

Abstract
Although most prostate cancer (PCa) cases are not life-threatening, approximately 293 000 men worldwide die annually due to PCa. These lethal cases are thought to be caused by coordinated genomic alterations that accumulate over time. Recent genome-wide analyses of DNA from subjects with PCa have revealed most, if not all, genetic changes in both germline and PCa tumor genomes. In this article, I first review the major, somatically acquired genomic characteristics of various subtypes of PCa. I then recap key findings on the relationships between genomic alterations and clinical parameters, such as biochemical recurrence or clinical relapse, metastasis and cancer-specific mortality. Finally, I outline the need for, and challenges with, validation of recent findings in prospective studies for clinical utility. It is clearer now than ever before that the landscape of somatically acquired aberrations in PCa is highlighted by DNA copy number alterations (CNAs) and TMPRSS2-ERG fusion derived from complex rearrangements, numerous single nucleotide variations or mutations, tremendous heterogeneity, and continuously punctuated evolution. Genome-wide CNAs, PTEN loss, MYC gain in primary tumors, and TP53 loss/mutation and AR amplification/mutation in advanced metastatic PCa have consistently been associated with worse cancer prognosis. With this recently gained knowledge, it is now an opportune time to develop DNA-based tests that provide more accurate patient stratification for prediction of clinical outcome, which will ultimately lead to more personalized cancer care than is possible at present.
Keywords: copy number, DNA markers, fusion, mutation, outcome, prognostic, prostate cancer, tumor genome
INTRODUCTION
Globally, PCa is the most common cancer among men, with an estimated 1 442 000 new cases in 2013.1,2 While most PCa patients have an indolent form of the disease that may not require treatment, about 10%–20% of men affected with PCa have an aggressive form of the disease that may progress to metastases and death, thus requiring more intensive therapies. The inability to reliably distinguish between these two forms of PCa early in the course of the disease has resulted in overtreatment of many and undertreatment of some cases.3,4,5 This is because all patients with PCa are treated similarly in most cases even though the underlying genetic causes of their PCa tumors are likely different.
With high-resolution microarrays and next-generation sequencing (NGS), significant advances have recently been made in genetic dissections of both germline and prostate tumor genome, which is characterized by diverse somatic mutations and pathway alterations6,7,8,9,10,11 derived from complex DNA rearrangements.12,13,14 Each tumor genome harbors genetic aberrations affecting numerous genes as the result of successive clonal expansion and punctuated evolution.13,15,16,17,18,19,20 While most of these affected genes may be neutral “passengers,” aberrations of multiple “drivers” cause increased cell proliferation and PCa progression.21,22,23 Moreover, chromothripsis and chromoplexy with catastrophic and chained genomic rearrangements,12,13,24,25 respectively, suggest that multiple cancer genes are often altered coordinately. These massive and coordinated genetic alterations affect the expression of many genes, which define the fate of PCa.
Delineating the genomic and molecular characteristics of various types of PCa at different stages can improve our ability to understand the mechanisms that drive reemergence as aggressive PCa from indolent tumors and progression of malignant tumors to advanced stages. Analyzing the relationships between genomic alterations and clinical parameters may lead to better molecular markers for more accurate patient stratification and personalized cancer care. Due to a broad coverage of topics by other articles in this special issue, this review primarily focuses on somatically acquired aberrations of genomic DNA in the tumor genome and highlights their potentials for use as molecular biomarkers for prognosis and prediction of clinical outcome.
LANDSCAPE OF GENOMIC ALTERATIONS IN CLINICALLY LOCALIZED PROSTATE CANCER
Detailed genomic alterations in PCa have been the subject of extensive reviews recently for their biological and therapeutic implications.26,27,28,29,30,31,32 This work aims to briefly highlight the major somatic alterations with potential to distinguish aggressive from nonaggressive PCa.
DNA copy number alterations
Studies on genome-wide analyses of PCa have revealed that DNA copy number alterations (CNAs) are a major component of the landscape alterations in the tumor genome. The most frequently detected CNAs in primary tumors include amplifications of genomic regions containing oncogenic MYC (8q24.21, 10%–40%), and deletions of DNA sequences harboring tumor suppressor genes such as NKX3-1 (8p21.2, 40%–70%), PTEN (10q23.31, 10%–40%), CDKN1B (12p13.1, 20%–30%), RB1 (13q14.2, 30%–50%), and TP53 (17p13.1, 20%–30%). In addition, CNAs of many other genes with various biological functions have also been reported in many cohorts.7,33,34,35,36 It is notable that the frequencies of these CNAs vary among different tumor samples, depending on cohort composition, tumor grade, degree of tumor cell heterogeneity, and pathological stage, thus reflecting the complex roles of these CNAs in PCa. However, the major CNA regions across the whole genome among different cohorts are remarkably similar (Figure 1), suggesting a core CNA commonality of most PCa.
Figure 1.

Significant genomic deletions (blue) and amplifications (red) in the tumor genomes from two independent cohorts of patients with clinically localized PCa. JHH: Johns Hopkins cohort, SWD: Swedish cohort.
With genomic deletions outnumbering gains, the majority of the deletions affecting tumor suppressor genes involves the loss of only one copy. It is speculated that either these genes are haploinsufficient or these hemizygous deletions are eventually complemented by additional genetic and/or epigenetic alterations in another copy of the gene during cancer progression. For example, NKX3-1 and LPL were reported to be affected by both deletion and methylation.37,38 The combination of a hemizygous germline frameshift in one copy and an acquired somatic deletion of the other copy was found to inactivate BRCA2.19,39 The combination of a somatic point mutation in one copy and a hemizygous deletion of another was reported to abolish PTEN and TP53.39,40
Homozygous deletions causing loss of both copies of the sequence have been observed in many genes. The most common homozygously deleted genes are PTEN (~15%) and CHD1 (~10%) in PCa.41 Others include BNIP3L (8p21.1), LRP1B (2q22.1), RB1 (13q14.2), USP10 (16q24.1), HTR3A (11q23.2), RYPB (3p13), MAP3K7 (6q15), TP53 (17p13.1), CDKN1B (12p13.1), and miR-15a/miR-16-1 (13q14.2) with much lower frequencies.41,42 While most gains are hemizygous, amplifications with more than two extra copies involved both chromosomes, primarily on 8q24 including MYC, are also reported in PCa.43
Distinct CNA profiles across the tumor genome have been observed among patients of different races. Castro et al. reported that CNAs at six regions in clinically localized tumors from 20 African American (AA) men resembled those in metastatic cancers from Caucasian American (CA) men.44 Rose et al. identified four CNAs with significantly higher frequencies in AA than in CA men.45 The frequencies of deletions between TMPRSS2 and ERG that create a fusion of these two genes, as well as PTEN loss, are found to be significantly lower in AA, Chinese, Japanese, and Korean PCa populations than in CA.46,47,48,49,50,51,52
Single nucleotide variations (SNVs)
Before NGS, targeted sequencing identified many point mutations (SNVs in somatic tissues) in selected genes thought to be biologically important for cancer development.6,7 It has been reported that PCa harbors the fewest point mutations (~0.33–1.4 per Mb) among the major human cancers.6,9 However, relatively high frequencies of SNVs at TP53 (12%), PTEN (7%), SPOP (7%), KRAS (4%), FOXA1 (3%), KMT2C (3%), EGFR (3%), and CTNNB1 (3%) have been documented (Figure 2). Systematic analyses of SNVs across the tumor genome using exome and whole genome NGS have revealed a comprehensive mutation landscape of clinically localized PCa.9,11,12,13,14,53 While the majority of these mutations is observed in a relatively small proportion of tumors, many genes, including SPOP, FOXA1, TP53, PTEN, CDKN1B, MED12, THSD7B, SCN11A, NIPA2, PIK3CA, ZNF595, C14orf49, CDC27, MLL3, KDM6A, and KIF5A, are considered to be significantly mutated. SPOP and TP53 are the most frequently mutated genes with 10%–15% across different cohorts in primary PCa.9,11
Figure 2.
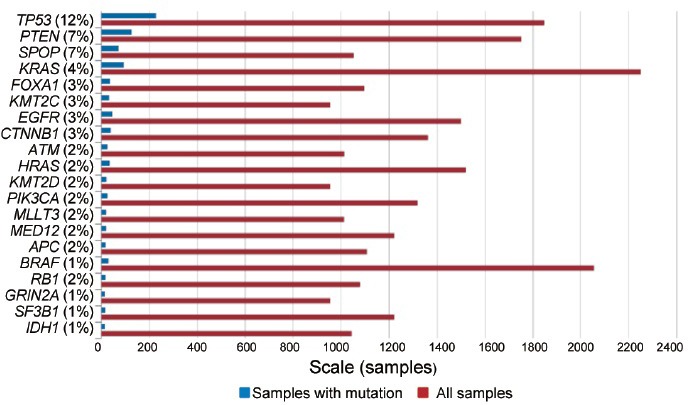
Top 20 genes mutated in prostate carcinoma as January 31, 2016 (http://www.sanger.ac.uk/genetics/CGP/cosmic).
The mutation landscape and biological implications of mutated genes in PCa have been reviewed extensively.27,28,30,31,32,54,55,56,57 Although drugs have been developed to target specific pathways of some mutations, such as the PI3K/Akt/mTOR signaling pathway in tumors with PTEN and PIK3CA mutations, these significantly mutated genes have not been systematically investigated for their clinical utility in prognosis and predication of cancer progression or lethal PCa. This is partially due to the lower frequencies of mutated genes (in comparison to CNAs), small cohorts used in PCa genomic studies, and tremendous heterogeneity of PCa. Notably, there were only two overlapping genes identified as being significantly mutated in two independent cohorts of primary tumors analyzed by exome sequencing.9,11 In addition, the detected frequency of gene mutations depends on the depth used in NGS and the sampled tissue location because of intra- and inter-tumor heterogeneity.16,18,19,53,58
Genomic subtypes of primary tumors
Since the discovery of TMPRSS2-ERG fusion transcripts caused by genomic DNA translocation59 and deletion,35,60 many studies have been carried out to investigate recurrent gene fusions and their roles in PCa (see reviews).61,62 The fusion between TMPRSS2 and E26 transformation-specific (ETS) family genes, observed in ~50% of PCa patients, is the most commonly acquired genetic alteration defining a distinct subtype of PCa. Moreover, there is a significant correlation between ERG rearrangements and PTEN loss. For example, Carver et al. observed a reduction or absence of PTEN expression in14 of 15 tumors with TMPRSS2-ERG fusion among 40 cases (P = 0.007).21 King et al. found that 14 of 17 tumors with PTEN deletion also harbored ERG rearrangements in 121 cases (P = 0.002).22 Analyzing specimens from 281 patients, Han et al. demonstrated a significant association between PTEN deletions and ERG rearrangements (P = 0.0008).63 Reid et al. observed a significant association between PTEN loss and ERG/ETV1 rearrangements among 308 patients (P < 0.001).64 Bismar et al. reported that 71% of homozygous and 44% of hemizygous PTEN deletions were concurrent with ERG rearrangements among 220 patients.65 Krohn et al. showed that PTEN deletion was significantly associated with ERG fusion among 2177 tumors (P = 0.0001).66 Recently, Qi et al. also reported a significant correlation between PTEN deletion and ERG rearrangements in a cohort of 176 Chinese PCa patients (P = 0.0008).67 These findings support the notion that a synergistic cooperation between PTEN deletion and ERG rearrangement drives the development and progression of this subtype of PCa.21,22,68,69
Another major subtype of PCa is characterized by CHD1 deletions and/or SPOP mutations in the tumor genome.8,9,41,70,71,72,73 Searching for homozygous deletions among a total of 244 primary tumors, Liu et al. discovered that CHD1 was second only to PTEN as the most frequent (~10%) homozygously deleted gene in PCa.41 More importantly, none of the 21 subjects with CHD1 homozygous deletions harbored a deletion from the 3’ end of TMPRSS2 to the 5’ end of ERG. Barbieri et al. found that somatic deletions at 5q21 including CHD1 significantly associated with SPOP mutation in a cohort with 112 tumors.9 Interestingly, all of the SPOP mutations were exclusively observed in the tumors without TMPRSS2-ERG fusion. Analyzing the relationship between CHD1 and ETS status in 13 studies with a total of 945 cancers, Grasso et al. demonstrated a significant negative correlation between loss of CHD1 and ETS rearrangements (P < 0.0001).8 Using fluorescence in situ hybridization (FISH) to analyze the status of CHD1 and ERG in 2093 tumors, Burkhardt et al. confirmed the significant negative association between CHD1 deletion and ERG fusion (P < 0.0001).71 Investigating SPOP mutations among 720 PCa samples from six international cohorts with Caucasian, AA, and Asian subjects, Blattner et al. further validated the inverse correlation between ERG rearrangements and SPOP mutations or CHD1 deletion.70
In addition to SPOP mutation, tumors with deletions of CHD1 are also enriched with deletions of LRP1B (2q22), PDE4D (5q11), MAP3K7 (6q15), FOXO3, and PRDM1 (6q21) and gain of COL1A2 (7q21).9,41 Loss of CHD1 has been reported to be associated with a larger number of homozygous deletions at other locations in the tumor genome41 and an increased number of CNAs.13 Using whole genome NGS, Baca et al. found that tumors with CHD1 deletions harbored significantly more intrachromosomal DNA rearrangements in the regions with low GC content. In contrast, tumors with ETS fusions contained more interchromosomal rearrangements in locations with active transcriptional hubs.13 Burkhardt et al. demonstrated that in vitro inactivation of CHD1 impaired androgen receptor (AR)-dependent transcription required for translocation, thereby preventing ERG rearrangements.71
GENOMIC ALTERATIONS IN ADVANCED METASTATIC PROSTATE CANCER
The majority of primary PCa cases is not life-threatening. However, approximately 293 000 men worldwide die annually from metastases of advanced PCa. While the initial driver of cancer cell dissemination from the prostate to other organs in untreated subjects is unknown, androgen deprivation therapy (ADT) apparently leads to metastatic castration-resistant prostate cancer (mCRPC) in most cases. Although a new generation of ADT agents, including enzalutamide74,75 and abiraterone acetate,76,77 has been proven to improve survival of patients with mCRPC, it is thought that cancers in nearly all affected subjects may eventually develop resistance to these new drugs via genomic alterations.39,40,77,78,79,80,81 Therefore, the genomic landscape of advanced PCa, in addition to those inherited from primary tumors, is also shaped, in part, by selective pressure applied to cancer cells from treatment. The landscape of advanced PCa, highlighted by increased alterations of AR, TP53, and PTEN7,8,10,17,19,39 and circulating tumor cells/DNA (CTC/ctDNA),82,83,84,85,86,87,88,89 has recently been reviewed for clinical and therapeutic implications.26,27,28,31,90,91,92,93,94,95 This section briefly outlines the major genomic aberrations and focuses on the genes that currently have potential for outcome prediction.
Lethal metastases are originally derived from primary tumors in the prostate although various subclones and multiclonal seeding have been documented in both clinically localized and distant metastatic PCa.16,17,18,19,20,40,53,58 Genome-wide comparative genomic hybridization (CGH) has demonstrated that advanced metastatic PCa harbors similar CNAs, to those seen in primary tumors, but at much higher frequencies.7,8,20,36,96 Analyzing DNA copy number in lethal PCa, Liu et al. reported that multiple distant metastases from anatomically distinct sites and tumors in the prostate shared a core profile of CNAs in the same subjects while each also accumulated a variable number of separate subclonally sustained changes.20 Exome sequencing mCRPC from 150 individuals, Robinson et al. found that more than 40% of tumors had genetic alterations, including 27% with homozygous deletion of, as well as a number of mutations in, PTEN.39 Comparing all gene-coding sequences between tumors and matched normal tissues, Barbieri et al. and Grasso et al. identified 12 and 9 significantly mutated genes in clinically localized9 and castration-resistant8 PCa, respectively. Importantly, PTEN and TP53 are the only genes significantly mutated in both of these two distinct types of PCa. To track the origin of lethal PCa, Haffner et al. analyzed three anatomically distinct autopsy metastases of liver, perigastric lymph node, and lung and nine lesions in the prostate obtained 17 years prior via radical prostatectomy (RP) from a subject who died of PCa.18 They discovered that the lethal clone was derived from a small, low-grade focus harboring PTEN deletion and TP53 mutation, rather than from the bulk, higher-grade primary cancer or from a lymph node metastasis that did not have these gene alterations. Using ultra-deep sequencing, Hong et al. demonstrated that PCa metastases were enriched with TP53 mutations that apparently drove subclonal expansion.19 Moreover, Ferraldeschi et al. found that loss of PTEN was associated with a shorter time on abiraterone treatment and worse patient survival for those with mCRPC,97 possibly via resistance to ADT.98 These findings emphasize the broad roles of these alterations in cancer initiation, progression, and treatment resistance.
AR amplification and mutation characterize 30%–80% of advanced metastatic PCa in various cohorts. Using FISH to analyze AR in transurethral resection of the prostate (TURP) from 23 subjects treated with ADT, Visakorpi et al. reported that 7 (30%) had AR amplification while none showed AR amplification from the same subjects before therapy.99 Correspondingly, Koivisto et al. found that 28% of therapy-resistant tumors, but none of the untreated primary tumors, contained AR amplification in tumor specimens obtained via biopsy and TURP from 54 subjects.100 Using SNP array to analyze multiple distant metastases from anatomically distinct sites in each subject from a cohort of 14 lethal PCa cases, Liu et al. found only two individuals harboring a normal single copy of AR in all metastatic sites. Seven subjects showed gains of two to eight copies and five subjects showed AR amplification ranging from 9 to 40 copies.20 In contrast, they observed no AR gain in primary tumors in a separate study of 228 untreated subjects from two independent cohorts.101 Similarly, Friedlander et al. reported that 11 of 15 of the metastatic samples harbored AR amplification.96 Using exome NGS to analyze 50 pre-treated mCRPCs obtained by rapid autopsy, Grasso et al. found 30 harboring AR amplification and/or protein-coding mutations.8 Moreover, Beltran et al. reported that 44% of 25 mCRPC cases had AR alterations.102 Robinson et al. observed 63% of subjects with AR amplification and/or missense mutations in a prospective study cohort of 150 treated mCRPC patients.39 These findings suggest that amplification and mutation of AR are primarily resulted from AR-related therapies.
Defects in DNA repair are apparently the initial cause of genome-wide alterations and subsequent genomic instability, with higher frequencies in advanced metastatic PCa. Biallelic BRCA2 inactivation is reported in more than 10% of mCRPC, with ~5% damaging germline mutations.39,102 Correspondingly, subjects carrying BRCA2 mutations have a significantly higher PCa-specific mortality rate and shorter median survival time.103,104,105,106 In addition, aberrations of the mismatch repair genes MSH2 and MSH6, which are associated with hypermutation, have been found at higher frequencies in advanced metastatic PCa cases39,107 although changes in MSH2 and MSH6 are rarely observed in untreated primary tumors.7,9,11 Pritchard et al. showed that 7 of 60 (12%) patients with advanced PCa had structural rearrangements in MSH2 and MSH6, with all being hypermutated and microsatellite instable.107 Robinson et al. reported that 2% of 150 patients with mCRPC harbored MSH2 alterations.39 Tracking the origin and driver of subclonal expansion in a longitudinal study, Hong et al. identified MSH2 alterations in both the original clone of a primary tumor and the clone of a shoulder metastasis.19 However, whether defects of these genes lead to the dissemination of a lethal clone is warranted for further investigation.
IMPLICATIONS OF DNA-BASED MARKERS FOR PROGNOSIS OF CLINICAL OUTCOME
With the unprecedented amounts of genomic data generated from the tumor genome as mentioned above, there is hope for identifying genomic markers that can distinguish aggressive from indolent PCa at the time of diagnosis, as well as genes that drive cancer progression. Because there is no consensus on the definition of aggressiveness, this section primarily focuses on DNA alterations associated with clinical outcome in PCa.
CNAs associated with biochemical recurrence (BCR)/clinical relapse of PCa
BCR/clinical relapse of PCa is the most common characteristic of clinical outcomes that have been investigated for association with CNAs.7,34,66,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130 The most frequently investigated candidate genes include PTEN on 10q23.31 and MYC on 8q24.21 (Table 1). Significant associations between PTEN loss and PCa recurrence have been reported in multiple cohorts.66,109,123,127,128 In other cohorts, loss of PTEN alone by FISH is not significantly associated with BCR111,130 whereas loss of PTEN by immunohistochemistry (IHC) is significantly associated with BCR.111,115,131,132,133,134,135,136 The inconsistency of these observations is likely derived from variations in cohort composition, including homozygous versus hemizygous deletions, and the interval to PCa recurrence after treatments such as prostatectomy and radiotherapy.
Table 1.
Significant CNAs associated with BCR/clinical relapse of PCa
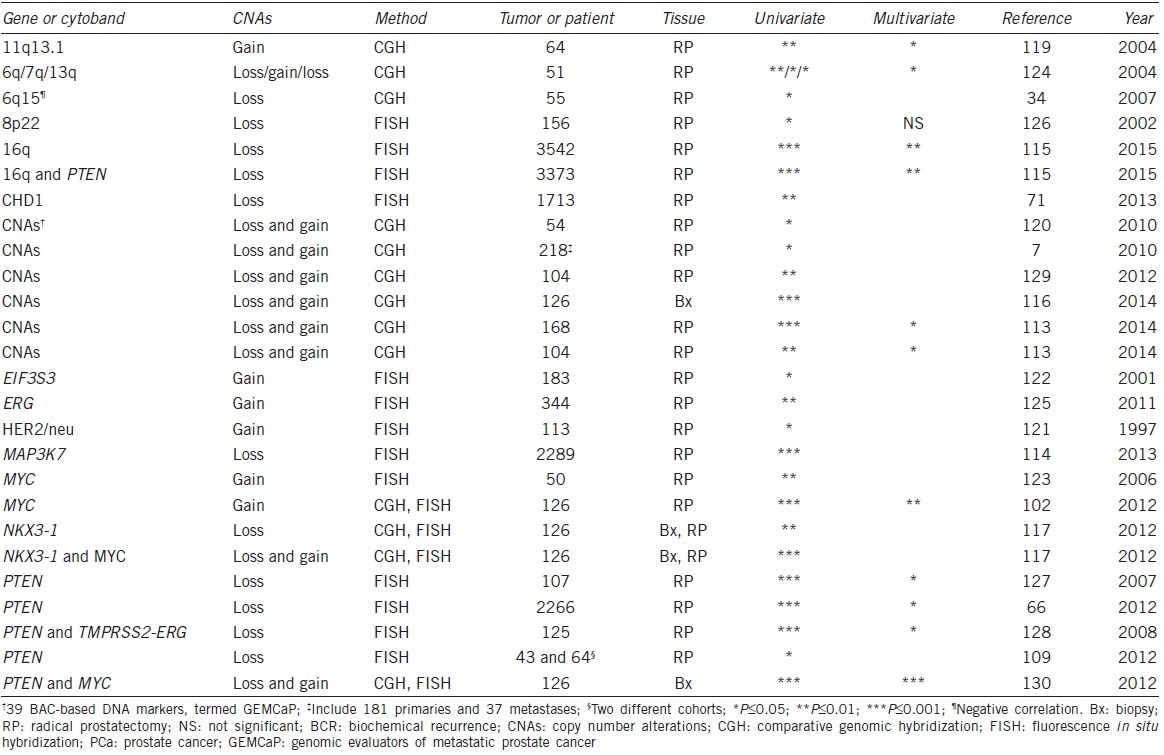
MYC gain, either itself or together with other CNAs, is found to be associated with an earlier recurrence of PCa.117,123,130 This is consistent with findings that patients with increased MYC expression have an earlier disease relapse.123,133,137,138 In addition, the combined gain of MYC and loss of either PTEN or NKX3.1 have been reported to be better prognostic predictors of PCa relapse after radiotherapy.117,130 Similarly, concurrent loss of PTEN and loss of 16q or fusion of TMPRSS2-ERG are reported to contribute to independent prognostic information regarding BCR.115,128
Genome-wide CNAs have been consistently reported to be associated with PCa relapse.7,66,113,116,120,124,129 Using an unsupervised hierarchical clustering of CNAs identified by the Agilent 244K CGH among 181 primary and 37 metastatic tumors, Taylor et al. classified PCa into 6 distinct groups, with some significantly associated with BCR.7 Extending this study with a contemporary cohort containing 104 patients, Hieronymus et al. demonstrated that CNA burden, measured as the percentage of tumor genome affected, predicted PCa recurrence independent of histopathological parameters.113 For example, patients with CNAs ≥1.34% had a 29%–38% risk of BCR within 5 years as opposed to a 13%–15% risk for those harboring CNAs <1.34%. Lalonde et al. reported that every 1% increase of CNAs in the tumor genome led to a 5%–8% decrease of 5-year BCR-free survival.116 These findings are consistent with Strohmeyer et al.‘s observation that the total number of deletions was significantly higher in patients with PCa relapse than in those without although no difference in total number of gains was observed between progressors and nonprogressors.124 Selected multiple genomic CNAs across the tumor genome are also demonstrated to be significantly associated with PCa relapse.116,120,129 Using a Genomic Evaluators of Metastatic Prostate Cancer (GEMCaP) score consisting of 39 CNAs, Paris et al. were able to increase the area under the receiver operating characteristic curve (AUC) up to 0.85 in prediction of PCa recurrence.120 A 100-locus CNA signature has been claimed to be prognostic for disease recurrence in patients treated with radiotherapy and prostatectomy.116 Moreover, it helps identify subjects (with an AUC of 0.68) who are most likely to fail initial treatment in 18 months with BCR, which has been considered a robust prognostic factor for PCa mortality after radiotherapy.139
Furthermore, CNA signature has been demonstrated to provide prognostic information beyond RNA-based profiling for PCa relapse.113,116 For example, Hieronymus et al. reported that CNAs outperformed an RNA-based cell cycle progression signature for BCR in Gleason 7 tumors while both of these signatures were independently associated with PCa relapse.113 In addition, CHD1 deletion is reported to be associated with BCR71,140 while there is no correlation between SPOP mutation and BCR.70 Systematic investigation on the relationship between CHD1 deletion or SPOP mutation and clinical outcomes, such as PCa recurrence, metastasis, and cancer-specific death, are apparently needed although knockdown of CHD1/MAP3K7 has been reported to alter cell morphology, increase cell invasiveness, and promote aggressive PCa.41,71,72,141
CNAs associated with metastasis or mortality of PCa
Metastasis is a more definitive clinical outcome parameter than BCR for lethal PCa because most patients with distant metastasis eventually die from the disease. However, the relationship between CNAs and metastasis has only been reported in a few studies. Paris et al. identified 40 CNAs in primary tumors that appear to be associated with metastasis.119 The percentage of CNAs in the tumor genome as a continuous variable is also reported to be significantly associated with metastasis in one (168 primary tumors) of two cohorts that Hieronymus et al. investigated.113 PTEN copy number loss is not associated with metastasis in this cohort although PTEN protein loss by IHC is observed to be associated with decreased time to PCa metastasis in another cohort.142 Conversely, using FISH, Krohn et al. found that PTEN hemizygous and homozygous deletions were significantly correlated with lymph node metastasis in a cohort with 1111 tumors.66
The association between CNAs and PCa-specific mortality has been investigated in a number of cohorts (Table 2).64,67,101,123,143,144,145,146,147,148,149 Using FISH to study the relationship between CNAs on chromosome 8 and clinical outcome, three independent groups found that gains of MYC were significantly associated with systematic disease progression and earlier PCa-specific death.123,146,147 Using the Affymetrix 6.0 SNP array to analyze normal and tumor DNA from 125 high-risk subjects, Liu et al. uncovered seven regions in the tumor genome that were significantly associated with lethal PCa.101 These included gains at 8q24.21 (MYC), 1q21.3 (ADAR), and 8q21.13 (TPD52) and deletions at 18q21.33 (SERPINB5), 16q24.1 (USP10), 10q23.31 (PTEN), and 17p13.1 (TP53). Among these CNAs, gains of MYC conferred the greatest risk of dying from PCa with an OR of 4.75, consistent with previous findings.
Table 2.
Significant CNAs associated with mortality of PCa
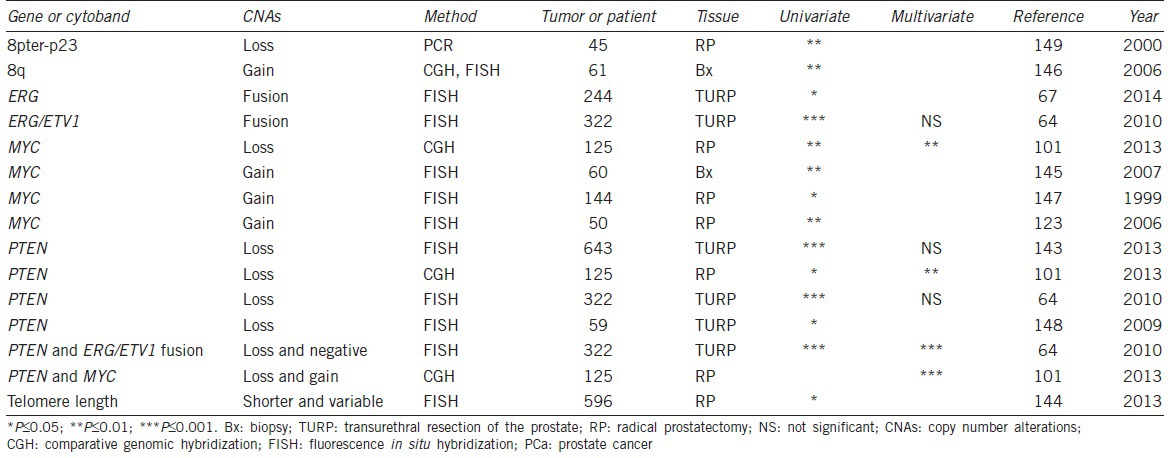
Significant association between PTEN deletion and PCa-specific death has been documented by four independent groups.64,101,143,148 In a cohort of 59 patients with hormone refractory PCa, Sircar et al. found that PTEN was deleted in 77% of the cases, including 25% homozygous, 34% hemizygous, and 18% mixture deletions, and correlated with cancer-specific mortality.148 In a TURP cohort with 643 patients, Cuzick et al. observed significant associations of both deletion and amplification of PTEN with PCa-specific death.143 Further analysis indicated that patients with homozygous deletions were at greater risk of dying from PCa. This is consistent with the finding that loss of PTEN expression is significantly associated with an increased risk of PCa-specific mortality.134,150 In addition, Reid et al. reported that patients harboring PTEN deletions in the absence of ERG/ETV rearrangements carried a significantly higher risk of dying from PCa in both univariate and multivariate analyses.64 Moreover, using a model that incorporated both genetic markers and clinicopathologic variables in a multivariate logistic regression analysis, Liu et al. showed that the CNAs of PTEN and MYC conferred additional independent prognostic information.101 Compared to patients without either PTEN or MYC alterations, patients with both alterations had a 53-fold higher risk for dying from the disease. This study is the first to demonstrate a stronger joint effect of PTEN and MYC on PCa-specific mortality. An important aspect of this study is the further confirmation of the joint effect of PTEN loss and MYC gain in 333 tumors from three additional distinct patient cohorts.101
Association between ETS fusions and clinical outcome of PCa
The relationship between TMPRSS2-ERG fusion and clinical outcome has been investigated extensively. However, conflicting, even opposite, results are reported. For example, using FISH and qPCR to analyze TURP samples from 111 patients in a watchful waiting cohort, Demichelis et al. identified 15% harboring ERG rearrangements associated with PCa-specific deaths.151 Analyzing TURP specimens from 445 conservatively managed patients, Attard et al. found that subjects harboring a duplication of the fusion together with interstitial deletion of ERG exhibited extremely poor survival after 8 years.152 Similarly, Qi et al. demonstrated that ERG rearrangements were associated with cancer-related death in 224 Chinese patients using TURP samples.67 These data are consistent with the findings that higher ERG expression, usually caused by fusion, is significantly associated with BCR, distant metastasis or PCa-specific mortality.153,154,155,156,157
Conversely, using FISH to analyze samples from 521 cases treated by RP, Gopalan et al. demonstrated that TMPRSS2-ERG rearrangement was not associated with BCR, metastasis or mortality.112 Toubaji et al. found that TMPRSS2-ERG fusion was not associated with PCa relapse in a cohort containing 172 patients with and 172 patients without recurrence after prostatectomy.125 FitzGerald et al. observed no correlation between ERG rearrangements and cancer-specific death in a population of 214 tumors analyzed by FISH.158 In addition, Fleischmann et al. reported that TMPRSS2-ERG fusion was correlated with favorable BCR-free survival in a cohort of 119 surgically treated tumors.159 Similarly, Saramaki et al. demonstrated that ERG fusion was associated with longer progression-free survival in 150 patients treated by prostatectomy.160 These results are consistent with the findings that ERG overexpression either is not associated with cancer relapse or mortality or is correlated with better BCR-free survival.161,162,163,164,165,166
The conflicting and opposite findings described above may be derived from a number of factors, including cohort composition, techniques used to detect the fusion, and other confounding alterations in the tumor samples. Because TMPRSS2-ERG fusion is significantly associated with PTEN loss that correlates with worse clinical outcomes as reviewed in previous sections, prognostic values of the fusion are likely confounded by PTEN status in various cohorts.140 Indeed, Reid et al. found that ERG rearrangement alone or PTEN loss alone was not correlated with patient survival in multivariate analyses. However, patients with PTEN loss in the absence of the fusion exhibited a significantly poorer cancer-specific survival rate than those harboring PTEN loss and ERG rearrangement.64
FUTURE PERSPECTIVES
Although many studies have been performed to explore the utility of DNA alterations for prognosis of clinical outcome via association, survival, and receiver operating characteristic analyses as reviewed above, none has been systematically validated in a Clinical Laboratory Improvement Amendments (CLIA) lab. In contrast, many new biomarkers, including RNA-based genomic predictors, have been validated for commercial use.167,168 Clinical application of genomic DNA-based prognostic markers has lagged expression-based molecular markers even though they hold some advantages such as robustness to degradation and remaining constant despite physiological and environmental fluctuations.
The clinical validity and utility of DNA-based alterations associated with PCa outcome need to be fully assessed by systematic investigations with proper design to address a number of confounding factors in prospective studies. These include (1) intra- and inter-tumor (focal) heterogeneity,16,18,19,53,58 (2) field effects of genomic alterations,16 (3) choice and availability of tissues (e.g., biopsy, TURP, RP, CTC/ctDNA, etc.) and sampling bias, (4) composition of patient cohort (e.g., untreated patients with primary tumors at active surveillance [AS] or RP, patients with advanced PCa treated various drugs), (5) stratifications of age, family history, race, other genomic changes, and environmental factors, and (6) technology for detecting alterations and standardization of experimental protocols.
Taking heterogeneity, for example, Cooper et al. identified mutations at high levels in morphologically normal tissues distant from the cancer site and multiple genetically distinct clones in a single tumor mass using NGS with an average depth of 10 000 Χ.16 They also documented a number of somatic mutations in the prostate that were not detected in tumor or blood samples from the same patient. Investigating tumor origin, Lindberg et al. observed no apparent common somatic mutations in different tumor foci of the same prostate among three of four patients.58 Similarly, Boutros et al. reported multiclonal tumor foci with no shared CNAs and very few shared SNVs in the prostate of the same individuals.53 These findings on heterogeneity complicate unbiased sampling for validating the clinical utilities of genomic biomarkers. Therefore, extensive retrospective and prospective investigations are warranted to track and validate driver alterations using longitudinal studies and clonal analyses,16,20,40,53 to assess their associations with clinical outcomes, and to assess their potential utilities for personalized prognosis.
Genome-wide CNAs, PTEN loss, MYC gain in primary tumors, and TP53 loss/mutation and AR amplification/mutation in advanced metastatic PCa have been consistently observed to be associated with poorer clinical outcome. Further investigations are necessary to validate their utilities as genomic markers for distinguishing aggressive PCa from indolent tumors, especially in AS patients, because RP was the major source of DNA used for discovery of the associations with BCR, metastasis, and mortality. Due to the complexity of genomic alterations, it is unlikely that a common set of genomic markers can be used for diagnosis of aggressive PCa, prognosis of outcome after surgery, and monitoring/prediction of adjuvant therapy. Therefore, a distinct set of genomic markers is needed for a particular subtype (e.g., ERG+ vs CHD−) of PCa at a specific stage (e.g., AS vs RP vs mCRPC) of the disease in an appropriate subpopulation (e.g., Caucasian vs African vs Asian men). While genome-wide analysis is feasible for RP patients due to ample amount of available tissues, targeted genetic analysis seems more practical using tissue samples obtained from biopsies and CTC/ctDNA samples for AS and mCRPC patients, respectively, until whole genome analysis becomes robust and cost effective.
In addition to association, AUC by receiver operating characteristic analysis and positive and negative predictive values of these genomic markers should be evaluated for prognostic accuracy and their utilities in clinical settings. Although new technological tools, such as NGS and high-throughput genotyping, are widely adapted by clinical labs, use of these tools for genomic prognosis/prediction of clinical outcome currently adds significant costs. Therefore, inexpensive/robust methods and simple/effective algorithms must be developed for common clinical practice until higher-cost technology with more complex analyses is proven necessary for added clinical benefits.
Furthermore, analytical validity of DNA-based genomic predictors in a given method must be assessed. This includes precision and reproducibility, accuracy and trueness, sensitivity and limit of detection, specificity, and interference. To maximize clinical benefit, various sets of cutoff criteria and algorithms for clinical prediction and analytical validity using genomic DNA profiling at different stages must be developed to minimize the risks of underestimating cancer progression, and vice versa.
CONCLUSIONS
Unprecedented amounts of data from recent genome-wide analyses of various tumor genomes now enable a better understanding of the relationships between specific genomic alterations and clinical characteristics. As a result, new DNA-based prognostic markers have been discovered with the potential for distinguishing aggressive PCa from indolent tumors and predicting clinical outcome. Validation of the utility of these genomic markers in prospective studies with proper experimental design is warranted before they can be used clinically as predictors of cancer outcomes. While DNA-based markers hold advantages over and complement expression-based markers, their real contributions to clinical practice and their net benefits to PCa patients must be the subjects of long-term prospective investigations in clinical settings.
COMPETING INTERESTS
The author has no competing interests to declare.
ACKNOWLEDGMENTS
This work was partially supported by Department of Defense grant W81XWH-12-1-0188 to WL. I would like to thank Carly Conran for editing this manuscript.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Fitzmaurice C, Dicker D, Pain A, Hamavid H, et al. Global Burden of Disease Cancer Collaboration. The global burden of cancer 2013. JAMA Oncol. 2015;1:505–27. doi: 10.1001/jamaoncol.2015.0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Delpierre C, Lamy S, Kelly-Irving M, Molinie F, Velten M, et al. Life expectancy estimates as a key factor in over-treatment: the case of prostate cancer. Cancer Epidemiol. 2013;37:462–8. doi: 10.1016/j.canep.2013.03.014. [DOI] [PubMed] [Google Scholar]
- 4.Lee YJ, Park JE, Jeon BR, Lee SM, Kim SY, et al. Is prostate-specific antigen effective for population screening of prostate cancer. A systematic review? Ann Lab Med. 2013;33:233–41. doi: 10.3343/alm.2013.33.4.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moyer VA. U.S. Preventive Services Task Force. Screening for prostate cancer: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2012;157:120–34. doi: 10.7326/0003-4819-157-2-201207170-00459. [DOI] [PubMed] [Google Scholar]
- 6.Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869–73. doi: 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- 7.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–43. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44:685–9. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumar A, White TA, MacKenzie AP, Clegg N, Lee C, et al. Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc Natl Acad Sci U S A. 2011;108:17087–92. doi: 10.1073/pnas.1108745108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindberg J, Mills IG, Klevebring D, Liu W, Neiman M, et al. The mitochondrial and autosomal mutation landscapes of prostate cancer. Eur Urol. 2013;63:702–8. doi: 10.1016/j.eururo.2012.11.053. [DOI] [PubMed] [Google Scholar]
- 12.Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–20. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, et al. Punctuated evolution of prostate cancer genomes. Cell. 2013;153:666–77. doi: 10.1016/j.cell.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weischenfeldt J, Simon R, Feuerbach L, Schlangen K, Weichenhan D, et al. Integrative genomic analyses reveal an androgen-driven somatic alteration landscape in early-onset prostate cancer. Cancer Cell. 2013;23:159–70. doi: 10.1016/j.ccr.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 15.Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469:356–61. doi: 10.1038/nature09650. [DOI] [PubMed] [Google Scholar]
- 16.Cooper CS, Eeles R, Wedge DC, Van Loo P, Gundem G, et al. Analysis of the genetic phylogeny of multifocal prostate cancer identifies multiple independent clonal expansions in neoplastic and morphologically normal prostate tissue. Nat Genet. 2015;47:367–72. doi: 10.1038/ng.3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JM, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520:353–7. doi: 10.1038/nature14347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haffner MC, Mosbruger T, Esopi DM, Fedor H, Heaphy CM, et al. Tracking the clonal origin of lethal prostate cancer. J Clin Invest. 2013;123:4918–22. doi: 10.1172/JCI70354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hong MK, Macintyre G, Wedge DC, Van Loo P, Patel K, et al. Tracking the origins and drivers of subclonal metastatic expansion in prostate cancer. Nat Commun. 2015;6:6605. doi: 10.1038/ncomms7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu W, Laitinen S, Khan S, Vihinen M, Kowalski J, et al. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat Med. 2009;15:559–65. doi: 10.1038/nm.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carver BS, Tran J, Gopalan A, Chen Z, Shaikh S, et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet. 2009;41:619–24. doi: 10.1038/ng.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.King JC, Xu J, Wongvipat J, Hieronymus H, Carver BS, et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat Genet. 2009;41:524–6. doi: 10.1038/ng.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ding Z, Wu CJ, Chu GC, Xiao Y, Ho D, et al. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature. 2011;470:269–73. doi: 10.1038/nature09677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu C, Wyatt AW, McPherson A, Lin D, McConeghy BJ, et al. Poly-gene fusion transcripts and chromothripsis in prostate cancer. Genes Chromosomes Cancer. 2012;51:1144–53. doi: 10.1002/gcc.21999. [DOI] [PubMed] [Google Scholar]
- 26.Attard G, Parker C, Eeles RA, Schröder F, Tomlins SA, et al. Prostate cancer. Lancet. 2016;387:70–82. doi: 10.1016/S0140-6736(14)61947-4. [DOI] [PubMed] [Google Scholar]
- 27.Barbieri CE, Bangma CH, Bjartell A, Catto JW, Culig Z, et al. The mutational landscape of prostate cancer. Eur Urol. 2013;64:567–76. doi: 10.1016/j.eururo.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beltran H, Rubin MA. New strategies in prostate cancer: translating genomics into the clinic. Clin Cancer Res. 2013;19:517–23. doi: 10.1158/1078-0432.CCR-12-1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dean M, Lou H. Genetics and genomics of prostate cancer. Asian J Androl. 2013;15:309–13. doi: 10.1038/aja.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang J, Wang JK, Sun Y. Molecular pathology of prostate cancer revealed by next-generation sequencing: opportunities for genome-based personalized therapy. Curr Opin Urol. 2013;23:189–93. doi: 10.1097/MOU.0b013e32835e9ef4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roychowdhury S, Chinnaiyan AM. Advancing precision medicine for prostate cancer through genomics. J Clin Oncol. 2013;31:1866–73. doi: 10.1200/JCO.2012.45.3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schoenborn JR, Nelson P, Fang M. Genomic profiling defines subtypes of prostate cancer with the potential for therapeutic stratification. Clin Cancer Res. 2013;19:4058–66. doi: 10.1158/1078-0432.CCR-12-3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lapointe J, Li C, Giacomini CP, Salari K, Huang S, et al. Genomic profiling reveals alternative genetic pathways of prostate tumorigenesis. Cancer Res. 2007;67:8504–10. doi: 10.1158/0008-5472.CAN-07-0673. [DOI] [PubMed] [Google Scholar]
- 35.Liu W, Chang B, Sauvageot J, Dimitrov L, Gielzak M, et al. Comprehensive assessment of DNA copy number alterations in human prostate cancers using affymetrix100K SNP mapping array. Genes Chromosomes Cancer. 2006;45:1018–32. doi: 10.1002/gcc.20369. [DOI] [PubMed] [Google Scholar]
- 36.Sun JS, Liu WN, Adams TS, Sun JL, Li XN, et al. DNA copy number alterations in prostate cancers: a combined analysis of published CGH studies. Prostate. 2007;67:692–700. doi: 10.1002/pros.20543. [DOI] [PubMed] [Google Scholar]
- 37.Asatiani E, Huang WX, Wang A, Rodriguez Ortner E, Cavalli LR, et al. Deletion, methylation, and expression of the NKX3.1 suppressor gene in primary human prostate cancer. Cancer Res. 2005;65:1164–73. doi: 10.1158/0008-5472.CAN-04-2688. [DOI] [PubMed] [Google Scholar]
- 38.Kim JW, Cheng Y, Liu W, Li T, Yegnasubramanian S, et al. Genetic and epigenetic inactivation of LPL gene in human prostate cancer. Int J Cancer. 2009;124:734–8. doi: 10.1002/ijc.23972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–28. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carreira S, Romanel A, Goodall J, Grist E, Ferraldeschi R, et al. Tumor clone dynamics in lethal prostate cancer. Sci Transl Med. 2014;6:254ra125. doi: 10.1126/scitranslmed.3009448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu W, Lindberg J, Sui G, Luo J, Egevad L, et al. Identification of novel CHD1-associated collaborative alterations of genomic structure and functional assessment of CHD1 in prostate cancer. Oncogene. 2012;31:3939–48. doi: 10.1038/onc.2011.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Porkka KP, Ogg EL, Saramaki OR, Vessella RL, Pukkila H, et al. The miR-15a-miR-16-1 locus is homozygously deleted in a subset of prostate cancers. Genes Chromosomes Cancer. 2011;50:499–509. doi: 10.1002/gcc.20873. [DOI] [PubMed] [Google Scholar]
- 43.Liu W, Xie CC, Zhu Y, Li T, Sun J, et al. Homozygous deletions and recurrent amplifications implicate new genes involved in prostate cancer. Neoplasia. 2008;10:897–907. doi: 10.1593/neo.08428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Castro P, Creighton CJ, Ozen M, Berel D, Mims MP, et al. Genomic profiling of prostate cancers from African American men. Neoplasia. 2009;11:305–12. doi: 10.1593/neo.81530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rose AE, Satagopan JM, Oddoux C, Zhou Q, Xu R, et al. Copy number and gene expression differences between African American and Caucasian American prostate cancer. J Transl Med. 2010;8:70. doi: 10.1186/1479-5876-8-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khani F, Mosquera JM, Park K, Blattner M, O’Reilly C, et al. Evidence for molecular differences in prostate cancer between African American and Caucasian men. Clin Cancer Res. 2014;20:4925–34. doi: 10.1158/1078-0432.CCR-13-2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee K, Chae JY, Kwak C, Ku JH, Moon KC. TMPRSS2-ERG gene fusion and clinicopathologic characteristics of Korean prostate cancer patients. Urology. 2010;76:1268–e7-13. doi: 10.1016/j.urology.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 48.Magi-Galluzzi C, Tsusuki T, Elson P, Simmerman K, LaFargue C, et al. TMPRSS2-ERG gene fusion prevalence and class are significantly different in prostate cancer of Caucasian, African-American and Japanese patients. Prostate. 2011;71:489–97. doi: 10.1002/pros.21265. [DOI] [PubMed] [Google Scholar]
- 49.Mao X, Yu Y, Boyd LK, Ren G, Lin D, et al. Distinct genomic alterations in prostate cancers in Chinese and western populations suggest alternative pathways of prostate carcinogenesis. Cancer Res. 2010;70:5207–12. doi: 10.1158/0008-5472.CAN-09-4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miyagi Y, Sasaki T, Fujinami K, Sano J, Senga Y, et al. ETS family-associated gene fusions in Japanese prostate cancer: analysis of 194 radical prostatectomy samples. Mod Pathol. 2010;23:1492–8. doi: 10.1038/modpathol.2010.149. [DOI] [PubMed] [Google Scholar]
- 51.Ren S, Peng Z, Mao JH, Yu Y, Yin C, et al. RNA-seq analysis of prostate cancer in the Chinese population identifies recurrent gene fusions, cancer-associated long noncoding RNAs and aberrant alternative splicings. Cell Res. 2012;22:806–21. doi: 10.1038/cr.2012.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xue L, Mao X, Ren G, Stankiewicz E, Kudahetti SC, et al. Chinese and western prostate cancers show alternate pathogenetic pathways in association with ERG status. Am J Cancer Res. 2012;2:736–44. [PMC free article] [PubMed] [Google Scholar]
- 53.Boutros PC, Fraser M, Harding NJ, de Borja R, Trudel D, et al. Spatial genomic heterogeneity within localized, multifocal prostate cancer. Nat Genet. 2015;47:736–45. doi: 10.1038/ng.3315. [DOI] [PubMed] [Google Scholar]
- 54.Barbieri CE, Demichelis F, Rubin MA. Molecular genetics of prostate cancer: emerging appreciation of genetic complexity. Histopathology. 2012;60:187–98. doi: 10.1111/j.1365-2559.2011.04041.x. [DOI] [PubMed] [Google Scholar]
- 55.Dong JT. Prevalent mutations in prostate cancer. J Cell Biochem. 2006;97:433–47. doi: 10.1002/jcb.20696. [DOI] [PubMed] [Google Scholar]
- 56.Febbo PG. Genomic approaches to outcome prediction in prostate cancer. Cancer. 2009;115:3046–57. doi: 10.1002/cncr.24350. [DOI] [PubMed] [Google Scholar]
- 57.Kopper L, Timar J. Genomics of prostate cancer: is there anything to “translate”? Pathol Oncol Res. 2005;11:197–203. doi: 10.1007/BF02893851. [DOI] [PubMed] [Google Scholar]
- 58.Lindberg J, Klevebring D, Liu W, Neiman M, Xu J, et al. Exome sequencing of prostate cancer supports the hypothesis of independent tumour origins. Eur Urol. 2013;63:347–53. doi: 10.1016/j.eururo.2012.03.050. [DOI] [PubMed] [Google Scholar]
- 59.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–8. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 60.Hermans KG, van Marion R, van Dekken H, Jenster G, van Weerden WM, et al. TMPRSS2:ERG fusion by translocation or interstitial deletion is highly relevant in androgen-dependent prostate cancer, but is bypassed in late-stage androgen receptor-negative prostate cancer. Cancer Res. 2006;66:10658–63. doi: 10.1158/0008-5472.CAN-06-1871. [DOI] [PubMed] [Google Scholar]
- 61.Kumar-Sinha C, Tomlins SA, Chinnaiyan AM. Recurrent gene fusions in prostate cancer. Nat Rev Cancer. 2008;8:497–511. doi: 10.1038/nrc2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rubin MA. ETS rearrangements in prostate cancer. Asian J Androl. 2012;14:393–9. doi: 10.1038/aja.2011.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Han B, Mehra R, Lonigro RJ, Wang L, Suleman K, et al. Fluorescence in situ hybridization study shows association of PTEN deletion with ERG rearrangement during prostate cancer progression. Mod Pathol. 2009;22:1083–93. doi: 10.1038/modpathol.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reid AH, Attard G, Ambroisine L, Fisher G, Kovacs G, et al. Molecular characterisation of ERG, ETV1 and PTEN gene loci identifies patients at low and high risk of death from prostate cancer. Br J Cancer. 2010;102:678–84. doi: 10.1038/sj.bjc.6605554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bismar TA, Yoshimoto M, Vollmer RT, Duan Q, Firszt M, et al. PTEN genomic deletion is an early event associated with ERG gene rearrangements in prostate cancer. BJU Int. 2011;107:477–85. doi: 10.1111/j.1464-410X.2010.09470.x. [DOI] [PubMed] [Google Scholar]
- 66.Krohn A, Diedler T, Burkhardt L, Mayer PS, De Silva C, et al. Genomic deletion of PTEN is associated with tumor progression and early PSA recurrence in ERG fusion-positive and fusion-negative prostate cancer. Am J Pathol. 2012;181:401–12. doi: 10.1016/j.ajpath.2012.04.026. [DOI] [PubMed] [Google Scholar]
- 67.Qi M, Yang X, Zhang F, Lin T, Sun X, et al. ERG rearrangement is associated with prostate cancer-related death in Chinese prostate cancer patients. PLoS One. 2014;9:e84959. doi: 10.1371/journal.pone.0084959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bismar TA, Yoshimoto M, Duan Q, Liu S, Sircar K, et al. Interactions and relationships of PTEN, ERG, SPINK1 and AR in castration-resistant prostate cancer. Histopathology. 2012;60:645–52. doi: 10.1111/j.1365-2559.2011.04116.x. [DOI] [PubMed] [Google Scholar]
- 69.Zong Y, Xin L, Goldstein AS, Lawson DA, Teitell MA, et al. ETS family transcription factors collaborate with alternative signaling pathways to induce carcinoma from adult murine prostate cells. Proc Natl Acad Sci U S A. 2009;106:12465–70. doi: 10.1073/pnas.0905931106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Blattner M, Lee DJ, O’Reilly C, Park K, MacDonald TY, et al. SPOP mutations in prostate cancer across demographically diverse patient cohorts. Neoplasia. 2014;16:14–20. doi: 10.1593/neo.131704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Burkhardt L, Fuchs S, Krohn A, Masser S, Mader M, et al. CHD1 is a 5q21 tumor suppressor required for ERG rearrangement in prostate cancer. Cancer Res. 2013;73:2795–805. doi: 10.1158/0008-5472.CAN-12-1342. [DOI] [PubMed] [Google Scholar]
- 72.Huang S, Gulzar ZG, Salari K, Lapointe J, Brooks JD, et al. Recurrent deletion of CHD1 in prostate cancer with relevance to cell invasiveness. Oncogene. 2012;31:4164–70. doi: 10.1038/onc.2011.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tereshchenko IV, Zhong H, Chekmareva MA, Kane-Goldsmith N, Santanam U, et al. ERG and CHD1 heterogeneity in prostate cancer: use of confocal microscopy in assessment of microscopic foci. Prostate. 2014;74:1551–9. doi: 10.1002/pros.22873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Beer TM, Tombal B. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. 2014;371:1755–6. doi: 10.1056/NEJMc1410239. [DOI] [PubMed] [Google Scholar]
- 75.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–97. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 76.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368:138–48. doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Azad AA, Volik SV, Wyatt AW, Haegert A, Le Bihan S, et al. Androgen receptor gene aberrations in circulating cell-free DNA: biomarkers of therapeutic resistance in castration-resistant prostate cancer. Clin Cancer Res. 2015;21:2315–24. doi: 10.1158/1078-0432.CCR-14-2666. [DOI] [PubMed] [Google Scholar]
- 79.Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013;3:1020–9. doi: 10.1158/2159-8290.CD-13-0226. [DOI] [PubMed] [Google Scholar]
- 80.Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide) Cancer Discov. 2013;3:1030–43. doi: 10.1158/2159-8290.CD-13-0142. [DOI] [PubMed] [Google Scholar]
- 81.Mostaghel EA, Marck BT, Plymate SR, Vessella RL, Balk S, et al. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res. 2011;17:5913–25. doi: 10.1158/1078-0432.CCR-11-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Scher HI, Heller G, Molina A, Attard G, Danila DC, et al. Circulating tumor cell biomarker panel as an individual-level surrogate for survival in metastatic castration-resistant prostate cancer. J Clin Oncol. 2015;33:1348–55. doi: 10.1200/JCO.2014.55.3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Friedlander TW, Ngo VT, Dong H, Premasekharan G, Weinberg V, et al. Detection and characterization of invasive circulating tumor cells derived from men with metastatic castration-resistant prostate cancer. Int J Cancer. 2014;134:2284–93. doi: 10.1002/ijc.28561. [DOI] [PubMed] [Google Scholar]
- 84.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra24. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Diamond E, Lee GY, Akhtar NH, Kirby BJ, Giannakakou P, et al. Isolation and characterization of circulating tumor cells in prostate cancer. Front Oncol. 2012;2:131. doi: 10.3389/fonc.2012.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stott SL, Lee RJ, Nagrath S, Yu M, Miyamoto DT, et al. Isolation and characterization of circulating tumor cells from patients with localized and metastatic prostate cancer. Sci Transl Med. 2010;2:25ra3. doi: 10.1126/scitranslmed.3000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Magbanua MJ, Sosa EV, Scott JH, Simko J, Collins C, et al. Isolation and genomic analysis of circulating tumor cells from castration resistant metastatic prostate cancer. BMC Cancer. 2012;12:78. doi: 10.1186/1471-2407-12-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Park S, Ang RR, Duffy SP, Bazov J, Chi KN, et al. Morphological differences between circulating tumor cells from prostate cancer patients and cultured prostate cancer cells. PLoS One. 2014;9:e85264. doi: 10.1371/journal.pone.0085264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014;159:176–87. doi: 10.1016/j.cell.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pantel K, Alix-Panabieres C. Real-time liquid biopsy in cancer patients: fact or fiction? Cancer Res. 2013;73:6384–8. doi: 10.1158/0008-5472.CAN-13-2030. [DOI] [PubMed] [Google Scholar]
- 91.Friedlander TW, Premasekharan G, Paris PL. Looking back, to the future of circulating tumor cells. Pharmacol Ther. 2014;142:271–80. doi: 10.1016/j.pharmthera.2013.12.011. [DOI] [PubMed] [Google Scholar]
- 92.Diaz LA, Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014;32:579–86. doi: 10.1200/JCO.2012.45.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bidard FC, Weigelt B, Reis-Filho JS. Going with the flow: from circulating tumor cells to DNA. Sci Transl Med. 2013;5:207ps14. doi: 10.1126/scitranslmed.3006305. [DOI] [PubMed] [Google Scholar]
- 94.Esmaeilsabzali H, Beischlag TV, Cox ME, Parameswaran AM, Park EJ. Detection and isolation of circulating tumor cells: principles and methods. Biotechnol Adv. 2013;31:1063–84. doi: 10.1016/j.biotechadv.2013.08.016. [DOI] [PubMed] [Google Scholar]
- 95.Haber DA, Velculescu VE. Blood-based analyses of cancer: circulating tumor cells and circulating tumor DNA. Cancer Discov. 2014;4:650–61. doi: 10.1158/2159-8290.CD-13-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Friedlander TW, Roy R, Tomlins SA, Ngo VT, Kobayashi Y, et al. Common structural and epigenetic changes in the genome of castration-resistant prostate cancer. Cancer Res. 2012;72:616–25. doi: 10.1158/0008-5472.CAN-11-2079. [DOI] [PubMed] [Google Scholar]
- 97.Ferraldeschi R, Nava Rodrigues D, Riisnaes R, Miranda S, Figueiredo I, et al. PTEN protein loss and clinical outcome from castration-resistant prostate cancer treated with abiraterone acetate. Eur Urol. 2015;67:795–802. doi: 10.1016/j.eururo.2014.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19:575–86. doi: 10.1016/j.ccr.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9:401–6. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- 100.Koivisto P, Kononen J, Palmberg C, Tammela T, Hyytinen E, et al. Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997;57:314–9. [PubMed] [Google Scholar]
- 101.Liu W, Xie CC, Thomas CY, Kim ST, Lindberg J, et al. Genetic markers associated with early cancer-specific mortality following prostatectomy. Cancer. 2013;119:2405–12. doi: 10.1002/cncr.27954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Beltran H, Yelensky R, Frampton GM, Park K, Downing SR, et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol. 2013;63:920–6. doi: 10.1016/j.eururo.2012.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Akbari MR, Wallis CJ, Toi A, Trachtenberg J, Sun P, et al. The impact of a BRCA2 mutation on mortality from screen-detected prostate cancer. Br J Cancer. 2014;111:1238–40. doi: 10.1038/bjc.2014.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gallagher DJ, Gaudet MM, Pal P, Kirchhoff T, Balistreri L, et al. Germline BRCA mutations denote a clinicopathologic subset of prostate cancer. Clin Cancer Res. 2010;16:2115–21. doi: 10.1158/1078-0432.CCR-09-2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Thorne H, Willems AJ, Niedermayr E, Hoh IM, Li J, et al. Decreased prostate cancer-specific survival of men with BRCA2 mutations from multiple breast cancer families. Cancer Prev Res (Phila) 2011;4:1002–10. doi: 10.1158/1940-6207.CAPR-10-0397. [DOI] [PubMed] [Google Scholar]
- 106.Tryggvadottir L, Vidarsdottir L, Thorgeirsson T, Jonasson JG, Olafsdottir EJ, et al. Prostate cancer progression and survival in BRCA2 mutation carriers. J Natl Cancer Inst. 2007;99:929–35. doi: 10.1093/jnci/djm005. [DOI] [PubMed] [Google Scholar]
- 107.Pritchard CC, Morrissey C, Kumar A, Zhang X, Smith C, et al. Complex MSH2 and MSH6 mutations in hypermutated microsatellite unstable advanced prostate cancer. Nat Commun. 2014;5:4988. doi: 10.1038/ncomms5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bott SR, Masters JR, Parkinson MC, Kirby RS, Feneley M, et al. Allelic imbalance and biochemical outcome after radical prostatectomy. Prostate Cancer Prostatic Dis. 2006;9:160–8. doi: 10.1038/sj.pcan.4500862. [DOI] [PubMed] [Google Scholar]
- 109.Choucair K, Ejdelman J, Brimo F, Aprikian A, Chevalier S, et al. PTEN genomic deletion predicts prostate cancer recurrence and is associated with low AR expression and transcriptional activity. BMC Cancer. 2012;12:543. doi: 10.1186/1471-2407-12-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.El Gammal AT, Bruchmann M, Zustin J, Isbarn H, Hellwinkel OJ, et al. Chromosome 8p deletions and 8q gains are associated with tumor progression and poor prognosis in prostate cancer. Clin Cancer Res. 2010;16:56–64. doi: 10.1158/1078-0432.CCR-09-1423. [DOI] [PubMed] [Google Scholar]
- 111.Fontugne J, Lee D, Cantaloni C, Barbieri CE, Caffo O, et al. Recurrent prostate cancer genomic alterations predict response to brachytherapy treatment. Cancer Epidemiol Biomarkers Prev. 2014;23:594–600. doi: 10.1158/1055-9965.EPI-13-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gopalan A, Leversha MA, Satagopan JM, Zhou Q, Al-Ahmadie HA, et al. TMPRSS2-ERG gene fusion is not associated with outcome in patients treated by prostatectomy. Cancer Res. 2009;69:1400–6. doi: 10.1158/0008-5472.CAN-08-2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hieronymus H, Schultz N, Gopalan A, Carver BS, Chang MT, et al. Copy number alteration burden predicts prostate cancer relapse. Proc Natl Acad Sci U S A. 2014;111:11139–44. doi: 10.1073/pnas.1411446111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kluth M, Hesse J, Heinl A, Krohn A, Steurer S, et al. Genomic deletion of MAP3K7 at 6q12-22 is associated with early PSA recurrence in prostate cancer and absence of TMPRSS2:ERG fusions. Mod Pathol. 2013;26:975–83. doi: 10.1038/modpathol.2012.236. [DOI] [PubMed] [Google Scholar]
- 115.Kluth M, Runte F, Barow P, Omari J, Abdelaziz ZM, et al. Concurrent deletion of 16q23 and PTEN is an independent prognostic feature in prostate cancer. Int J Cancer. 2015;137:2354–63. doi: 10.1002/ijc.29613. [DOI] [PubMed] [Google Scholar]
- 116.Lalonde E, Ishkanian AS, Sykes J, Fraser M, Ross-Adams H, et al. Tumour genomic and microenvironmental heterogeneity for integrated prediction of 5-year biochemical recurrence of prostate cancer: a retrospective cohort study. Lancet Oncol. 2014;15:1521–32. doi: 10.1016/S1470-2045(14)71021-6. [DOI] [PubMed] [Google Scholar]
- 117.Locke JA, Zafarana G, Ishkanian AS, Milosevic M, Thoms J, et al. NKX3.1 haploinsufficiency is prognostic for prostate cancer relapse following surgery or image-guided radiotherapy. Clin Cancer Res. 2012;18:308–16. doi: 10.1158/1078-0432.CCR-11-2147. [DOI] [PubMed] [Google Scholar]
- 118.Oxley JD, Winkler MH, Gillatt DA, Peat DS. Her-2/neu oncogene amplification in clinically localised prostate cancer. J Clin Pathol. 2002;55:118–20. doi: 10.1136/jcp.55.2.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Paris PL, Andaya A, Fridlyand J, Jain AN, Weinberg V, et al. Whole genome scanning identifies genotypes associated with recurrence and metastasis in prostate tumors. Hum Mol Genet. 2004;13:1303–13. doi: 10.1093/hmg/ddh155. [DOI] [PubMed] [Google Scholar]
- 120.Paris PL, Weinberg V, Albo G, Roy R, Burke C, et al. A group of genome-based biomarkers that add to a Kattan nomogram for predicting progression in men with high-risk prostate cancer. Clin Cancer Res. 2010;16:195–202. doi: 10.1158/1078-0432.CCR-09-0948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ross JS, Sheehan CE, Hayner-Buchan AM, Ambros RA, Kallakury BV, et al. Prognostic significance of HER-2/neu gene amplification status by fluorescence in situ hybridization of prostate carcinoma. Cancer. 1997;79:2162–70. doi: 10.1002/(sici)1097-0142(19970601)79:11<2162::aid-cncr14>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 122.Saramaki O, Willi N, Bratt O, Gasser TC, Koivisto P, et al. Amplification of EIF3S3 gene is associated with advanced stage in prostate cancer. Am J Pathol. 2001;159:2089–94. doi: 10.1016/S0002-9440(10)63060-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sato H, Minei S, Hachiya T, Yoshida T, Takimoto Y. Fluorescence in situ hybridization analysis of c-myc amplification in stage TNM prostate cancer in Japanese patients. Int J Urol. 2006;13:761–6. doi: 10.1111/j.1442-2042.2006.01399.x. [DOI] [PubMed] [Google Scholar]
- 124.Strohmeyer DM, Berger AP, Moore DH, 2nd, Bartsch G, Klocker H, et al. Genetic aberrations in prostate carcinoma detected by comparative genomic hybridization and microsatellite analysis: association with progression and angiogenesis. Prostate. 2004;59:43–58. doi: 10.1002/pros.20028. [DOI] [PubMed] [Google Scholar]
- 125.Toubaji A, Albadine R, Meeker AK, Isaacs WB, Lotan T, et al. Increased gene copy number of ERG on chromosome 21 but not TMPRSS2-ERG fusion predicts outcome in prostatic adenocarcinomas. Mod Pathol. 2011;24:1511–20. doi: 10.1038/modpathol.2011.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tsuchiya N, Slezak JM, Lieber MM, Bergstralh EJ, Jenkins RB. Clinical significance of alterations of chromosome 8 detected by fluorescence in situ hybridization analysis in pathologic organ-confined prostate cancer. Genes Chromosomes Cancer. 2002;34:363–71. doi: 10.1002/gcc.10064. [DOI] [PubMed] [Google Scholar]
- 127.Yoshimoto M, Cunha IW, Coudry RA, Fonseca FP, Torres CH, et al. FISH analysis of 107 prostate cancers shows that PTEN genomic deletion is associated with poor clinical outcome. Br J Cancer. 2007;97:678–85. doi: 10.1038/sj.bjc.6603924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yoshimoto M, Joshua AM, Cunha IW, Coudry RA, Fonseca FP, et al. Absence of TMPRSS2:ERG fusions and PTEN losses in prostate cancer is associated with a favorable outcome. Mod Pathol. 2008;21:1451–60. doi: 10.1038/modpathol.2008.96. [DOI] [PubMed] [Google Scholar]
- 129.Yu YP, Song C, Tseng G, Ren BG, LaFramboise W, et al. Genome abnormalities precede prostate cancer and predict clinical relapse. Am J Pathol. 2012;180:2240–8. doi: 10.1016/j.ajpath.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zafarana G, Ishkanian AS, Malloff CA, Locke JA, Sykes J, et al. Copy number alterations of c-MYC and PTEN are prognostic factors for relapse after prostate cancer radiotherapy. Cancer. 2012;118:4053–62. doi: 10.1002/cncr.26729. [DOI] [PubMed] [Google Scholar]
- 131.Barnett CM, Heinrich MC, Lim J, Nelson D, Beadling C, et al. Genetic profiling to determine risk of relapse-free survival in high-risk localized prostate cancer. Clin Cancer Res. 2014;20:1306–12. doi: 10.1158/1078-0432.CCR-13-1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chaux A, Peskoe SB, Gonzalez-Roibon N, Schultz L, Albadine R, et al. Loss of PTEN expression is associated with increased risk of recurrence after prostatectomy for clinically localized prostate cancer. Mod Pathol. 2012;25:1543–9. doi: 10.1038/modpathol.2012.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Antonarakis ES, Keizman D, Zhang Z, Gurel B, Lotan TL, et al. An immunohistochemical signature comprising PTEN, MYC, and Ki67 predicts progression in prostate cancer patients receiving adjuvant docetaxel after prostatectomy. Cancer. 2012;118:6063–71. doi: 10.1002/cncr.27689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.McCall P, Witton CJ, Grimsley S, Nielsen KV, Edwards J. Is PTEN loss associated with clinical outcome measures in human prostate cancer? Br J Cancer. 2008;99:1296–301. doi: 10.1038/sj.bjc.6604680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Halvorsen OJ, Haukaas SA, Akslen LA. Combined loss of PTEN and p27 expression is associated with tumor cell proliferation by Ki-67 and increased risk of recurrent disease in localized prostate cancer. Clin Cancer Res. 2003;9:1474–9. [PubMed] [Google Scholar]
- 136.Leinonen KA, Saramaki OR, Furusato B, Kimura T, Takahashi H, et al. Loss of PTEN is associated with aggressive behavior in ERG-positive prostate cancer. Cancer Epidemiol Biomarkers Prev. 2013;22:2333–44. doi: 10.1158/1055-9965.EPI-13-0333-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hawksworth D, Ravindranath L, Chen Y, Furusato B, Sesterhenn IA, et al. Overexpression of C-MYC oncogene in prostate cancer predicts biochemical recurrence. Prostate Cancer Prostatic Dis. 2010;13:311–5. doi: 10.1038/pcan.2010.31. [DOI] [PubMed] [Google Scholar]
- 138.Yang G, Timme TL, Frolov A, Wheeler TM, Thompson TC. Combined c-Myc and caveolin-1 expression in human prostate carcinoma predicts prostate carcinoma progression. Cancer. 2005;103:1186–94. doi: 10.1002/cncr.20905. [DOI] [PubMed] [Google Scholar]
- 139.Buyyounouski MK, Pickles T, Kestin LL, Allison R, Williams SG. Validating the interval to biochemical failure for the identification of potentially lethal prostate cancer. J Clin Oncol. 2012;30:1857–63. doi: 10.1200/JCO.2011.35.1924. [DOI] [PubMed] [Google Scholar]
- 140.Steurer S, Mayer PS, Adam M, Krohn A, Koop C, et al. TMPRSS2-ERG fusions are strongly linked to young patient age in low-grade prostate cancer. Eur Urol. 2014;66:978–81. doi: 10.1016/j.eururo.2014.06.027. [DOI] [PubMed] [Google Scholar]
- 141.Rodrigues LU, Rider L, Nieto C, Romero L, Karimpour-Fard A, et al. Coordinate loss of MAP3K7 and CHD1 promotes aggressive prostate cancer. Cancer Res. 2015;75:1021–34. doi: 10.1158/0008-5472.CAN-14-1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lotan TL, Gurel B, Sutcliffe S, Esopi D, Liu W, et al. PTEN protein loss by immunostaining: analytic validation and prognostic indicator for a high risk surgical cohort of prostate cancer patients. Clin Cancer Res. 2011;17:6563–73. doi: 10.1158/1078-0432.CCR-11-1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cuzick J, Yang ZH, Fisher G, Tikishvili E, Stone S, et al. Prognostic value of PTEN loss in men with conservatively managed localised prostate cancer. Br J Cancer. 2013;108:2582–9. doi: 10.1038/bjc.2013.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Heaphy CM, Yoon GS, Peskoe SB, Joshu CE, Lee TK, et al. Prostate cancer cell telomere length variability and stromal cell telomere length as prognostic markers for metastasis and death. Cancer Discov. 2013;3:1130–41. doi: 10.1158/2159-8290.CD-13-0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ribeiro FR, Henrique R, Martins AT, Jeronimo C, Teixeira MR. Relative copy number gain of MYC in diagnostic needle biopsies is an independent prognostic factor for prostate cancer patients. Eur Urol. 2007;52:116–25. doi: 10.1016/j.eururo.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 146.Ribeiro FR, Jeronimo C, Henrique R, Fonseca D, Oliveira J, et al. 8q gain is an independent predictor of poor survival in diagnostic needle biopsies from prostate cancer suspects. Clin Cancer Res. 2006;12:3961–70. doi: 10.1158/1078-0432.CCR-05-1977. [DOI] [PubMed] [Google Scholar]
- 147.Sato K, Qian J, Slezak JM, Lieber MM, Bostwick DG, et al. Clinical significance of alterations of chromosome 8 in high-grade, advanced, nonmetastatic prostate carcinoma. J Natl Cancer Inst. 1999;91:1574–80. doi: 10.1093/jnci/91.18.1574. [DOI] [PubMed] [Google Scholar]
- 148.Sircar K, Yoshimoto M, Monzon FA, Koumakpayi IH, Katz RL, et al. PTEN genomic deletion is associated with p-Akt and AR signalling in poorer outcome, hormone refractory prostate cancer. J Pathol. 2009;218:505–13. doi: 10.1002/path.2559. [DOI] [PubMed] [Google Scholar]
- 149.Washburn JG, Wojno KJ, Dey J, Powell IJ, Macoska JA. 8pter-p23 deletion is associated with racial differences in prostate cancer outcome. Clin Cancer Res. 2000;6:4647–52. [PubMed] [Google Scholar]
- 150.Mithal P, Allott E, Gerber L, Reid J, Welbourn W, et al. PTEN loss in biopsy tissue predicts poor clinical outcomes in prostate cancer. Int J Urol. 2014;21:1209–14. doi: 10.1111/iju.12571. [DOI] [PubMed] [Google Scholar]
- 151.Demichelis F, Fall K, Perner S, Andren O, Schmidt F, et al. TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene. 2007;26:4596–9. doi: 10.1038/sj.onc.1210237. [DOI] [PubMed] [Google Scholar]
- 152.Attard G, Clark J, Ambroisine L, Fisher G, Kovacs G, et al. Duplication of the fusion of TMPRSS2 to ERG sequences identifies fatal human prostate cancer. Oncogene. 2008;27:253–63. doi: 10.1038/sj.onc.1210640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Barwick BG, Abramovitz M, Kodani M, Moreno CS, Nam R, et al. Prostate cancer genes associated with TMPRSS2-ERG gene fusion and prognostic of biochemical recurrence in multiple cohorts. Br J Cancer. 2010;102:570–6. doi: 10.1038/sj.bjc.6605519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Berg KD, Vainer B, Thomsen FB, Roder MA, Gerds TA, et al. ERG protein expression in diagnostic specimens is associated with increased risk of progression during active surveillance for prostate cancer. Eur Urol. 2014;66:851–60. doi: 10.1016/j.eururo.2014.02.058. [DOI] [PubMed] [Google Scholar]
- 155.Hagglof C, Hammarsten P, Stromvall K, Egevad L, Josefsson A, et al. TMPRSS2-ERG expression predicts prostate cancer survival and associates with stromal biomarkers. PLoS One. 2014;9:e86824. doi: 10.1371/journal.pone.0086824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Nam RK, Sugar L, Yang W, Srivastava S, Klotz LH, et al. Expression of the TMPRSS2:ERG fusion gene predicts cancer recurrence after surgery for localised prostate cancer. Br J Cancer. 2007;97:1690–5. doi: 10.1038/sj.bjc.6604054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Spencer ES, Johnston RB, Gordon RR, Lucas JM, Ussakli CH, et al. Prognostic value of ERG oncoprotein in prostate cancer recurrence and cause-specific mortality. Prostate. 2013;73:905–12. doi: 10.1002/pros.22636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.FitzGerald LM, Agalliu I, Johnson K, Miller MA, Kwon EM, et al. Association of TMPRSS2-ERG gene fusion with clinical characteristics and outcomes: results from a population-based study of prostate cancer. BMC Cancer. 2008;8:230. doi: 10.1186/1471-2407-8-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Fleischmann A, Saramaki OR, Zlobec I, Rotzer D, Genitsch V, et al. Prevalence and prognostic significance of TMPRSS2-ERG gene fusion in lymph node positive prostate cancers. Prostate. 2014;74:1647–54. doi: 10.1002/pros.22882. [DOI] [PubMed] [Google Scholar]
- 160.Saramaki OR, Harjula AE, Martikainen PM, Vessella RL, Tammela TL, et al. TMPRSS2:ERG fusion identifies a subgroup of prostate cancers with a favorable prognosis. Clin Cancer Res. 2008;14:3395–400. doi: 10.1158/1078-0432.CCR-07-2051. [DOI] [PubMed] [Google Scholar]
- 161.Dal Pra A, Lalonde E, Sykes J, Warde F, Ishkanian A, et al. TMPRSS2-ERG status is not prognostic following prostate cancer radiotherapy: implications for fusion status and DSB repair. Clin Cancer Res. 2013;19:5202–9. doi: 10.1158/1078-0432.CCR-13-1049. [DOI] [PubMed] [Google Scholar]
- 162.Hoogland AM, Jenster G, van Weerden WM, Trapman J, van der Kwast T, et al. ERG immunohistochemistry is not predictive for PSA recurrence, local recurrence or overall survival after radical prostatectomy for prostate cancer. Mod Pathol. 2012;25:471–9. doi: 10.1038/modpathol.2011.176. [DOI] [PubMed] [Google Scholar]
- 163.Kim SH, Kim SH, Joung JY, Lee GK, Hong EK, et al. Overexpression of ERG and wild-type PTEN are associated with favorable clinical prognosis and low biochemical recurrence in prostate cancer. PLoS One. 2015;10:e0122498. doi: 10.1371/journal.pone.0122498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kimura T, Furusato B, Miki J, Yamamoto T, Hayashi N, et al. Expression of ERG oncoprotein is associated with a less aggressive tumor phenotype in Japanese prostate cancer patients. Pathol Int. 2012;62:742–8. doi: 10.1111/pin.12006. [DOI] [PubMed] [Google Scholar]
- 165.Minner S, Enodien M, Sirma H, Luebke AM, Krohn A, et al. ERG status is unrelated to PSA recurrence in radically operated prostate cancer in the absence of antihormonal therapy. Clin Cancer Res. 2011;17:5878–88. doi: 10.1158/1078-0432.CCR-11-1251. [DOI] [PubMed] [Google Scholar]
- 166.Pettersson A, Graff RE, Bauer SR, Pitt MJ, Lis RT, et al. The TMPRSS2:ERG rearrangement, ERG expression, and prostate cancer outcomes: a cohort study and meta-analysis. Cancer Epidemiol Biomarkers Prev. 2012;21:1497–509. doi: 10.1158/1055-9965.EPI-12-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sartori DA, Chan DW. Biomarkers in prostate cancer: what's new? Curr Opin Oncol. 2014;26:259–64. doi: 10.1097/CCO.0000000000000065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Boström PJ, Bjartell AS, Catto JW, Eggener SE, Lilja H, et al. Genomic predictors of outcome in prostate cancer. Eur Urol. 2015;68:1033–44. doi: 10.1016/j.eururo.2015.04.008. [DOI] [PubMed] [Google Scholar]