubMed] [Google Scholar]
- 28. Guerder S, Joncker N, Mahiddine K, Serre L. Dendritic cells in tolerance and autoimmune diabetes. Curr Opin Immunol 2013; 25:670–5. [DOI] [PubMed] [Google Scholar]
- 29. Paraskevas S, Maysinger D, Wang R, Duguid TP, Rosenberg L. Cell loss in isolated human islets occurs by apoptosis. Pancreas 2000; 20:270–6. [DOI] [PubMed] [Google Scholar]
- 30. Sahraoui A, Kloster‐Jensen K, Ueland T, Korsgren O, Foss A, Scholz H. Anakinra and tocilizumab enhance survival and function of human islets during culture: implications for clinical islet transplantation. Cell Transplant 2014; 23:1199–211. [DOI] [PubMed] [Google Scholar]
- 31. Vader LW, de Ru A, van der Wal Y et al Specificity of tissue transglutaminase explains cereal toxicity in celiac disease. J Exp Med 2002; 195:643–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Robinson NE, Robinson ZW, Robinson BR et al Structure‐dependent nonenzymatic deamidation of glutaminyl and asparaginyl pentapeptides. J Pept Res 2004; 63:426–36. [DOI] [PubMed] [Google Scholar]
- 33. Wright HT. Nonenzymatic deamidation of asparaginyl and glutaminyl residues in proteins. Crit Rev Biochem Mol Biol 1991; 26:1–52. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Cytokine levels in resting and activated CD4 T cell supernatants. Supernatant from resting (white bars) and activated (black bars) glutamic acid decarboxylase (GAD65)‐restricted T helper cells was measured by Luminex cytokine array.