

Summary
During chronic inflammation, interleukin (IL)‐22 expression is up‐regulated in both CD4 and CD8 T cells, exerting a protective role in infections. However, in autoimmunity, IL‐22 appears to have either a protective or a pathogenic role in a variety of murine models of autoimmunity and, by extrapolation, in humans. It is not clear whether IL‐22 itself mediates inflammation or is a by‐product of inflammation. We have taken advantage of the dominant negative form of transforming growth factor beta receptor type II (dnTGF‐βRII) mice that develop both inflammatory bowel disease and autoimmune cholangitis and studied the role and the biological function of IL‐22 by generating IL‐22–/– dnTGF‐βRII mice. Our data suggest that the influence of IL‐22 on autoimmunity is determined in part by the local microenvironment. In particular, IL‐22 deficiency exacerbates tissue injury in inflammatory bowel disease, but has no influence on either the hepatocytes or cholangiocytes in the same model. These data take on particular significance in the previously defined effects of IL‐17A, IL‐12p40 and IL‐23p19 deficiency and emphasize that, in colitis, there is a dominant role of IL‐23/T helper type 17 (Th17) signalling. Furthermore, the levels of IL‐22 are IL‐23‐dependent. The use of cytokine therapy in patients with autoimmune disease has significant potential, but must take into account the overlapping and often promiscuous effects that can theoretically exacerbate inflammation.
Keywords: cholangitis, colitis, IL‐22, IL‐23, primary biliary cirrhosis
Introduction
Interleukin (IL)‐22 is a cytokine of the IL‐10 family that is produced preferentially by T helper type 17 (Th17) cells. IL‐22 receptors are not expressed on immune cells, but expressed on various tissue cells including liver and intestine 1, thus providing signalling from the immune system to tissue. During chronic inflammation, IL‐22 is up‐regulated highly by both CD4 and CD8 T cells and exerts a protective role against infections 2, 3, 4. Recently, however, emerging evidence indicates that IL‐22 is involved in the development and pathogenesis of autoimmune diseases, including systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), Sjögren's syndrome (SS) and autoimmune hepatitis (AIH) 5, 6, 7. Indeed, aberrant expression of IL‐22 in targeted tissues has been observed in several autoimmune diseases 6, but it is not clear whether IL‐22 itself mediates the inflammation or is a by‐product of the chronic inflammation. In animal models of autoimmune disease, analysis of IL‐22‐deficient mice or inhibition of IL‐22 protein demonstrated that IL‐22 plays either a pathogenic or protective role 8, 9. For example, IL‐22 knock‐out mice were less susceptible to collagen‐induced arthritis than wild‐type mice 10, suggesting that IL‐22 mediates inflammation. Conversely, IL‐22 prevented tissue injury in mice with inflammatory bowel disease 11, 12, and hepatocytes from mice deficient in IL‐22 are highly sensitive to autoimmune hepatitis 9. We postulate that a protective versus a pathological effect of IL‐22 in autoimmune disease may depend upon the distinct tissue microenvironment.
We have reported that mice transgenic for the dominant negative form of transforming growth factor beta receptor type II (dnTGF‐βRII), under the control of the CD4 promoter, develop inflammatory bowel disease (IBD) spontaneously 13. In addition, dnTGF‐βRII mice develop intrahepatic bile duct‐targeted autoimmune disease with substantial similarity to human primary biliary cholangitis (PBC), which is an organ‐specific autoimmune disease characterized by destruction of intrahepatic small bile duct epithelial cells 14. Deletion of IL‐12p40 in dnTGF‐βRII mice, which results in deficiency of both IL‐12 and IL‐23, leads to marked diminution of inflammation in both the liver and the colon, indicating that a highly dysregulated Th1/Th17 cell response leads to the development of autoimmune disease 15. Deletion of IL‐23p19 in dnTGF‐βRII mice resulted in a marked reduction of the Th17 cell population followed by prevention of colitis, but not cholangitis, suggesting a dominant role of IL‐23/Th17 signalling in the pathogenesis of IBD 16. However, deletion of IL‐17A did not suppress either colitis or cholangitis in the dnTGF‐βRII mice, implying that IL‐17A is not necessary for disease development 16. These results prompted us to hypothesize that IL‐22 may play a pathological role in the development of autoimmune disease, as Th17 cells are also a major producer of IL‐22. We demonstrate herein that levels of IL‐22 were increased significantly in dnTGF‐βRII mice and its production is IL‐23‐dependent. To understand further the role of IL‐22 in the dnTGF‐βRII mice model, we generated IL‐22–/– dnTGF‐βRII mice by back‐crossing IL‐22–/– C57/BL6 mice to dnTGF‐βRII mice and evaluated the effect of IL‐22 in the pathogenesis of disease. We demonstrated that depletion of IL‐22 did not affect the severity of cholangitis significantly, but exacerbated colitis compared with the control dnTGF‐βRII mice. In addition, deletion of IL‐22 elicited an enhanced Th1 response. Our data suggest that IL‐22 is a potential protective factor against colitis in this chronic inflammatory model.
Materials and methods
Animals
The dnTGF‐βRII and IL‐22–/– colony on a C57BL/6 background [B6.Cg‐Tg (CD4‐TGF‐βRII)16Flv/J] was maintained at the University of California at Davis (Davis, CA, USA). The C57BL/6 background IL‐23p19–/– mice were generous gifts from Dr Frederic J. de Sauvage (Genentech, South San Francisco, CA, USA). IL‐23p19–/– dnTGF‐βRII mice were generated as described previously 16. For generating IL‐22–/– dnTGF‐βRII mice, male dnTGF‐βRII mice were mated with female IL‐22–/– mice to obtain male IL‐22+/– dnTGF‐βRII mice, which were subsequently back‐crossed with female IL‐22–/– mice to obtain IL‐22–/– dnTGF‐βRII mice. The parental dnTGF‐βRII and the derived IL‐22–/–dnTGF‐βRII mice were genotyped at 3–4 weeks of age to confirm the dnTGF‐βRII and IL‐22–/– genes in their genomic DNA 9, 14. All mice were fed with sterile rodent Helicobacter Medicated Dosing System (three‐drug combination) diets (Bio‐Serv, Frenchtown, NJ, USA) and maintained in individually ventilated cages under specific pathogen‐free conditions. Sulfatrim (Hi‐tech Pharmacal, Amityville, NY, USA) was delivered through drinking water. At 24 weeks of age, animals were killed to collect sera, spleen, liver and colon tissues. The experimental protocols were approved by the University of California Animal Care and Use Committee.
Histopathology
Immediately upon killing, the liver and colon were excised from dnTGF‐βRII mice, fixed in 4% paraformaldehyde, embedded in paraffin, cut into 4‐µm sections, deparaffinized and stained with haematoxylin and eosin (H&E). Pathological changes were evaluated using light microscopy 14, 15. The liver histopathology was graded as: 0, no inflammation (or bile duct damage); 1, mild inflammation (or bile duct damage); 2, moderate inflammation (or bile duct damage); and 3, severe inflammation (or bile duct damage) 15, 16. The colon histopathology was graded as: 0, no significant changes; 1, minimal scattered mucosal inflammatory cell infiltrates, with or without minimal epithelial hyperplasia; 2, mild scattered to diffuse inflammatory cell infiltrates, sometimes extending into the submucosa and associated with erosions, with mild to moderate epithelial hyperplasia and mild to moderate mucin depletion from goblet cells; 3, moderate inflammatory cell infiltrates that were sometimes transmural, with moderate to severe epithelial hyperplasia and mucin depletion; and 4, marked inflammatory cell infiltrates that were often transmural and associated with crypt abscesses and occasional ulceration, with marked epithelial hyperplasia, mucin depletion and loss of intestinal glands 16, 17, 18.
Flow cytometry
Mononuclear cells (MNCs) were isolated from spleen, liver and mesenteric lymph nodes (MLNs), as described previously 16, 18, 19. For cell surface staining, 1 × 106 MNCs were resuspended in staining buffer [0·2% bovine serum albumin (BSA), 0·04% ethylenediamine tetraacetic acid (EDTA) and 0·05% sodium azide in phosphate‐buffered saline (PBS)], divided into 25‐µl aliquots, and incubated with anti‐mouse FcR blocking reagent (eBioscience, San Diego, CA, USA) for 15 min at 4°C. Cells were washed and stained for 30 min at 4°C with cocktails containing combinations of fluorochrome‐conjugated monoclonal antibodies (mAbs) for cell surface markers CD4, CD8α, CD44, CD62L, natural killer (NK)1·1, Gr‐1 (BioLegend, San Diego, CA, USA) and T cell receptor (TCR)‐β, CD11b, CD19 (eBioscience). To evaluate T cell activation, mAbs for CD44 and CD62L (BioLegend) were used. IgG isotype antibodies with matching conjugates (all from BioLegend) were used in parallel as negative controls. The cells were then washed with PBS containing 0·2% BSA (PBS–BSA). For intracellular cytokine staining, 1 × 106 cells were resuspended in 10% fetal bovine serum (FBS) RPMI and stimulated with leucocyte activation cocktail in the presence of BD GolgiPlug (BD Pharmingen, San Diego, CA, USA) at 37°C for 4 h. The cells were washed once with PBS–BSA and then stained for surface CD4, CD8, NK1·1 and TCR‐β, fixed and permeabilized with BD Cytofix/Cytoperm Solution (BD Biosciences, San Diego, CA, USA). The cells were stained for intracellular interferon (IFN)‐γ (BioLegend), as described previously 29. A fluorescence activated cell sorter (FACS)can flow cytometer (BD Immunocytometry Systems, San Jose, CA, USA), upgraded for detection of five colours by Cytek Development (Fremont, CA, USA), was used to acquire data, which were analysed with Cellquest PRO software (BD Immunocytometry Systems).
Evaluation of serum anti‐mitochondrial antibodies (AMA)
Serum anti‐mitochondrial antibodies (AMAs) were detected using the enzyme‐linked immune‐sorbent assay (ELISA) based on recombinant human pyruvate dehydrogenase multi‐enzyme complex (PDC‐E2), as described previously 14, 20. The serum samples were tested at a dilution of 1 : 250. Immunoreactivity was determined by measuring the optical density at 450 nm after incubation with 100 µl of tetramethylbenzidine substrate (BD Biosciences) for 30 min.
Inflammatory cytokine analysis
For analysis of cytokines secreted from cultured T cells, CD4 and CD8 T cells were isolated from spleen MNCs with CD4 (L3T4) and CD8 (Ly‐2) MicroBeads (Miltenyi Biotec Inc., Auburn, CA, USA), respectively. Aliquots of 1·0 × 105 CD4 or CD8 T cells were cultured in 96‐well round‐bottomed plates in 200 µl of RPMI‐1640 supplemented with 10% heat‐inactivated FBS (gibco‐Invitrogen Corp., Grand Island, NY, USA), 100 µg/ml streptomycin, 100 U/ml penicillin and 0·5 µg/ml each of anti‐CD3 (BioLegend) and anti‐CD28 (BioLegend). The cultures were incubated for 72 h at 37°C in a humidified 5% CO2 incubator, then centrifuged to collect supernatants. Similar experiments were performed using MNCs isolated freshly from livers of mice.
Levels of IL‐17A, tumour necrosis factor (TNF)‐α, IL‐6, IL‐10, IL‐4, IL‐2 and IFN‐γ in serum or cell culture supernatant were measured with a cytokine bead array assay using the mouse T helper type 1 (Th1)/Th2/Th17 cytokine kit (BD Biosciences). Levels of IL‐22 were measured using the Quantikine mouse mouse/rat IL‐22 immunoassay kit (R&D Systems, Minneapolis, MN, USA).
Statistical analysis
Two‐tailed unpaired t‐test, one‐way analysis of variance (anova) followed by Bonferroni's multiple‐comparisons test or χ2 test were used for different analyses as appropriate. P‐values < 0·05 were considered statistically significant.
Results
IL‐22 is elevated and IL‐23‐dependent in dnTGF‐βRII mice
Our previous data demonstrated that in addition to IFN‐γ, the level of IL‐22 is increased significantly in dnTGF‐βRII mice 16, 21. To investigate whether IL‐22 expression is induced by IL‐23, we compared serum levels of IL‐22 and IFN‐γ in IL‐23p19–/– dnTGF‐βRII mice with age‐matched dnTGF‐βRII mice. Deletion of IL‐23p19 in the dnTGF‐βRII mice did not affect the level of IFN‐γ but suppressed the production of IL‐22 significantly (Fig. 1a). Hepatic IL‐22 was also decreased significantly in IL‐23p19–/– dnTGF‐βRII liver compared to dnTGF‐βRII mice, reduced to a level comparable to control C57BL/6 mice (Fig. 1b). In contrast, the level of colonic IL‐22 was not affected significantly by depletion of IL‐23p19 in dnTGF‐βRII mice (Fig. 1b). Given that IL‐22 production is not changed in IL‐12p35–/– dnTGF‐βRII mice (as we demonstrated previously) 21, these data indicate that elevated production of IL‐22 in dnTGF‐βRII mice is dependent upon IL‐23.
Figure 1.
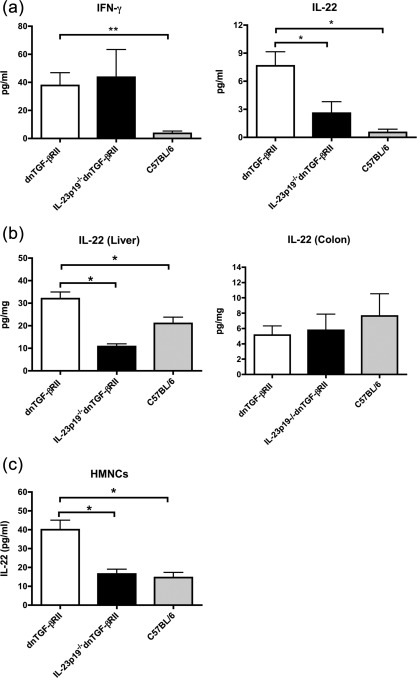
Expression of interleukin (IL)‐22 in dominant negative form of transforming growth factor beta receptor type II (dnTGF‐βRII), IL‐23p19–/– dnTGF‐βRII and C57BL/6 mice. (a) Levels of interferon (IFN)‐γ and IL‐22 in serum of dnTGF‐βRII mice, IL‐23p19–/– dnTGF‐βRII mice and control C57BL/6 mice. (b) IL‐22 level in whole protein lysates of liver and colon from dnTGF‐βRII mice, IL‐23p19–/– dnTGF‐βRII mice and control C57BL/6 mice. Each group in (a) and (b) included 10–11 mice. (c) Concentration of IL‐22 secreted from cultured hepatic mononuclear cells (MNCs). MNCs were isolated from 24‐week‐old dnTGF‐βRII mice, IL‐23p19–/– dnTGF‐βRII mice and control C57BL/6 mice. Cells were cultured with anti‐CD3 and CD28 antibodies for 3 days at 37°C. Each group included three mice. The level of IL‐22 was measured by an enzyme‐linked immunosorbent assay (ELISA). Data are expressed as mean ± standard error of the mean (s.e.m.). *P < 0·05; **P < 0·01 determined by analysis of variance (anova) followed by Bonferroni's multiple‐comparisons test.
To determine further whether the high level of IL‐22 in liver of dnTGF‐βRII mice is due to the infiltrating T cells, we assessed the cytokine production by ex‐vivo stimulation of hepatic mononuclear cells (HMNCs), isolated from dnTGF‐βRII, IL‐23p19–/– dnTGF‐βRII mice and C57BL/6 control littermates. Figure 1c shows that IL‐22 production by HMNCs increased significantly in dnTGF‐βRII mice compared to C57BL/6 mice.
Depletion of IL‐22 did not affect the severity of cholangitis in dnTGF‐βRII mice
To address whether IL‐22 is involved in the pathogenesis of autoimmune cholangitis in dnTGF‐βRII mice, we back‐crossed IL‐22–/– mice with dnTGF‐βRII mice to generate IL‐22–/– dnTGF‐βRII mice. The pathological changes in livers of mice were evaluated at 24 weeks of age. As shown in Fig. 2, the levels of portal inflammation and bile duct damage were not affected by deletion of the IL‐22 gene, while the lobular inflammation was more severe in IL‐22–/– dnTGF‐βRII mice than that of age‐matched dnTGF‐βRII mice (Fig. 2b).
Figure 2.
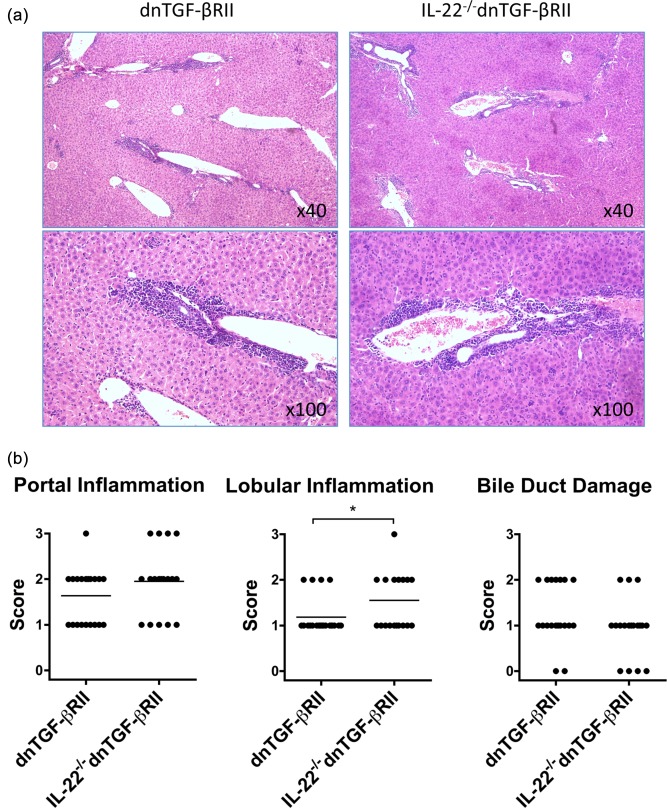
Live histopathology in interleukin (IL)‐22–/– dominant negative form of transforming growth factor beta receptor type II (dnTGF‐βRII) mice. (a) Representative haematoxylin and eosin staining of liver sections. (b) Pathological score of portal inflammation, lobular inflammation and bile duct damage in IL‐22–/– dnTGF‐βRII (n = 20) compared to parental dnTGF‐βRII mice (n = 22). The lobular inflammation score was higher in IL‐22–/– dnTGF‐βRII mice than in dnTGF‐βRII mice. *P < 0·05 determined using χ2 test.
Depletion of IL‐22 exacerbates colitis in dnTGF‐βRII mice
As 24‐week‐old dnTGF‐βRII mice also exhibited chronic inflammatory disorders in their intestines 13, 15, 16, we next compared the severity of colitis in 24‐week‐old IL‐22–/– dnTGF‐βRII mice with age‐matched dnTGF‐βRII mice. As shown in Fig. 3a, the mouse body and colon weights, which are correlated inversely or positively with severity of colitis, respectively, were decreased significantly in IL‐22–/– dnTGF‐βRII mice. More importantly, the levels of colon inflammation and epithelial cell dysplasia were much higher in the colons of IL‐22–/– dnTGF‐βRII mice than dnTGF‐βRII mice (Fig. 3b,c). In addition, a relatively higher incidence of colonic ulcer was also observed in IL‐22–/– dnTGF‐βRII mice (Fig. 3c).
Figure 3.
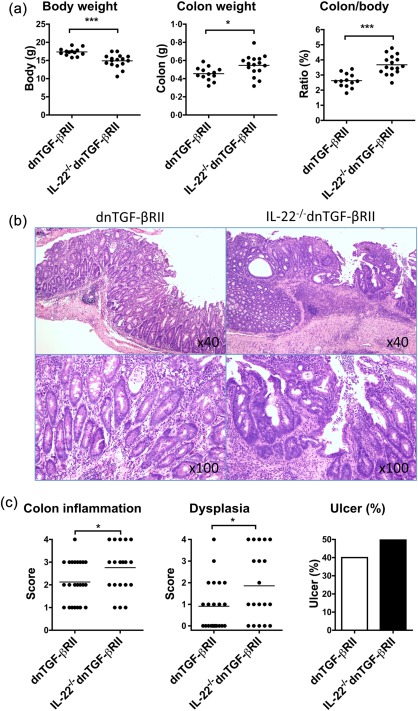
Exacerbation of colitis in interleukin (IL)‐22–/– dominant negative form of transforming growth factor beta receptor type II (dnTGF‐βRII) mice compared to parental dnTGF‐βRII mice. (a) Body and colon weight of IL‐22–/– dnTGF‐βRII mice (n = 13) compared to parental dnTGF‐βRII mice (n = 16). Data are expressed as the mean ± standard error of the mean (s.e.m.). *P < 0·05; ***P < 0·001 determined using two‐tailed unpaired t‐test. (b) Representative histological staining of colon sections. (c) The levels of colon inflammation, colonic dysplasia and ulcers in IL‐22–/– dnTGF‐βRII mice. *P < 0·05 determined using χ2 test.
Increased Th1 response in IL‐22–/– dnTGF‐βRII mice
To address the role of IL‐22 on T cell activation in dnTGF‐βRII mice, mononuclear cells were isolated from the spleen, liver and MLNs of 24‐week‐old mice. The cells were stimulated with phorbol myristate acetate (PMA) and ionomycin and quantified for IFN‐γ expression by intracellular staining. The frequency of IFN‐γ‐producing cells was increased significantly in both CD4 and CD8 T cells from IL‐22–/– dnTGF‐βRII mice regardless of the organs from which cells were isolated (Fig. 4a). Similar results were observed in T cells from the mice at the age of 8 weeks, which was before the onset of colitis (data not shown). These data indicate that depletion of IL‐22 promotes the capability of T cells to produce IFN‐γ in dnTGF‐βRII mice.
Figure 4.
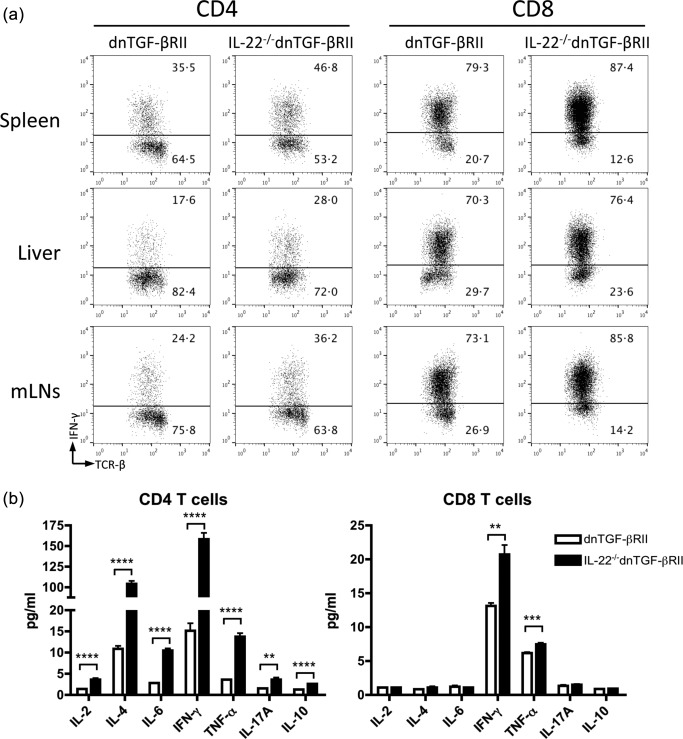
Activation of T cells in interleukin (IL)‐22–/– dominant negative form of transforming growth factor beta receptor type II (dnTGF‐βRII) mice. (a) Frequency of interferon (IFN)‐γ expressing cells within CD4 and CD8 T cell populations isolated from spleen, liver and mesenteric lymph nodes (MLNs) of IL‐22–/–dnTGF‐βRII mice compared with parental dnTGF‐βRII mice. (b) Production of inflammatory cytokines by CD4 and CD8 T cells stimulated with anti‐CD3/CD28 antibodies for 3 days. Each group included three to five mice aged 24 weeks. *P < 0·05; **P < 0·01; ***P < 0·001; ****P < 0·0001 determined using two‐tailed unpaired t‐test.
To evaluate further the function of T cells in inflammatory cytokine production, we isolated CD4 T and CD8 T cells from the spleen of 24‐week‐old IL‐22−/− dnTGF‐βRII mice and age‐matched dnTGF‐βRII mice. The cells were cultured with anti‐CD3/CD28 antibody for 3 days and the supernatants were analysed for concentrations of Th1, Th2 and Th17 cytokines. As shown in Fig. 4b, the levels of secreted IFN‐γ were increased significantly in both CD4 and CD8 T cells from IL‐22–/– dnTGF‐βRII mice compared to those from dnTGF‐βRII mice. Notably, CD4 T cells from IL‐22–/– dnTGF‐βRII mice produced approximately 10‐fold more IFN‐γ and IL‐4 than those from dnTGF‐βRII mice. Although to a much lesser extent, there was also an increase in other cytokines secreted by IL‐22–/– dnTGF‐βRII CD4 T cells, including IL‐2, IL‐6, IL‐10, IL‐17A and TNF‐α (Fig. 4b). In CD8 T cells, only IFN‐γ and TNF‐α were higher in IL‐22–/– dnTGF‐βRII mice than in dnTGF‐βRII mice (Fig. 4b). Compared to CD4 T cells, CD8 T cells produced much less IFN‐γ in IL‐22–/– dnTGF‐βRII mice (ratio of CD4/CD8, IL‐22–/– dnTGF‐βRII versus dnTGF‐βRII: 7·7 versus 1·2). These data suggest that deletion of IL‐22 in dnTGF‐βRII mice enhances the production of Th1‐dominant inflammatory cytokines from the T cells.
We next assessed whether deficiency of IL‐22 causes a change in distribution of immune cells in diseased mice. The frequency of immune cell subpopulations in liver and spleen of IL‐22–/– dnTGF‐βRII mice was compared to that of dnTGF‐βRII mice. The total number of mononuclear cells in liver and spleen did not exhibit differences between the two group mice (data not shown). However, as shown in Fig. 5a, the percentages of CD4 T cells and Gr‐1+/CD11b+ granulocytes were higher, whereas the percentage of NK T cells was much lower, in the spleens of IL‐22–/– dnTGF‐βRII mice than dnTGF‐βRII mice. Analysis of intrahepatic mononuclear cells revealed an increase of CD4 T cells, but not CD8 T cells, in IL‐22–/– dnTGF‐βRII mice (Fig. 5a). In addition, the frequencies of effector (CD44+CD62L–) CD4 and CD8 T cells was not significantly different between IL‐22–/– dnTGF‐βRII and dnTGF‐βRII mice (Fig. 5b), indicating that deletion of the IL‐22 gene did not affect the T cell activation. Because CD8 T cells are the major inflammatory cells destroying biliary epithelial cells, as we demonstrated previously 22, an accumulation of CD4 T cells might be responsible for the higher degree of parenchymal inflammation in the liver of IL‐22–/– dnTGF‐βRII mice.
Figure 5.
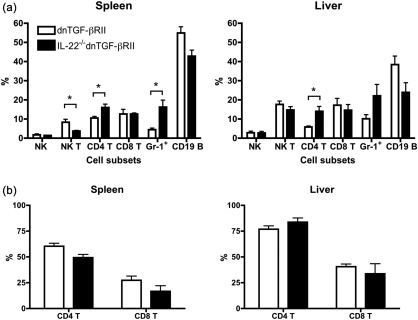
Flow cytometric analysis of the mononuclear cells (MNCs) in spleen and liver of interleukin (IL)‐22–/– dominant negative form of transforming growth factor beta receptor type II (dnTGF‐βRII) mice. (a) Comparison of frequency of immune cell subpopulations in liver and spleen of IL‐22–/– dnTGF‐βRII mice with that of dnTGF‐βRII mice. (b) The frequency of effector (CD44+CD62L−) CD4 and CD8 T cells in liver and spleen of IL‐22–/– dnTGF‐βRII mice. Each group includes five mice. Data are expressed as the mean ± standard error of the mean (s.e.m.). *P < 0·05 determined using two‐tailed unpaired t‐test.
Depletion of IL‐22 did not alter AMA production in dnTGF‐βRII mice
To assess whether IL‐22 regulates autoantibody production, the levels of anti‐PDC‐E2 antibodies were measured in serum of IL‐22–/– dnTGF‐βRII mice. There were no significant differences in the levels of AMA between the IL‐22–/– dnTGF‐βRII and dnTGF‐βRII mice at 24 weeks of age (Fig. 6).
Figure 6.
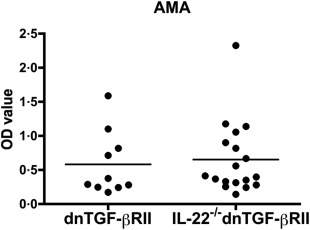
Serum levels of anti‐mitochondrial antibodies (AMA). Serum lever of anti‐ pyruvate dehydrogenase multi‐enzyme complex (PDC‐E2) antibodies in interleukin (IL)‐22–/– dominant negative form of transforming growth factor beta receptor type II (dnTGF‐βRII) mice (n = 18) were compared with parental dnTGF‐βRII mice (n = 10) by a enzyme‐linked immunosorbent assay (ELISA). There was no significant different between the two groups. Horizontal bars represent median values.
Discussion
IL‐22 is important in maintaining mucosal barrier function and tissue homeostasis through its pro‐ and anti‐inflammatory effects 23, 24. IL‐22‐producing CD4 T cells are reduced significantly in actively inflamed tissues compared to both normal tissue and healthy controls in ulcerative colitis patients 25. The depletion of IL‐22 is associated with specific alteration to the bacterial communities of the mucosal microbiota 25. Using animal models of gene knock‐out or inhibition of IL‐22 protein, it has been demonstrated that IL‐22 plays either an inflammatory role or a protective role in the pathogenesis of different autoimmune diseases, which varies depending on the specific disease model 10, 11, 26. In this study utilizing the dnTGF‐βRII mouse model, which develops both colitis and autoimmune cholangitis induced by spontaneously activated T cells, we demonstrated that the depletion of IL‐22 did not affect significantly the severity of portal inflammation and small bile duct damage while mesenchymal inflammation was elevated in their livers. In contrast, the severity of colitis was higher in IL‐22–/– dnTGF‐βRII mice than that in dnTGF‐βRII mice, suggesting a potential protective role of IL‐22 on colitis in this chronic inflammatory model, while the efficiency on cholangitis seems inadequate.
Compared to CD8 T cells, IFN‐γ‐producing CD4 T cells were increased more significantly in IL‐22–/– dnTGF‐βRII mice (Fig. 4), suggesting that the major function of IL‐22 is suppressing the generation of IFN‐γ‐producing CD4 T cells. Indeed, neutralization of IL‐22 resulted in a significant increase of Th1 response and reduced severity of disease significantly in the collagen‐induced arthritis animal model 27. We have demonstrated previously that transfer of CD8 T cells from the dnTGF‐βRII mice into Rag1–/– recipients resulted in autoimmune cholangitis similar to that in the donor mice, while transfer of CD4 T cells led predominantly to colitis 22. These data indicate that CD8 T cells are the major pathogenic effector for development of cholangitis, whereas CD4 T cells are involved potentially in the induction of IBD. Our results in this study are in agreement with this notion. More importantly, the partial protective effect of IL‐22 in dnTGF‐βRII mice may be mediated by suppression of autoactivated IFN‐γ‐producing CD4 T cells.
The pathogenic roles of the IL‐12/Th1 and IL‐23/Th17 signalling pathways have been addressed in the dnTGF‐βRII mouse model by analysis of colitis and cholangitis in different signalling‐deficient mouse strains. Several studies have demonstrated that IL‐12/Th1 immunity is necessary and sufficient for the development of cholangitis in dnTGF‐βRII mice, whereas the IL‐23/Th17 signalling pathway is required for the development of colitis 14, 15, 16, 21, 28, 29. However, we should also note that both IL‐17A and IL‐22 are the major cytokines produced by IL‐23‐triggered Th17 cells 30, 31. As we reported previously, deletion of the IL‐17A gene in dnTGF‐βRII mice does not affect the severity of either cholangitis or colitis 16, indicating that IL‐17 is not a key factor in the pathogenic IL‐23/Th17 axis in the development of colon disease of the dnTGF‐βRII mice. In the current study, we demonstrated further that genetic deletion of IL‐22 did not suppress but, instead, exacerbated colitis in dnTGF‐βRII mice, while the mouse cholangitis was not affected significantly (Figs 2 and 3). Our data indicate that IL‐22 is not a pathogenic factor for the development of IBD in dnTGF‐βRII mice. Therefore, which signalling driven by IL‐23 contributes to the induction of colitis needs to be explored further. In fact, the production of several inflammatory cytokines such as TNF‐α, IL‐2, IL‐6 and IL‐10 are suppressed in IL‐23p19–/– dnTGF‐βRII mice 16, whereas the expression of these cytokines are increased in IL‐22–/– dnTGF‐βRII mice (Fig. 4). These cytokines may contribute to autoimmunity targeted towards intestine epithelial cells. In addition, the contribution of IL‐17 to colitis cannot be excluded, as the production of IL‐17 by CD4 T cells from IL‐22–/– dnTGF‐βRII mice was elevated, as shown in Fig. 4.
Recently, we have examined the role of IL‐22 in an inducible animal model of cholangitis, in which mice developed PBC‐like disease after immunization with xenobiotic mimic, i.e. 2‐octynoic acid (2OA)‐BSA 32, 33, 34. We demonstrated that 2OA‐BSA‐immunized IL‐22–/– mice had significantly decreased cholangitis compared to wild‐type mice 32. Although the IFN‐γ‐ and IL‐17A‐producing T cells were not affected in the IL‐22–/– mice after immunization 32, it appears that IL‐22 plays a proinflammatory role in the induction of cholangitis. In the current study, however, depletion of IL‐22 in dnTGF‐βRII mice did not change the extent of cholangitis in liver while the colitis was exacerbated. These contradictory effects of IL‐22 on cholangitis are probably dependent upon a different disease model. In addition, deletion of the IL‐22 gene or over‐expression of IL‐22 in mice may cause distinct effects on disease development. For example, over‐expression of IL‐22 in mice with IL‐22 expressing adeno‐associated virus vector (AAV‐IL‐22) reduced significantly the portal inflammatory response and biliary cell damage in xenobiotics‐immunized mice. The potential role of IL‐22 is not only in protecting mice from autoimmune cholangitis, but also in treating animals with established portal inflammation 35. The levels of Th1 cytokines such as IFN‐γ and TNF‐α were also decreased significantly in livers of the AAV‐IL‐22‐treated mice 35. These data show clearly the protective role of IL‐22 on cholangitis, although such effects are not clear in the dnTGF‐βRII mouse model, as we demonstrated in the current study. Additionally, we should note that IL‐22 also exacerbated chronic liver inflammation as exhibited, for example, in acetaminophen‐induced liver injury and the hepatitis B virus (HBV) transgenic mouse model 36, 37.
These data take on significance in PBC in which, thus far, there has not been a successful translation of the critical immunological and molecular data in both human and murine models towards new therapy 38, 39, 40, 41, 42, 43, 44, 45, 46. In addition, we should note that further attention should be placed not only on the local tissue microenvironment, but also on innate immunity, not only in PBC but also in other autoimmune diseases 35, 47, 48, 49. We recognize that IL‐22 has both pro‐ and anti‐inflammatory functions, which will be a key issue for development of a novel therapeutic approach based on this molecule.
Disclosure
None.
Acknowledgements
The authors thank Dr Chen‐Yen Yang for taking care of the animal ecology and technical support in this experiment. We also thank Ms Nikki Phipps for support in preparing this article. Funding was provided by National Institutes of Health grant, DK090019.
References
- 1. Kotenko SV, Izotova LS, Mirochnitchenko OV et al Identification of the functional interleukin‐22 (IL‐22) receptor complex: the IL‐10R2 chain (IL‐10Rbeta) is a common chain of both the IL‐10 and IL‐22 (IL‐10‐related T cell‐derived inducible factor, IL‐TIF) receptor complexes. J Biol Chem 2001; 276:2725–32. [DOI] [PubMed] [Google Scholar]
- 2. Aujla SJ, Kolls JK. IL‐22: a critical mediator in mucosal host defense. J Mol Med (Berl) 2009; 87:451–4. [DOI] [PubMed] [Google Scholar]
- 3. Ortega C, Fernandez AS, Carrillo JM et al IL‐17‐producing CD8+ T lymphocytes from psoriasis skin plaques are cytotoxic effector cells that secrete Th17‐related cytokines. J Leukoc Biol 2009; 86:435–43. [DOI] [PubMed] [Google Scholar]
- 4. Eyerich S, Eyerich K, Pennino D et al Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J Clin Invest 2009; 119:3573–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang X, Zheng SG. Interleukin‐22: a likely target for treatment of autoimmune diseases. Autoimmun Rev 2014; 13:615–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pan HF, Li XP, Zheng SG, Ye DQ. Emerging role of interleukin‐22 in autoimmune diseases. Cytokine Growth Factor Rev 2013; 24:51–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pan CX, Tang J, Wang XY, Wu FR, Ge JF, Chen FH. Role of interleukin‐22 in liver diseases. Inflamm Res 2014; 63:519–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sugimoto K, Ogawa A, Mizoguchi E et al IL‐22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest 2008; 118:534–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Karow M, Flavell RA. Interleukin‐22 but not interleukin‐17 provides protection to hepatocytes during acute liver inflammation. Immunity 2007; 27:647–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Geboes L, Dumoutier L, Kelchtermans H et al Proinflammatory role of the Th17 cytokine interleukin‐22 in collagen‐induced arthritis in C57BL/6 mice. Arthritis Rheum 2009; 60:390–5. [DOI] [PubMed] [Google Scholar]
- 11. Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin‐22 protects mice from inflammatory bowel disease. Immunity 2008; 29:947–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Radaeva S, Sun R, Pan HN, Hong F, Gao B. Interleukin 22 (IL‐22) plays a protective role in T cell‐mediated murine hepatitis: IL‐22 is a survival factor for hepatocytes via STAT3 activation. Hepatology 2004; 39:1332–42. [DOI] [PubMed] [Google Scholar]
- 13. Gorelik L, Flavell RA. Abrogation of TGF‐beta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity 2000; 12:171–81. [DOI] [PubMed] [Google Scholar]
- 14. Oertelt S, Lian ZX, Cheng CM et al Anti‐mitochondrial antibodies and primary biliary cirrhosis in TGF‐beta receptor II dominant‐negative mice. J Immunol 2006; 177:1655–60. [DOI] [PubMed] [Google Scholar]
- 15. Yoshida K, Yang GX, Zhang W et al Deletion of interleukin‐12p40 suppresses autoimmune cholangitis in dominant negative transforming growth factor beta receptor type II mice. Hepatology 2009; 50:1494–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ando Y, Yang GX, Tsuda M et al The immunobiology of colitis and cholangitis in interleukin‐23p19 and interleukin‐17a deleted dominant negative form of transforming growth factor beta receptor type II mice. Hepatology 2012; 56:1418–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moritoki Y, Lian ZX, Lindor K et al B‐cell depletion with anti‐CD20 ameliorates autoimmune cholangitis but exacerbates colitis in transforming growth factor‐beta receptor II dominant negative mice. Hepatology 2009; 50:1893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moritoki Y, Zhang W, Tsuneyama K et al B cells suppress the inflammatory response in a mouse model of primary biliary cirrhosis. Gastroenterology 2009; 136:1037–47. [DOI] [PubMed] [Google Scholar]
- 19. Lian ZX, Okada T, He XS et al Heterogeneity of dendritic cells in the mouse liver: identification and characterization of four distinct populations. J Immunol 2003; 170:2323–30. [DOI] [PubMed] [Google Scholar]
- 20. Dhirapong A, Yang GX, Nadler S et al Therapeutic effect of cytotoxic T lymphocyte antigen 4/immunoglobulin on a murine model of primary biliary cirrhosis. Hepatology 2013; 57:708–15.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tsuda M, Zhang W, Yang GX et al Deletion of interleukin (IL)‐12p35 induces liver fibrosis in dominant‐negative TGF‐beta receptor type II mice. Hepatology 2013; 57:806–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang GX, Lian ZX, Chuang YH et al Adoptive transfer of CD8(+) T cells from transforming growth factor beta receptor type II (dominant negative form) induces autoimmune cholangitis in mice. Hepatology 2008; 47:1974–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL‐22. Nat Immunol 2011; 12:383–90. [DOI] [PubMed] [Google Scholar]
- 24. Pickert G, Neufert C, Leppkes M et al STAT3 links IL‐22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med 2009; 206:1465–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Leung JM, Davenport M, Wolff MJ et al IL‐22‐producing CD4+ cells are depleted in actively inflamed colitis tissue. Mucosal Immunol 2014; 7:124–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sonnenberg GF, Monticelli LA, Alenghat T et al Innate lymphoid cells promote anatomical containment of lymphoid‐resident commensal bacteria. Science 2012; 336:1321–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Justa S, Zhou X, Sarkar S. Endogenous IL‐22 plays a dual role in arthritis: regulation of established arthritis via IFN‐gamma responses. PLOS ONE 2014; 9:e93279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ando Y, Yang GX, Kenny TP et al Overexpression of microRNA‐21 is associated with elevated pro‐inflammatory cytokines in dominant‐negative TGF‐beta receptor type II mouse. J Autoimmun 2013; 41:111–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kawata K, Yang GX, Ando Y et al Clonality, activated antigen‐specific CD8 T cells, and development of autoimmune cholangitis in dnTGF‐betaRII mice. Hepatology 2013; 58:1094–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of T(H)17 cells. Nature 2008; 453:1051–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bettelli E, Carrier Y, Gao W et al Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006; 441:235–8. [DOI] [PubMed] [Google Scholar]
- 32. Kawata K, Tsuda M, Yang GX et al Identification of potential cytokine pathways for therapeutic intervention in murine primary biliary cirrhosis. PLOS ONE 2013; 8:e74225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wakabayashi K, Lian ZX, Leung PS et al Loss of tolerance in C57BL/6 mice to the autoantigen E2 subunit of pyruvate dehydrogenase by a xenobiotic with ensuing biliary ductular disease. Hepatology 2008; 48:531–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wakabayashi K, Yoshida K, Leung PS et al Induction of autoimmune cholangitis in non‐obese diabetic (NOD).1101 mice following a chemical xenobiotic immunization. Clin Exp Immunol 2009; 155:577–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hsueh YH, Chang YN, Loh CE, Gershwin ME, Chuang YH. AAV‐IL‐22 modifies liver chemokine activity and ameliorates portal inflammation in murine autoimmune cholangitis. J Autoimmun 2016; 66:89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhao J, Zhang Z, Luan Y et al Pathological functions of interleukin‐22 in chronic liver inflammation and fibrosis with hepatitis B virus infection by promoting T helper 17 cell recruitment. Hepatology 2014; 59:1331–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Feng D, Wang Y, Wang H et al Acute and chronic effects of IL‐22 on acetaminophen‐induced liver injury. J Immunol 2014; 193:2512–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Beuers U, Gershwin ME. Unmet challenges in immune‐mediated hepatobiliary diseases. Clin Rev Allergy Immunol 2015; 48:127–31. [DOI] [PubMed] [Google Scholar]
- 39. Floreani A, Franceschet I, Perini L, Cazzagon N, Gershwin ME, Bowlus CL. New therapies for primary biliary cirrhosis. Clin Rev Allergy Immunol 2015; 48:263–72. [DOI] [PubMed] [Google Scholar]
- 40. Huang W, Kachapati K, Adams D et al Murine autoimmune cholangitis requires two hits: cytotoxic KLRG1(+) CD8 effector cells and defective T regulatory cells. J Autoimmun 2014; 50:123–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Katsumi T, Tomita K, Leung PS, Yang GX, Gershwin ME, Ueno Y. Animal models of primary biliary cirrhosis. Clin Rev Allergy Immunol 2015; 48:142–53. [DOI] [PubMed] [Google Scholar]
- 42. Lleo A, Zhang W, McDonald WH et al Shotgun proteomics: identification of unique protein profiles of apoptotic bodies from biliary epithelial cells. Hepatology 2014; 60:1314–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang L, Sun Y, Zhang Z et al CXCR5+ CD4+ T follicular helper cells participate in the pathogenesis of primary biliary cirrhosis. Hepatology 2015; 61:627–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang YH, Yang W, Yang JB et al Systems biologic analysis of T regulatory cells genetic pathways in murine primary biliary cirrhosis. J Autoimmun 2015; 59:26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yao Y, Yang W, Yang YQ et al Distinct from its canonical effects, deletion of IL‐12p40 induces cholangitis and fibrosis in interleukin‐2Ralpha(‐/‐) mice. J Autoimmun 2014; 51:99–108. [DOI] [PubMed] [Google Scholar]
- 46. Zhang J, Zhang W, Leung PS et al Ongoing activation of autoantigen‐specific B cells in primary biliary cirrhosis. Hepatology 2014; 60:1708–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang CY, Ma X, Tsuneyama K et al IL‐12/Th1 and IL‐23/Th17 biliary microenvironment in primary biliary cirrhosis: implications for therapy. Hepatology 2014; 59:1944–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hudspeth K, Donadon M, Cimino M et al Human liver‐resident CD56(bright)/CD16(neg) NK cells are retained within hepatic sinusoids via the engagement of CCR5 and CXCR6 pathways. J Autoimmun 2016; 66:40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shimoda S, Hisamoto S, Harada K et al Natural killer cells regulate T cell immune responses in primary biliary cirrhosis. Hepatology 2015; 62:1817–27. [DOI] [PMC free article] [PubMed] [Google Scholar]