

Abstract
Prostate cancer is a clinically heterogeneous disease, with some men having indolent disease that can safely be observed, while others have aggressive, lethal disease. Over the past decade, researchers have begun to unravel some of the genomic heterogeneity that contributes to these varying clinical phenotypes. Distinct molecular sub-classes of prostate cancer have been identified, and the uniqueness of these sub-classes has been leveraged to predict clinical outcomes, design novel biomarkers for prostate cancer diagnosis, and develop novel therapeutics. Recent work has also elucidated the temporal and spatial heterogeneity of prostate cancer, helping us understand disease pathogenesis, response to therapy, and progression. New genomic techniques have provided us with a window into the remarkable clinical and genomic heterogeneity of prostate cancer, and this new perspective will increasingly impact patient care.
Keywords: cell biology, ERG, genomics, molecular heterogeneity, prostate cancer, sequencing, serine peptidase inhibitor, Kazal type 1, SPOP, tumor profiling
INTRODUCTION
Understanding the clinical heterogeneity seen in prostate cancer (PCa), where some men have indolent disease that may never need treatment while others have lethal disease, is one of the greatest challenges faced by urologists and oncologists. Prostate cancer is fundamentally a genetic disease, driven by genomic instability causing the activation of oncogenes and the inactivation of tumor suppressors. Recent work has begun to unravel some of the genomic heterogeneity in prostate cancer and to define distinct molecular sub-classes of the disease. Leveraging this genomic heterogeneity may fundamentally change how we diagnose and manage prostate cancer in the decades to come. This review will not only examine the heterogeneity of prostate cancer at multiple levels, including clinical and pathological, but also primarily focus on the molecular heterogeneity of prostate cancer. We will discuss the implications of this heterogeneity for patient care across disease states, ranging from low-risk clinically localized prostate cancer to lethal castration-resistant disease.
PROSTATE CANCER PATHOLOGICAL HETEROGENEITY
The histologic spectrum of prostate cancer ranges from its precursor lesion, high-grade prostatic intra-epithelial neoplasia (HgPIN), to de-differentiated cancer. HgPIN is characterized by cellular proliferation within pre-existing glands with cytological changes mimicking cancer.1 The presence of HgPIN is certainly associated with the presence of prostate cancer, but the definitive development of PCa from HgPIN is much less clear. Historically, it was believed that adenocarcinoma would develop in most men with HgPIN within 10 years,1 and therefore men found to have HgPIN on biopsy generally underwent closer surveillance than men with no evidence of disease, with repeat biopsy typically recommended. However, more recent data suggest a lower risk of HgPIN progression, particularly with the standardization of biopsies consisting of more cores, limiting undersampling.2 Importantly, HgPIN harboring specific molecular alterations (such as ERG overexpression) may be more associated with subsequent cancer detection, implying considerable heterogeneity in behavior even within HgPIN sub-classes.3
Historically, perhaps, the most powerful correlate to prostate cancer behavior is the histologic Gleason scoring system. Gleason grading was devised in the 1960s and 70s by Donald F Gleason and the Veterans Administration Cooperative Urological Research Group, and uses the pattern of carcinoma cells in Hematoxylin and Eosin stained sections to generate a histologic score.4 Gleason grade, combined with other clinical factors such as prostate-specific antigen (PSA), age, clinical stage, MRI findings, number of positive cores, and percentage of each core that contains cancer has been incorporated into models which attempt to predict the behavior of prostate cancer. The prognostic ability of these models has led to the stratification of treatment according to risk category. In the absence of other adverse features, it is widely believed that men with isolated Gleason 6 disease may have a low risk of disease progression, and therefore do not require treatment when they are closely monitored.5 In contrast, men with localized disease with a Gleason score of 7 or above are usually recommended for local treatment (either radiation or surgery), while men with particularly high-risk clinical or pathologic features may benefit from multimodality therapy.6
Despite local treatment with surgery or radiation, some patients’ cancers will progress again in a variable fashion, with a mix of local recurrence, in a nodal basin, or as a distant metastasis to bone or other sites. In terms of metastatic disease, it is well known that localized prostate cancer nearly universally responds to androgen deprivation therapy (ADT), while over time, almost all tumors will develop resistance to ADT and become castrate-resistant prostate cancer (CRPC).7 Some of these cancers will then acquire neuroendocrine features, which is associated with a poor response to treatment and prognosis.8
DEFINING MOLECULAR SUB-CLASSES OF PCA
Over the past 10 years, our understanding of PCa genomics has changed dramatically. The availability of next-generation sequencing technologies has allowed researchers to classify prostate cancers by their multiple levels of molecular signatures, exposing genomic, transcriptomic, and epigenetic heterogeneity. Distinct molecular sub-classes have emerged, with the potential to transform the disease from a poorly understood, heterogeneous disease with a highly variable clinical course to a collection of more homogeneous molecular sub-classes.
ETS family members
In 2005, a series of landmark papers discovered fusions of the 5’ untranslated region (UTR) of the TMPRSS2 gene with the ETS family transcription factor family members.9,10,11,12 This discovery provided the framework for organizing prostate cancers into those with ETS rearrangements and those without those re-arrangements. The most common ETS family re-arrangement is TMPRSS2:ERG which has now been identified in approximately half of prostate cancers and accounts for 90% of ETS family fusions.9,13,14 Fusions of other ETS family members, including ETV1, ETV4, ETV5, and FLII have been identified.10,15,16,17 These re-arrangements result in overexpression of the ETS family transcription factors which confer a neoplastic phenotype.18 The original report of the fusion products, which has subsequently been validated in other cohorts, found that fusions between ETV1 and ERG appear to be largely mutually exclusive.10,12,15 Several other 5’ partners have also subsequently been identified, most notably a fusion product involving the ETS family member ELK4 to SLC45A3 in 5%–10% of prostate cancers.19,20
ETS fusion seems to be an early event in carcinogenesis and has been detected in HgPIN.21,22,23 ERG re-arrangements when detected in HgPIN have also been detected in the adjoining prostate cancer and appear to precede other mutations.23,24,25 ERG re-arranged cancer is rarely identified distant from cancer foci in prostatectomy specimens, suggesting that ERG is important for the transition from HgPIN to cancer.22,24 Indeed, ERG re-arrangements in biopsy specimens with HgPIN have been shown to be predictive of the development of prostate cancer (53% vs 35%).3
Multiple studies have demonstrated that ETS-positive cancers have distinct features at a number of levels. These show a distinct gene expression signature from ETS-negative cancers.18,26,27 In addition, ETS fusion-positive tumors have distinct copy number aberrations and a distinct pattern of genomic re-arrangements involving chains of balanced re-arrangements, a phenomenon described as “chromoplexy.”28,29,30 Mice engineered to overexpress ERG or ETV1 under androgen regulation develop neoplastic prostate lesions similar to PIN,31 and ERG overexpression accelerates prostate cancer pathogenesis when combined with deletions in PTEN.12,32,33
Clinically, there is some evidence to support that ETS-rearranged cancers being more aggressive than cancers without these re-arrangements.14 This observation is largely derived from two studies from active surveillance cohorts of men diagnosed with prostate cancer on transurethral resection of the prostate (TURP). In both studies, men with TMPRSS2-ERG rearranged cancers had an increased risk of prostate cancer death.34,35 In addition, ERG-rearranged tumors have been demonstrated to have an increased risk of progression while on active surveillance.36 Importantly, it has been demonstrated that a urine test from TMPRSS2:ERG in conjunction with PCA3 is able to stratify patients undergoing active surveillance to determine their risk of having higher Gleason scores or a larger tumor volume.37 Analysis of prostatectomy specimens, however, has yielded conflicting results regarding the relative aggressiveness of ETS-rearranged cancers.9,14,38,39,40,41 ETS re-arrangements also appear to be more common in peripheral zone tumors than transition zone tumors.42,43,44
SPINK1
Using the same Cancer Outlier Profile Analysis (COPA) used to define ETS gene re-arrangements, Tomlins et al. identified a second sub-class of prostate cancers, which overexpress Serine peptidase inhibitor, Kazal type 1 (SPINK1).45 SPINK1 outlier expression has been identified in ~10% of prostate cancers, and appears to be mutually exclusive from ERG re-arrangements.45 Interestingly, patients harboring these tumors were found to have a shorter time to biochemical recurrence than patients who do not overexpress SPINK1. SPINK1 outlier status, independent of Gleason score, lymph node status, surgical margin status, seminal vesicle invasion, extracapsular extension, and preoperative PSA, has been shown to be a significant predictor of clinical recurrence.45 SPINK1 is an extracellular secreted protein and therefore is amenable to both therapeutic targeting and noninvasive diagnosis.45,46,47 Indeed, studies using antibodies against SPINK1 in mouse prostate cancer xenografts have identified SPINK1 as a likely target in patients harboring SPINK1+/ETS − tumors.46
SPOP/CHD1
Recurrent mutations in the SPOP gene are found in 5%–15% of tumors, making it the most common point mutation in prostate cancer.48,49 SPOP encodes the substrate-binding sub-unit of a Cullin-based E3 ubiquitin ligase, and mutations affect conserved residues in the structurally defined substrate-binding cleft. SPOP mutation appears to occur exclusively in tumors without ERG re-arrangement and constitute a unique sub-class of prostate cancer.48 SPOP mutations have been identified in HgPIN adjacent to adenocarcinoma, and likely represent early events in the natural history of prostate cancer.48 SPOP mutant tumors have been found to have recurrent somatic deletions at 5q21 at the CHD1 locus.48,49 CHD1 is an ATP-dependent chromatin-remodeling enzyme, and the genomic locus is deleted in ~5%–10% of prostate cancers.50,51 A recent study found no association between SPOP mutation and clinical or pathological parameters.49 However, others have reported that mutations and decreased expression of the SPOP gene are associated with worse progression-free survival.52 Functionally, SPOP mutation has been shown to modulate carcinogenesis by preventing the degradation of oncogenic factors including ERG and the androgen receptor.53,54,55,56,57 Importantly, our group recently demonstrated that SPOP mutation alters DNA double-strand break (DSB) repair, is associated with genomic instability, and sensitizes to DNA-damaging agents such as PARP inhibitors.58
HETEROGENEITY BETWEEN PROSTATE CANCER CLONES
Primary prostate cancer
Primary prostate cancer has relatively few genomic aberrations compared to other cancers. Recent work found the mutation rate to be ~0.9 per Mb, which is similar to that observed in acute myeloid leukemia and breast cancer, but 7 to 15-fold lower than rates reported for small cell lung cancer and melanoma.59 Primary tumors display a wide range of copy number alteration levels, from tumors appearing metastatic-like in whole chromosome, chromosome arm, and focal amplifications and deletions, to those with fundamentally diploid genomes.15
The identification of multiple distinct loci of cancer with varying histologic grades has led to the idea of prostate cancer as a multifocal disease. It is estimated that ~80% of prostate cancers contain more than one focus of disease.60,61,62 Multiple studies have shown heterogeneous foci of PIN within the same prostate associated with carcinoma with similar aberrations.25,63 ETS fusion is both an early event in carcinogenesis and is thought to be maintained throughout prostate cancer progression, making it an excellent marker to assess for clonality.12,14,64,65
Detailed spatial sampling and sequencing of prostate tumors has identified significant heterogeneity within multifocal tumors in the same patient (Figure 1).61,66,67 Using a combination of exome sequencing and copy number alteration profiling of 23 loci from five patients, Boutros et al. identified both divergent tumor evolution in multifocal cancer as well as tumors of independent clonal origin in three of those patients.61 Using whole genome sequencing, examining 12 cancer specimens and three normal specimens from three men, Cooper et al. identified mutations present at high levels in morphologically normal tissue, sometimes distant from the cancer.67 In addition, subsets of mutations were shared by normal and malignant tissues and between different ERG lineages, with multiple distinct ERG fusions noted within a single cancer nodule.67 These results were thought to be consistent with “field effects” underlying the evolution of prostate cancer with subsequent branching evolution and cancer clone mixing. Additional studies will be necessary to establish the pathogenicity of this “field effect” or whether this observation is a reflection of the expected accumulation of germline mutations during normal development. Finally, a recent effort found that categorizing men into four categories based on genomic subtype and including measurements of tumor hypoxia was able to accurately predict the outcome following local therapy.68
Figure 1.
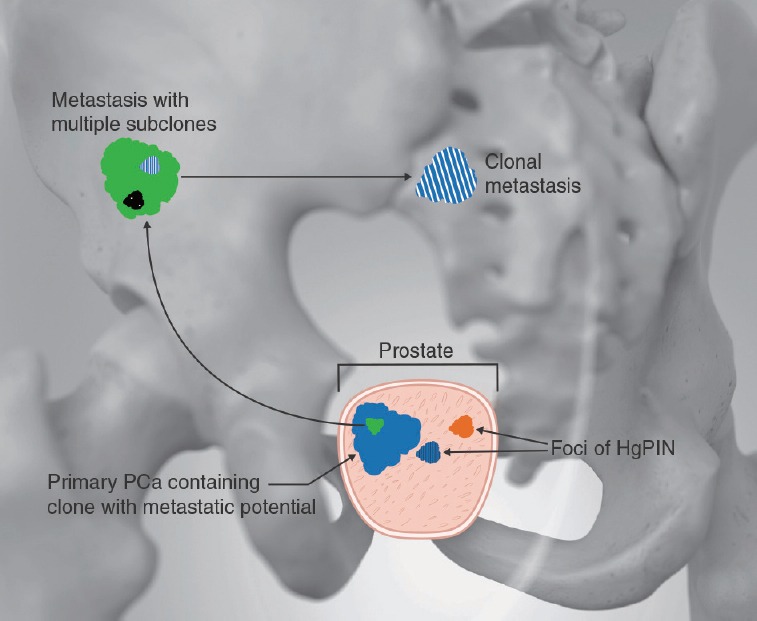
Illustration depicting prostate cancer heterogeneity. Within the prostate are multiple distinct clones of HgPIN. One of these clones (e.g., one with ETS rearrangement) is adjacent to the primary prostate cancer. Within the primary cancer, a clone with metastatic potential developed which metastasized to the iliac wing. Within this metastasis, multiple subclones developed, one of which has reached a second metastatic site in the sacrum.
Metastatic prostate cancer
Metastatic prostate cancer arises from rare subclones that attain metastatic potential. This “seed and soil” hypothesis, where rare cells acquire metastatic potential has been supported in a number of studies.69,70,71 Interestingly, a recent study of longitudinally collected primary and metastatic samples identified one metastasis comprising three distinct clones derived from two separate waves of metastasis from the prostate.70
The most commonly mutated gene in metastatic PCA is the androgen receptor (AR).15,72 AR mutations occur almost exclusively in mCRPC, and are virtually never seen in localized disease.48,73 After ERG and AR, the most common alterations involve the inactivation of PTEN in approximately ~40% of cancers resulting in activation of the PI3K pathway.6,74 PTEN copy number loss is consistently associated with aggressive disease features, cancer progression, and the development of castration-resistant disease.74,75,76 PTEN alterations are enriched in the ERG-rearranged subclass of PCA, and in this class, in particular, PTEN loss may be associated with more aggressive disease.32,33,77,78,79 Interestingly, PTEN deletion appears to occur after ERG rearrangement in these tumors, further complicating intratumoral heterogeneity and the search for the “lethal” subclone that leads to CRPC. Other common genetic alterations include p53 mutation (40%), RB loss (28%), aberrations in BRCA1, BRCA2, and ATM (19%), and MYC amplification also commonly occur in metastatic lesions.6
The heterogeneity in mutation status between patients has already begun to be exploited for therapeutic benefit. Our institution recently published the results of whole exome sequencing in 154 tumor-normal pairs from 97 patients with metastatic prostate cancer, 94% of whom were found to have alterations which were potentially actionable, similar to the 89% of actionable mutations previously published.6,80 Five percent of these patients actually received therapy based on these results.80
Between metastatic foci
Characterization of lethal prostate cancer by whole genome sequencing of 51 sites from 10 patients found metastasis-to-metastasis spread to be common, either through de novo monoclonal seeding of daughter metastases or transfer of multiple tumor clones between metastatic sites. Importantly, lesions affecting tumor suppressor genes usually occurred as single events while mutations in genes involved in AR signaling commonly derived from events in different metastases.69
By sequencing circulating tumor DNA, Carreira et al. examined the tumor clone dynamics of 106 sequential plasma samples, CRPC tumor biopsies, and precastration tumor cores in 16 ERG-positive patients. Multiple independent clones were differentially expressed in circulation in metastatic disease (Figure 2). Importantly, treatment with abiraterone and enzalutamide in men who subsequently progressed was correlated with the emergence of clones harboring mutations in AR that are activated by glucocorticoids.81
Figure 2.
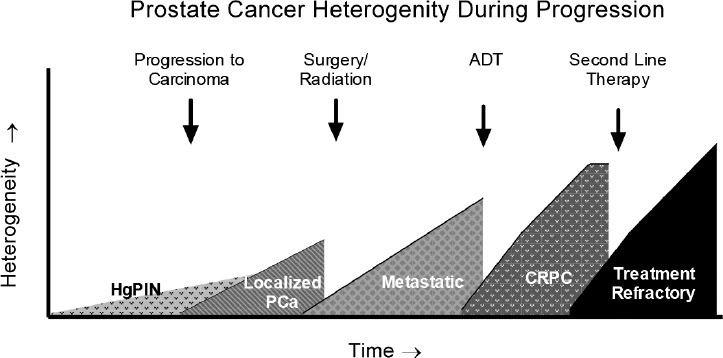
Graphical representation of tumor clonal diversity over time and in response to therapy. Multiple clones of HgPIN develop, only a few of which go on to become adenocarcinoma with subsequent clonal evolution throughout progression.
CLINICAL IMPLICATIONS
Localized disease
The high degree of heterogeneity in individuals has wide ranging implications for prostate cancer diagnosis and treatment. In terms of diagnosis, the genomic heterogeneity seen in localized prostate cancer challenges the idea of a “dominant lesion,” which is defined solely by size or histologic criteria, being largely responsible for a patient's clinical course. Molecular heterogeneity seen in PCa may instead suggest that genomic features, rather than size or histology alone, will determine the biology of disease.82 This raises questions about the underlying assumptions of both standard template biopsy, as well as image-guided targeted biopsies. Template biopsy, commonly utilizing 12–14 cores, is based on probabilistic sampling of the prostate to detect larger lesions. Given the spatial heterogeneity of prostate cancer, sampling of one larger lesion does not necessarily provide insight into the other lesions that are present. Similarly, targeted biopsy assumes that the MRI-visible lesion is clinically the most relevant, which also may not be the case that ongoing studies are crucial to validate this idea. This type of heterogeneity is also problematic for personalized genomic testing, as prognostic information may be clouded by a lack of adequate sampling.
Similarly, tumor heterogeneity is particularly problematic, given the increasing acceptance of focal therapy for prostate cancer. As discussed above, prostate cancer is often multifocal, however even the surrounding normal tissue can harbor clonal mutations.67 Furthermore, the marked heterogeneity seen in localized disease re-inforces how little understood prostate cancer pathogenesis is and how difficult it is to predict who requires therapy. This is because even if we are able to determine which mutations confer worse prognosis, the ability to sample all clones continues to be problematic.
Metastatic disease
Multiple reports have demonstrated polyclonal sub-populations in metastatic foci, as well as heterogeneity within a single focus and between foci. The dynamics of resistant clones in response to therapy suggest that preexistent clonal populations are responsible for resistance to therapy and disease progression. This has important implications for how we treat metastatic disease, as using multiple concurrent agents may be preferable to single-agent therapy as these will target multiple populations, similar to the rationale for highly active antiretroviral therapy for human immunodeficiency virus treatment. Indeed, recent work has demonstrated that combination of docetaxel and ADT was superior to ADT alone, perhaps because of this reason.83
In addition, there is increasing evidence that local treatment with radiation or surgery of lymph node-positive or oligo-metastatic disease may be beneficial in prostate cancer.84,85,86,89,88,89,90,91,92 While the data to this point are limited to case series, ongoing clinical trials will help to further elucidate this question. One hypothesis for how local control may benefit patients is the clearance of foci harboring resistant or more aggressive clones even if some cancer cells remain.
CONCLUSIONS
The past decade has brought new insights into the genomic pathogenesis of prostate cancer. The discovery of sub-classes of prostate cancer categorized by ETS rearrangement, SPOP mutation, and SPINK1 overexpression has paved the way for novel insights into the diagnosis, prognosis, and treatment of the disease. The molecular characterization of prostate cancer has also allowed examination of the spatial and temporal heterogeneity of prostate cancer with previously unobtainable resolution. These findings have important implications for prostate cancer screening and diagnosis. They also provide insights into the treatment of disease, and will likely be the basis for future therapeutic approaches.
AUTHOR CONTRIBUTIONS
JS and CEB reviewed the relevant literature and wrote the manuscript. All authors read and approved the final manuscript.
COMPETING INTEREST
All authors declare no competing financial interests.
ACKNOWLEDGMENTS
The authors thank the patients and families who have made advancements in prostate cancer research possible, and many collaborators who have helped drive these studies.
REFERENCES
- 1.Bostwick DG, Liu L, Brawer MK, Qian J. High-grade prostatic intraepithelial neoplasia. Rev Urol. 2004;6:171–9. [PMC free article] [PubMed] [Google Scholar]
- 2.Klink JC, Miocinovic R, Magi Galluzzi C, Klein EA. High-grade prostatic intraepithelial neoplasia. Korean J Urol. 2012;53:297–303. doi: 10.4111/kju.2012.53.5.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park K, Dalton JT, Narayanan R, Barbieri CE, Hancock ML, et al. TMPRSS2:ERG gene fusion predicts subsequent detection of prostate cancer in patients with high-grade prostatic intraepithelial neoplasia. J Clin Oncol. 2014;32:206–11. doi: 10.1200/JCO.2013.49.8386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Humphrey PA. Gleason grading and prognostic factors in carcinoma of the prostate. Mod Pathol. 2004;17:292–306. doi: 10.1038/modpathol.3800054. [DOI] [PubMed] [Google Scholar]
- 5.Tosoian JJ, Trock BJ, Landis P, Feng Z, Epstein JI, et al. Active surveillance program for prostate cancer: an update of the Johns Hopkins experience. J Clin Oncol. 2011;29:2185–90. doi: 10.1200/JCO.2010.32.8112. [DOI] [PubMed] [Google Scholar]
- 6.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–28. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wyatt AW, Mo F, Wang Y, Collins CC. The diverse heterogeneity of molecular alterations in prostate cancer identified through next-generation sequencing. Asian J Androl. 2013;15:301–8. doi: 10.1038/aja.2013.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Terry S, Beltran H. The many faces of neuroendocrine differentiation in prostate cancer progression. Front Oncol. 2014;4:60. doi: 10.3389/fonc.2014.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perner S, Demichelis F, Beroukhim R, Schmidt FH, Mosquera JM, et al. TMPRSS2:ERG fusion-associated deletions provide insight into the heterogeneity of prostate cancer. Cancer Res. 2006;66:8337–41. doi: 10.1158/0008-5472.CAN-06-1482. [DOI] [PubMed] [Google Scholar]
- 10.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–8. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 11.Tomlins SA, Mehra R, Rhodes DR, Smith LR, Roulston D, et al. TMPRSS2:ETV4 gene fusions define a third molecular subtype of prostate cancer. Cancer Res. 2006;66:3396–400. doi: 10.1158/0008-5472.CAN-06-0168. [DOI] [PubMed] [Google Scholar]
- 12.Chad CB, Arul MC, Scott AT. ETS Fusion Genes in Prostate Cancer Brenner. In: Tindall DJ, editor. Prostate Cancer-Biochemistry, Molecular Biology and Genetics. New York: Springer (Imprint); 2013. pp. 139–83. [Google Scholar]
- 13.Pettersson A, Graff RE, Bauer SR, Pitt MJ, Lis RT, et al. The TMPRSS2:ERG rearrangement, ERG expression, and prostate cancer outcomes: a cohort study and meta-analysis. Cancer Epidemiol Biomarkers Prev. 2012;21:1497–509. doi: 10.1158/1055-9965.EPI-12-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tomlins SA, Bjartell A, Chinnaiyan AM, Jenster G, Nam RK, et al. ETS gene fusions in prostate cancer: from discovery to daily clinical practice. Eur Urol. 2009;56:275–86. doi: 10.1016/j.eururo.2009.04.036. [DOI] [PubMed] [Google Scholar]
- 15.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Helgeson BE, Tomlins SA, Shah N, Laxman B, Cao Q, et al. Characterization of TMPRSS2:ETV5 and SLC45A3:ETV5 gene fusions in prostate cancer. Cancer Res. 2008;68:73–80. doi: 10.1158/0008-5472.CAN-07-5352. [DOI] [PubMed] [Google Scholar]
- 17.Paulo P, Barros-Silva JD, Ribeiro FR, Ramalho-Carvalho J, Jeronimo C, et al. FLI1 is a novel ETS transcription factor involved in gene fusions in prostate cancer. Genes Chromosomes Cancer. 2012;51:240–9. doi: 10.1002/gcc.20948. [DOI] [PubMed] [Google Scholar]
- 18.Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448:595–9. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- 19.Maher CA, Kumar-Sinha C, Cao X, Kalyana-Sundaram S, Han B, et al. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458:97–101. doi: 10.1038/nature07638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rickman DS, Pflueger D, Moss B, VanDoren VE, Chen CX, et al. SLC45A3-ELK4 is a novel and frequent erythroblast transformation-specific fusion transcript in prostate cancer. Cancer Res. 2009;69:2734–8. doi: 10.1158/0008-5472.CAN-08-4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perner S, Mosquera JM, Demichelis F, Hofer MD, Paris PL, et al. TMPRSS2-ERG fusion prostate cancer: an early molecular event associated with invasion. Am J Surg Pathol. 2007;31:882–8. doi: 10.1097/01.pas.0000213424.38503.aa. [DOI] [PubMed] [Google Scholar]
- 22.Park K, Tomlins SA, Mudaliar KM, Chiu YL, Esgueva R, et al. Antibody-based detection of ERG rearrangement-positive prostate cancer. Neoplasia. 2010;12:590–8. doi: 10.1593/neo.10726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cerveira N, Ribeiro FR, Peixoto A, Costa V, Henrique R, et al. TMPRSS2-ERG gene fusion causing ERG overexpression precedes chromosome copy number changes in prostate carcinomas and paired HGPIN lesions. Neoplasia. 2006;8:826–32. doi: 10.1593/neo.06427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mosquera JM, Perner S, Genega EM, Sanda M, Hofer MD, et al. Characterization of TMPRSS2-ERG fusion high-grade prostatic intraepithelial neoplasia and potential clinical implications. Clin Cancer Res. 2008;14:3380–5. doi: 10.1158/1078-0432.CCR-07-5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Furusato B, Tan SH, Young D, Dobi A, Sun C, et al. ERG oncoprotein expression in prostate cancer: clonal progression of ERG-positive tumor cells and potential for ERG-based stratification. Prostate Cancer Prostatic Dis. 2010;13:228–37. doi: 10.1038/pcan.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Setlur SR, Mertz KD, Hoshida Y, Demichelis F, Lupien M, et al. Estrogen-dependent signaling in a molecularly distinct subclass of aggressive prostate cancer. J Natl Cancer Inst. 2008;100:815–25. doi: 10.1093/jnci/djn150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–43. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Demichelis F, Rubin MA. TMPRSS2-ETS fusion prostate cancer: biological and clinical implications. J Clin Pathol. 2007;60:1185–6. doi: 10.1136/jcp.2007.046557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, et al. Punctuated evolution of prostate cancer genomes. Cell. 2013;153:666–77. doi: 10.1016/j.cell.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barbieri CE, Rubin MA. Genomic rearrangements in prostate cancer. Curr Opin Urol. 2015;25:71–6. doi: 10.1097/MOU.0000000000000129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tomlins SA, Laxman B, Varambally S, Cao X, Yu J, et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia. 2008;10:177–88. doi: 10.1593/neo.07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carver BS, Tran J, Gopalan A, Chen Z, Shaikh S, et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet. 2009;41:619–24. doi: 10.1038/ng.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.King JC, Xu J, Wongvipat J, Hieronymus H, Carver BS, et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat Genet. 2009;41:524–6. doi: 10.1038/ng.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Demichelis F, Fall K, Perner S, Andren O, Schmidt F, et al. TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene. 2007;26:4596–9. doi: 10.1038/sj.onc.1210237. [DOI] [PubMed] [Google Scholar]
- 35.Attard G, Clark J, Ambroisine L, Fisher G, Kovacs G, et al. Duplication of the fusion of TMPRSS2 to ERG sequences identifies fatal human prostate cancer. Oncogene. 2008;27:253–63. doi: 10.1038/sj.onc.1210640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Monecke T, Buschmann J, Neumann P, Wahle E, Ficner R. Crystal structures of the novel cytosolic 5’-nucleotidase IIIB explain its preference for m7GMP. PLoS One. 2014;9:e90915. doi: 10.1371/journal.pone.0090915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin DW, Newcomb LF, Brown EC, Brooks JD, Carroll PR, et al. Urinary TMPRSS2:ERG and PCA3 in an active surveillance cohort: results from a baseline analysis in the Canary Prostate Active Surveillance Study. Clin Cancer Res. 2013;19:2442–50. doi: 10.1158/1078-0432.CCR-12-3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheville JC, Karnes RJ, Therneau TM, Kosari F, Munz JM, et al. Gene panel model predictive of outcome in men at high-risk of systemic progression and death from prostate cancer after radical retropubic prostatectomy. J Clin Oncol. 2008;26:3930–6. doi: 10.1200/JCO.2007.15.6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gopalan A, Leversha MA, Satagopan JM, Zhou Q, Al-Ahmadie HA, et al. TMPRSS2-ERG gene fusion is not associated with outcome in patients treated by prostatectomy. Cancer Res. 2009;69:1400–6. doi: 10.1158/0008-5472.CAN-08-2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tu JJ, Rohan S, Kao J, Kitabayashi N, Mathew S, et al. Gene fusions between TMPRSS2 and ETS family genes in prostate cancer: frequency and transcript variant analysis by RT-PCR and FISH on paraffin-embedded tissues. Mod Pathol. 2007;20:921–8. doi: 10.1038/modpathol.3800903. [DOI] [PubMed] [Google Scholar]
- 41.Saramaki OR, Harjula AE, Martikainen PM, Vessella RL, Tammela TL, et al. TMPRSS2:ERG fusion identifies a subgroup of prostate cancers with a favorable prognosis. Clin Cancer Res. 2008;14:3395–400. doi: 10.1158/1078-0432.CCR-07-2051. [DOI] [PubMed] [Google Scholar]
- 42.Guo CC, Zuo G, Cao D, Troncoso P, Czerniak BA. Prostate cancer of transition zone origin lacks TMPRSS2-ERG gene fusion. Mod Pathol. 2009;22:866–71. doi: 10.1038/modpathol.2009.57. [DOI] [PubMed] [Google Scholar]
- 43.Falzarano SM, Navas M, Simmerman K, Klein EA, Rubin MA, et al. ERG rearrangement is present in a subset of transition zone prostatic tumors. Mod Pathol. 2010;23:1499–506. doi: 10.1038/modpathol.2010.150. [DOI] [PubMed] [Google Scholar]
- 44.Braun M, Scheble VJ, Menon R, Scharf G, Wilbertz T, et al. Relevance of cohort design for studying the frequency of the ERG rearrangement in prostate cancer. Histopathology. 2011;58:1028–36. doi: 10.1111/j.1365-2559.2011.03862.x. [DOI] [PubMed] [Google Scholar]
- 45.Tomlins SA, Rhodes DR, Yu J, Varambally S, Mehra R, et al. The role of SPINK1 in ETS rearrangement-negative prostate cancers. Cancer Cell. 2008;13:519–28. doi: 10.1016/j.ccr.2008.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ateeq B, Tomlins SA, Laxman B, Asangani IA, Cao Q, et al. Therapeutic targeting of SPINK1-positive prostate cancer. Sci Transl Med. 2011;3:72ra17. doi: 10.1126/scitranslmed.3001498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laxman B, Morris DS, Yu J, Siddiqui J, Cao J, et al. A first-generation multiplex biomarker analysis of urine for the early detection of prostate cancer. Cancer Res. 2008;68:645–9. doi: 10.1158/0008-5472.CAN-07-3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44:685–9. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blattner M, Lee DJ, O’Reilly C, Park K, MacDonald TY, et al. SPOP mutations in prostate cancer across demographically diverse patient cohorts. Neoplasia. 2014;16:14–20. doi: 10.1593/neo.131704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu W, Lindberg J, Sui G, Luo J, Egevad L, et al. Identification of novel CHD1-associated collaborative alterations of genomic structure and functional assessment of CHD1 in prostate cancer. Oncogene. 2012;31:3939–48. doi: 10.1038/onc.2011.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang S, Gulzar ZG, Salari K, Lapointe J, Brooks JD, et al. Recurrent deletion of CHD1 in prostate cancer with relevance to cell invasiveness. Oncogene. 2012;31:4164–70. doi: 10.1038/onc.2011.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garcia-Flores M, Casanova-Salas I, Rubio-Briones J, Calatrava A, Dominguez-Escrig J, et al. Clinico-pathological significance of the molecular alterations of the SPOP gene in prostate cancer. Eur J Cancer. 2014;50:2994–3002. doi: 10.1016/j.ejca.2014.08.009. [DOI] [PubMed] [Google Scholar]
- 53.Theurillat JP, Udeshi ND, Errington WJ, Svinkina T, Baca SC, et al. Prostate cancer. Ubiquitylome analysis identifies dysregulation of effector substrates in SPOP-mutant prostate cancer. Science. 2014;346:85–9. doi: 10.1126/science.1250255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.An J, Ren S, Murphy SJ, Dalangood S, Chang C, et al. Truncated ERG oncoproteins from TMPRSS2-ERG fusions are resistant to SPOP-mediated proteasome degradation. Mol Cell. 2015;59:904–16. doi: 10.1016/j.molcel.2015.07.025. [DOI] [PubMed] [Google Scholar]
- 55.Gan W, Dai X, Lunardi A, Li Z, Inuzuka H, et al. SPOP promotes ubiquitination and degradation of the ERG oncoprotein to suppress prostate cancer progression. Mol Cell. 2015;59:917–30. doi: 10.1016/j.molcel.2015.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Geng C, Rajapakshe K, Shah SS, Shou J, Eedunuri VK, et al. Androgen receptor is the key transcriptional mediator of the tumor suppressor SPOP in prostate cancer. Cancer Res. 2014;74:5631–43. doi: 10.1158/0008-5472.CAN-14-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.An J, Wang C, Deng Y, Yu L, Huang H. Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants. Cell Rep. 2014;6:657–69. doi: 10.1016/j.celrep.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boysen G, Barbieri CE, Prandi D, Blattner M, Chae SS, et al. SPOP mutation leads to genomic instability in prostate cancer. Elife. 2015;4 doi: 10.7554/eLife.09207. pii: e09207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–20. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Villers A, McNeal JE, Freiha FS, Stamey TA. Multiple cancers in the prostate. Morphologic features of clinically recognized versus incidental tumors. Cancer. 1992;70:2313–8. doi: 10.1002/1097-0142(19921101)70:9<2313::aid-cncr2820700917>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 61.Boutros PC, Fraser M, Harding NJ, de Borja R, Trudel D, et al. Spatial genomic heterogeneity within localized, multifocal prostate cancer. Nat Genet. 2015;47:736–45. doi: 10.1038/ng.3315. [DOI] [PubMed] [Google Scholar]
- 62.Ruijter ET, van de Kaa CA, Schalken JA, Debruyne FM, Ruiter DJ. Histological grade heterogeneity in multifocal prostate cancer. Biological and clinical implications. J Pathol. 1996;180:295–9. doi: 10.1002/(SICI)1096-9896(199611)180:3<295::AID-PATH663>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 63.Bostwick DG, Shan A, Qian J, Darson M, Maihle NJ, et al. Independent origin of multiple foci of prostatic intraepithelial neoplasia: comparison with matched foci of prostate carcinoma. Cancer. 1998;83:1995–2002. doi: 10.1002/(sici)1097-0142(19981101)83:9<1995::aid-cncr16>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 64.Mehra R, Tomlins SA, Yu J, Cao X, Wang L, et al. Characterization of TMPRSS2-ETS gene aberrations in androgen-independent metastatic prostate cancer. Cancer Res. 2008;68:3584–90. doi: 10.1158/0008-5472.CAN-07-6154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Attard G, Swennenhuis JF, Olmos D, Reid AH, Vickers E, et al. Characterization of ERG, AR and PTEN gene status in circulating tumor cells from patients with castration-resistant prostate cancer. Cancer Res. 2009;69:2912–8. doi: 10.1158/0008-5472.CAN-08-3667. [DOI] [PubMed] [Google Scholar]
- 66.Lindberg J, Klevebring D, Liu W, Neiman M, Xu J, et al. Exome sequencing of prostate cancer supports the hypothesis of independent tumour origins. Eur Urol. 2013;63:347–53. doi: 10.1016/j.eururo.2012.03.050. [DOI] [PubMed] [Google Scholar]
- 67.Cooper CS, Eeles R, Wedge DC, Van Loo P, Gundem G, et al. Analysis of the genetic phylogeny of multifocal prostate cancer identifies multiple independent clonal expansions in neoplastic and morphologically normal prostate tissue. Nat Genet. 2015;47:367–72. doi: 10.1038/ng.3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lalonde E, Ishkanian AS, Sykes J, Fraser M, Ross-Adams H, et al. Tumour genomic and microenvironmental heterogeneity for integrated prediction of 5-year biochemical recurrence of prostate cancer: a retrospective cohort study. Lancet Oncol. 2014;15:1521–32. doi: 10.1016/S1470-2045(14)71021-6. [DOI] [PubMed] [Google Scholar]
- 69.Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JM, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520:353–7. doi: 10.1038/nature14347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hong MK, Macintyre G, Wedge DC, Van Loo P, Patel K, et al. Tracking the origins and drivers of subclonal metastatic expansion in prostate cancer. Nat Commun. 2015;6:6605. doi: 10.1038/ncomms7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu W, Laitinen S, Khan S, Vihinen M, Kowalski J, et al. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat Med. 2009;15:559–65. doi: 10.1038/nm.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Beltran H, Yelensky R, Frampton GM, Park K, Downing SR, et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol. 2013;63:920–6. doi: 10.1016/j.eururo.2012.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Barbieri CE, Bangma CH, Bjartell A, Catto JW, Culig Z, et al. The mutational landscape of prostate cancer. Eur Urol. 2013;64:567–76. doi: 10.1016/j.eururo.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bostrom PJ, Bjartell AS, Catto JW, Eggener SE, Lilja H, et al. Genomic predictors of outcome in prostate cancer. Eur Urol. 2015;68:1033–44. doi: 10.1016/j.eururo.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 75.Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev. 2010;24:1967–2000. doi: 10.1101/gad.1965810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Saal LH, Johansson P, Holm K, Gruvberger-Saal SK, She QB, et al. Poor prognosis in carcinoma is associated with a gene expression signature of aberrant PTEN tumor suppressor pathway activity. Proc Natl Acad Sci U S A. 2007;104:7564–9. doi: 10.1073/pnas.0702507104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yoshimoto M, Joshua AM, Cunha IW, Coudry RA, Fonseca FP, et al. Absence of TMPRSS2:ERG fusions and PTEN losses in prostate cancer is associated with a favorable outcome. Mod Pathol. 2008;21:1451–60. doi: 10.1038/modpathol.2008.96. [DOI] [PubMed] [Google Scholar]
- 78.Leinonen KA, Saramaki OR, Furusato B, Kimura T, Takahashi H, et al. Loss of PTEN is associated with aggressive behavior in ERG-positive prostate cancer. Cancer Epidemiol Biomarkers Prev. 2013;22:2333–44. doi: 10.1158/1055-9965.EPI-13-0333-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Squire JA. TMPRSS2-ERG and PTEN loss in prostate cancer. Nat Gene. 2009;41:509–10. doi: 10.1038/ng0509-509. [DOI] [PubMed] [Google Scholar]
- 80.Beltran H, Eng K, Mosquera JM, Sigaras A, Romanel A, et al. Whole-exome sequencing of metastatic cancer and biomarkers of treatment response. JAMA Oncol. 2015;1:466–74. doi: 10.1001/jamaoncol.2015.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Carreira S, Romanel A, Goodall J, Grist E, Ferraldeschi R, et al. Tumor clone dynamics in lethal prostate cancer. Sci Transl Med. 2014;6:254ra125. doi: 10.1126/scitranslmed.3009448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Haffner MC, Mosbruger T, Esopi DM, Fedor H, Heaphy CM, et al. Tracking the clonal origin of lethal prostate cancer. J Clin Invest. 2013;123:4918–22. doi: 10.1172/JCI70354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sweeney CJ, Chen YH, Carducci M, Liu G, Jarrard DF, et al. Chemohormonal therapy in metastatic hormone-sensitive prostate cancer. N Engl J Med. 2015;373:737–46. doi: 10.1056/NEJMoa1503747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Reeves F, Costello AJ. Is there a place for cytoreduction in metastatic prostate cancer? BJU Int. 2015 doi: 10.1111/bju.13323. doi: 10.1111/bju.13323. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 85.Ploussard G, Almeras C, Briganti A, Giannarini G, Hennequin C, et al. Management of node only recurrence after primary local treatment for prostate cancer: a systematic review of the literature. J Urol. 2015;194:983–8. doi: 10.1016/j.juro.2015.04.103. [DOI] [PubMed] [Google Scholar]
- 86.Heidenreich A, Pfister D, Porres D. Cytoreductive radical prostatectomy in patients with prostate cancer and low volume skeletal metastases: results of a feasibility and case-control study. J Urol. 2015;193:832–8. doi: 10.1016/j.juro.2014.09.089. [DOI] [PubMed] [Google Scholar]
- 87.Karnes RJ, Murphy CR, Bergstralh EJ, DiMonte G, Cheville JC, et al. Salvage lymph node dissection for prostate cancer nodal recurrence detected by 11C-choline positron emission tomography/computerized tomography. J Urol. 2015;193:111–6. doi: 10.1016/j.juro.2014.08.082. [DOI] [PubMed] [Google Scholar]
- 88.Decaestecker K, De Meerleer G, Lambert B, Delrue L, Fonteyne V, et al. Repeated stereotactic body radiotherapy for oligometastatic prostate cancer recurrence. Radiat Oncol. 2014;9:135. doi: 10.1186/1748-717X-9-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.De Bari B, Alongi F, Buglione M, Campostrini F, Briganti A, et al. Salvage therapy of small volume prostate cancer nodal failures: a review of the literature. Crit Rev Oncol Hematol. 2014;90:24–35. doi: 10.1016/j.critrevonc.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 90.Tilki D, Mandel P, Seeliger F, Kretschmer A, Karl A, et al. Salvage lymph node dissection for nodal recurrence of prostate cancer after radical prostatectomy. J Urol. 2015;193:484–90. doi: 10.1016/j.juro.2014.08.096. [DOI] [PubMed] [Google Scholar]
- 91.Abdollah F, Briganti A, Montorsi F, Stenzl A, Stief C, et al. Contemporary role of salvage lymphadenectomy in patients with recurrence following radical prostatectomy. Eur Urol. 2015;67:839–49. doi: 10.1016/j.eururo.2014.03.019. [DOI] [PubMed] [Google Scholar]
- 92.Suardi N, Gandaglia G, Gallina A, Di Trapani E, Scattoni V, et al. Long-term outcomes of salvage lymph node dissection for clinically recurrent prostate cancer: results of a single-institution series with a minimum follow-up of 5 years. Eur Urol. 2015;67:299–309. doi: 10.1016/j.eururo.2014.02.011. [DOI] [PubMed] [Google Scholar]