

Abstract
Prostate cancer cells demonstrate a remarkable “addiction” to androgen receptor (AR) signaling in all stages of disease progression. As such, suppression of AR signaling remains the therapeutic goal in systemic treatment of prostate cancer. A number of molecular alterations arise in patients treated with AR-directed therapies. These molecular alterations may indicate the emergence of treatment resistance and may be targeted for the development of novel agents for prostate cancer. The presence of functional androgen receptor splice variants may represent a potential explanation for resistance to abiraterone and enzalutamide, newer AR-directed agents developed to treat metastatic castration-resistant prostate cancer (mCRPC). In the last 8 years, many androgen receptor splice variants have been identified and characterized. Among these, androgen receptor splice variant-7 (AR-V7) has been investigated extensively. In AR-V7, the entire COOH-terminal ligand-binding domain of the canonical AR is truncated and replaced with a variant-specific peptide of 16 amino acids. Functionally, AR-V7 is capable of mediating constitutive nuclear localization and androgen receptor signaling in the absence of androgens, or in the presence of enzalutamide. In this review, we will focus on clinical translational studies involving detection/measurement of AR-V7. Methods have been developed to detect AR-V7 in clinical mCRPC specimens. AR-V7 can be reliably measured in both tissue and circulating tumor cells derived from mCRPC patients, making it possible to conduct both cross-sectional and longitudinal clinical correlative studies. Current evidence derived from studies focusing on detection of AR-V7 in mCRPC support its potential clinical utility as a treatment selection marker.
Keywords: androgen receptor, androgen receptor splice variant-7 (AR-V7), castration-resistance prostate cancer, circulating tumor cells, treatment selection
INTRODUCTION
In 1941, Huggins and Hodges demonstrated the clinical efficacy of hormonal manipulation for the treatment of patients with metastatic prostate cancer.1 In their initial study, published in the first issue of Cancer Research,1 a serum biomarker named prostatic acid phosphatase (PAP) was used to assess the therapeutic response and track disease progression. Today, androgen deprivation therapy (ADT) involving surgical or chemical castrations is the standard of care for patients with advanced prostate cancer, and serum levels of prostate-specific antigen (PSA) are widely used as indicators of treatment response and disease progression. In men with metastatic prostate cancer treated with ADT, progression to castration-resistant prostate cancer (CRPC) almost always occurs after a variable period of clinical response,2 although notable exceptions have been reported in the literature indicating that ADT may lead to complete remission and long-term survival (>10 years) in a small subset of metastatic prostate cancer patients.3 Similarly, although serum PSA as a biomarker plays an important role in the assessment of therapeutic response and disease progression following ADT, it has very limited prognostic and predictive utility in men with metastatic prostate cancer and is not a marker of resistance to ADT.4,5
Mainly driven by compelling evidence supporting the importance of sustained androgen receptor (AR) signaling in CRPC, new AR-targeting therapies have been developed to treat mCRPC patients. It is an established concept that AR signaling remains a key driver of mCRPC.6,7 Under castrate levels of circulating androgens, AR signaling is primarily sustained by non-gonadal androgens as well as overexpression of the AR protein. This conceptual framework was corroborated by findings from early genome-wide studies8 and has directly contributed to the successful clinical development of CRPC therapies designed to further suppress ligand synthesis (abiraterone)9,10 or block the AR in the setting of AR overexpression (enzalutamide).11,12 Abiraterone and enzalutamide have been approved by the FDA to treat metastatic CRPC (mCRPC) on the basis of survival benefit, with or without prior treatment with chemotherapy.9,10,11,12
Abiraterone is a selective CYP17A1 inhibitor that effectively ablates both circulating (including adrenal-derived) and intra-tumor androgen levels.13,14,15 Enzalutamide is an AR antagonist that potently suppresses AR signaling due to its high-affinity binding to ARs.16,17 These two drugs have expanded the management options for patients with mCRPC and improved the disease outcome;18,19 indeed, they have rapidly changed the clinical landscape of prostate cancer treatment. Multiple ongoing clinical trials are likely to lead to broader indications for their use earlier in disease chronology.20 However, a significant subset of CRPC patients demonstrate primary resistance to the two agents, and nearly all treated patients eventually develop acquired resistance during the course of therapy.9,10,11,12,16,21 Although there is a clear clinical benefit of knowing whether the patient is resistant before therapy is given, no biomarker is currently available to indicate primary or acquired resistance for treatment selection.
It is now clear that the majority of patients who develop resistance to abiraterone and enzalutamide progress with rising PSA levels, suggesting a return of AR signaling despite these potent inhibitors of AR signaling.22 Activation of aberrant AR function may drive drug resistance. One candidate AR aberration is AR splice variants (AR-Vs). AR-Vs are truncated AR molecules lacking the ligand-binding domain (LBD), the intended target of all existing AR-directed therapies including abiraterone and enzalutamide, yet retains the N-terminal domain and DND-binding domain that are necessary for AR function. AR-Vs may mediate constitutively active AR signaling in the absence of ligands or in the presence of enzalutamide.20,22,23 Although many AR-Vs have been discovered and characterized, the clinical relevance for the vast majority of them remain unclear. AR-V7, the most frequently and abundantly expressed AR-V in mCRPC, has been more extensively investigated. In this review, we will discuss the pertinent literature supporting AR-V7 as a treatment selection marker in the setting of mCRPC patients treated with abiraterone and enzalutamide.
AR-V7 STRUCTURE
Previous studies published by members of our research team and collaborators have documented the discovery, characterization, and clinical relevance of a number of AR-Vs.16,20,22,24,25,26,27,28,29 On the basis of these findings, AR-V7 was identified as one of the most important AR splice variants due to its relative expression frequency and abundance, transcriptional activity, and the presence of a corresponding protein product that can be detected in mCRPC. Figure 1 (adapted from Figure 1 in reference 20) illustrates the key structural differences between the full-length AR and AR-V7. The human AR gene has eight canonical exons, color-coded in relation to the AR protein domains they encode, including the N-terminal domain (NTD) (cyan), DNA-binding domain (DBD) (yellow), the hinge region (green), and the ligand-binding domain (LBD) (purple). AR-V7 mRNA retains the first three canonical exons, followed by variant-specific cryptic exon 3 (CE3) within intron 3 (Figure 1). Splicing of CE3 results in a LBD-truncated AR-V7 protein due to premature translation termination after 16 variant-specific amino acids. These structural differences between the full-length AR transcript (AR-FL) and AR-V7 (Figure 1) allow for the specific detection of AR-FL and AR-V7. For specific detection of AR-FL and AR-V7 mRNA, RT-PCR can be performed using primer sets spanning specific splice junctions. For specific detection of AR-FL and AR-V7 protein, we have made an antibody recognizing peptide sequences specific to AR-V7 (Figure 1).
Figure 1.
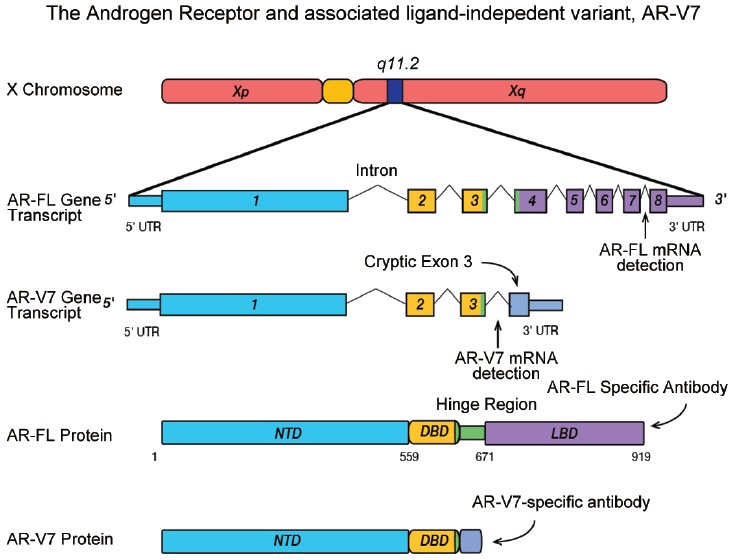
Transcript structures for the full-length AR and the splice variant AR-V7. Peptide positions are marked according to GRCh36/hg18 human genome sequences (not drawn to scale). (Adapted from Figure 1, Reference 20).
AR-V7 FUNCTION
Jenster et al. first demonstrated constitutive AR signaling activity for AR deletion mutants that retain the NH2-terminal transactivating domain and DNA-binding domain, but lack the entire ligand-binding domain.30 Many of the AR-Vs are structurally similar to the deletion mutant described in the study, but differ in the short variant-specific peptide sequence at the COOH-terminal end.27 As such, all AR-Vs lacking the ligand-binding domain are expected to have the potential to mediate constitutively active AR activity. However, the AR-Vs differ in their nuclear localization efficiency, and their transcriptional activities may be cell-context specific.26 We have previously categorized AR-Vs into three different groups on the basis of their nuclear localization efficiencies and transcriptional activities in different cell lines:26,27 constitutively active AR-Vs (e.g., AR-V7, ARv567ES), conditionally active AR-Vs (e.g., AR-V1, AR-V9), and inactive AR-Vs (e.g., AR-V13, AR-V14). For AR-V7, earlier studies have demonstrated, unequivocally, that its nuclear localization and genomic functions do not depend on the presence of the ligand or the full-length AR.24,25,26,31,32
The transcriptional output of AR-V7 function has also been investigated in detail.24,32 In our initial study, we observed that while forced AR-V7 expression can restore the expression of androgen-induced genes suppressed by androgen deprivation in LNCaP cells, many androgen-induced genes are not fully “rescued” by AR-V7.25 In addition, AR-V7 is capable of inducing a unique expression signature including genes involved in cell cycle progression.24
It is well established that the growth rate of prostate cancer cells is not proportional to the strength of AR signaling. AR-positive prostate cancer cells demonstrate a biphasic pattern of cellular response under increasing concentration of androgens. In androgen-responsive prostate cancer cells, suppressed cell growth is expected when androgen levels are too high or too low. The optimal cell-growth stimulating concentration is at the medium range, but may vary according to AR levels. One interpretation for AR genomic function is that AR-V7 expression in mCRPC may confer an optimal growth condition by inducing the expression of a unique set of genes while maintaining a minimally essential AR signature. Indeed, AR-Vs, in general, were found to have weaker genomic activity than AR-FL.33 While further investigations are needed, this interpretation is supported by findings from clinical correlative studies. In these studies, AR-V7 expression is increased after AR-FL signaling is maximally suppressed,24,34 and increased AR-V7 is associated with increased cell and tumor growth rate as well as worse clinical outcome in the context of weaker AR-FL signaling.24,31,35,36
Although AR-V7 is constitutively active and its activity does not require AR-FL, in clinical mCRPC specimens, AR-V7 was often found to co-exist with AR-FL, and the expression of AR-V7 is often less abundant than that of AR-FL.27 While AR-FL is known to form homodimers, whether AR-V7 function requires dimerization with AR-FL or itself remains controversial.37 Some studies have detected the dimerization of AR-V7 and AR-FL,38,39,40 suggesting that AR-V7 functions in mCRPC may be partly mediated by the dimer. If indeed AR-FL/AR-V7 heterodimerization is an essential step for AR-V7 function in certain cellular contexts, treatment with enzalutamide or other AR inhibitors may suppress AR-V7 function as observed in a cell line model.41 However, in other studies, AR-FL/AR-V7 heterodimer formation was not detected in prostate cancer cells expressing endogenous AR-V7.31,42,43 Due to the potential treatment implications for tumors expressing both AR-V7 and AR-FL, more studies are needed to further investigate the role of AR-FL/AR-V7 heterodimer formation. Thorough investigation in relevant cell line models followed by validation in clinical specimens may help address this important question.
DETECTION OF AR-V7 IN mCRPC
A number of methods have been used to detect AR-V7 in mCRPC specimens. These methods are summarized in Table 1. Because mature AR-V7 mRNA has a novel and unique splice junction between Exon 3 (E3) and cryptic exon 3 (CE3) within the canonical AR intron 3 sequence, it can be reliably detected and quantified by RT-PCR using primers that target the AR-V7-specific E3/CE3 junction. Samples suitable for RT-PCR analysis may include RNA extracted from flash frozen or FFPE surgical specimens, mCRPC biopsies, mCRPC autopsies, or freshly isolated circulating tumor cells (CTCs) from mCRPC patients. In our initial study reporting the discovery and characterization of AR-V7, its mRNA expression was found to be approximately 20-fold higher in mCRPC autopsies when compared with high-risk hormone naïve surgical prostate tumor specimens.25 In addition, higher levels of AR-V7 mRNA are generally accompanied by elevated expression of AR-FL, the expression of which is increased by about 10-fold in mCRPC patients.25 Because AR-FL and AR-V7 almost always coexist, and the abundance of AR-V7 is often substantially lower than that of AR-FL, a number of studies have measured their relative abundance by quantifying the ratio of AR-V7/AR-FL in different specimens, including primary tumors, mCRPC autopsies, mCRPC biopsies, and CTCs from mCRPC patients,25,36,41,44,45 using RT-PCR or RNA-Seq methods. In our study analyzing CTC cells isolated from mCRPC patients, the median ratio of AR-V7/AR-FL by RT-PCR was 21%.36 In addition, we have shown that AR-V7/AR-FL ratios in mCRPC patients undergoing treatment with abiraterone or enzalutamide may range from 12% to 25% by RNA-Seq in a few selected mCRPC autopsies.36 Other studies using mCRPC biopsies have estimated the ratio to be about 5%.45 However, the treatment histories associated with these biopsies were largely unknown. We have also previously reported the detection of AR-V7 in bulk tissue samples from primary prostate tumors as well as noncancerous prostate tissues by RT-PCR.25 Recently, a large-scale RNA-Seq study of primary prostate tumor confirmed the presence of AR-V7 in approximately 50% of specimens.44 However, comparisons of AR-V7 levels across the primary and mCRPC tumors were not made in these two large-scale RNA-Seq studies.44,45 While more studies are needed to precisely quantify the relative AR-V7 and AR-FL levels in mCRPC, current data collectively suggest that AR-V7 levels in mCRPC may be roughly equivalent to AR-FL levels in untreated high-risk primary tumors, assuming a 10-fold increase of AR-FL and a 5%–20% ratio of AR-V7/AR-FL in mCRPC. Therefore, although AR-V7 is often expressed at a lower abundance relative to AR-FL, we do not consider AR-V7 to be a low-abundance transcript because robust detection can be readily achieved in mCRPC specimens.
Table 1.
Detection methods for AR-V7 in mCRPC
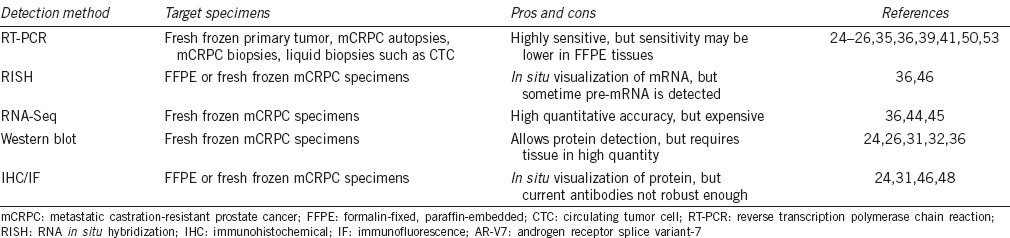
AR-V7 mRNA may also be detected by RNA in situ hybridization (RISH). RISH detection provides an added advantage by allowing visualization of AR-V7 transcripts in clinical specimens. In situ detection methods may also reveal differential expression in the tissue context and represent important tools with which to study AR expression heterogeneity. The feasibility of using RISH for the detection of AR-V7 has been demonstrated in both mCRPC biopsies and mCRPC autopsies.36,46 Because probe design for RISH requires a cDNA sequence up to 800 bp in length, we used cryptic exon 3 (CE3) sequence as the surrogate for AR-V7. The use of CE3 for probe design will exclude the possibility of nonspecific detection of other AR molecules containing upstream Exons (e.g., Exons 2 and 3), but may result in the detection of nuclear AR-V7 pre-mRNA in addition to cytoplasmic mature AR-V7 transcript. Further improvement in the sensitivity of the RISH method may address this technical limitation and allow for in situ detection of splice junctions that are only present in the mature cytoplasmic AR-V7 transcript.
The vast majority of the AR-Vs contain premature stop codons that have the potential to induce mRNA degradation through the nonsense-mediated decay (NMD) mechanism.47 It is therefore critical to demonstrate the expression of the corresponding translated product (i.e., protein) in an endogenous setting in order to determine the functional relevance. AR-V7 remains the only AR-V for which a protein product can be consistently detected in clinical specimens by both Western blot and immunohistochemical (IHC) methods. Using an AR-V7-specific mouse monoclonal antibody, the approximately 75–80 kD AR-V7 band can be reliably detected using Western blot in both prostate cancer cell lines and CRPC tissue specimens known to have elevated AR-V7 mRNA expression.24,25,26,36 The antibody has also been used for AR-V7 detection by immunofluorescence and by IHC. Within CRPC tissue specimens, predominantly, nuclear AR-V7 signal was detected in tumor cells but not in normal or stromal tissues by IHC.24 Different AR-V7-specific antibodies for IHC studies have been reported in the literature.24,31,46 Although these studies showed some promise, a common limitation with AR-V7 IHC is that the detection methods may not be a robust enough due to suboptimal detection sensitivity/specificity. The N-C subtraction method, employing antibodies recognizing AR-NTD and AR-LBD, has been used as an alternative approach to estimate the presence and abundance of LBD-truncated AR variants.48 More aggressive CRPC tumors were found to have higher ratios of AR-NTD/AR-LBD.48 Although the findings are consistent with the role of AR-Vs in CRPC, more studies are warranted to confirm the analytical validity of this approach and to determine whether the excess AR-NTD detected in CRPC specimens can be attributed to the expression of AR-Vs.
NONINVASIVE AR-V7 DETECTION IN CTCS
Using the commercially available AdnaTest36,49 CTC isolation platform, we have developed standard operating procedures for a test detecting AR-FL and AR-V7 mRNA, and performed extensive internal validation and quality control studies to determine its robustness to detect AR-FL/AR-V7 in blood samples. In AdnaTest, CTCs in 5 ml of blood are captured by magnetic beads coated with an optimized antibody combination, and RT-PCR is performed for the detection of mRNA markers of prostate tumor cells for a positive or negative CTC detection call. The CTC-positive samples are examined for the expression of AR-FL and AR-V7 using quantitative RT-PCR designed for specific detection of AR-FL and AR-V7.24,36 This laboratory-developed, RNA-based CTC AR-FL/AR-V7 test has been thoroughly evaluated and internally validated with standard quality control measures implemented. Samples collected from healthy volunteers and CTC-negative mCRPC patients were negative for AR-FL and AR-V7, excluding the possibility of false-positive detection, and confirming that contaminating leukocytes do not contribute to the AR-FL or AR-V7 signals. The test also consistently detected both AR-FL and AR-V7 in blood samples spiked with 5 LNCaP95 cells (positive for both AR-FL and AR-V7).
We have utilized this assay to test CTC-positive blood samples from prospectively enrolled patients with metastatic CRPC initiating standard-of-care treatment with either enzalutamide or abiraterone.36 In this study, we examined associations between baseline (i.e., prior to initiation of the therapies) AR-V7 detection status and treatment outcome measured by PSA response rates as well as clinical/radiographic-progression. This pilot study enrolled a total of 71 patients, 62 of which had detectable CTC and were evaluated for both AR-FL and AR-V7. Among the 62 patients, 31 were treated with enzalutamide and another 31 were treated with abiraterone. In the enzalutamide-treated group, 12 men (38.7%) were AR-V7-positive at baseline. These AR-V7-positive patients had substantially inferior PSA response rates (0% vs 52.6%) and shorter progression-free survival rates (PFS) (median: 2.1 vs 6.1 months) when compared to the 19 men who were AR-V7-negative. In the abiraterone group, 6 men (19.4%) were determined to be AR-V7-positive at baseline. Similarly, the AR-V7-positive patients treated with abiraterone had inferior PSA response rates (0% vs 68.0%) and shorter PFS (median: 2.3 months vs not reached). Importantly, significant associations were maintained after adjusting for clinical variables including prior treatments as well as expression of AR-FL. On the basis of this preliminary finding, we propose that AR-V7 may be developed as a predictive marker for treatment selection in mCRPC patients receiving abiraterone and enzalutamide. Additional prospective studies are currently ongoing to evaluate the clinical utility of this blood-based predictive biomarker.
To determine whether AR-V7 is also a relevant marker in mCRPC treated with taxane chemotherapies, we performed a second prospective study50 evaluating the association between AR-V7 detection at baseline and treatment outcome. In this study, we enrolled a total of 43 mCRPC patients, 37 of which had detectable CTC and were evaluated for both AR-FL and AR-V7. Most of these 37 patients beginning treatment with docetaxel (n = 30) or cabazitaxel (n = 7) had previously received abiraterone and/or enzalutamide. Interestingly, PSA responses and progression-free survival rates were not significantly different between AR-V7-positive (n = 17) and AR-V7-negative patients (n = 20) (41% vs 65%). Combining data from the 62 abiraterone- and enzalutamide-treated patients, we evaluated the treatment outcome by treatment type in AR-V7-positive and AR-V7-negative patients.50 In AR-V7-positive mCRPC patients, PSA response rates were higher, and progression-free survival was longer in taxane-treated men compared to enzalutamide- or abiraterone-treated men (41% vs 0%), while in AR-V7-negative mCRPC patients, these outcome measures did not appear to differ by treatment type. These findings are preliminary due to the study's small sample size. If confirmed, however, the data support the clinical utility of an AR-V7 blood test for treatment selection in mCRPC patients. Findings from a few smaller studies published recently51,52 are indeed in line with our findings.36,50
AR-V7 BEFORE AND AFTER TREATMENT
In both studies evaluating the clinical utility of CTC AR-V7 detection in mCRPC patients,36,50 all AR-V7-positive samples were also concurrently positive for AR-FL. In addition, consistent with tissue-based studies, AR-FL demonstrated greater abundance in the majority of the samples. In men with detectable AR-V7 at baseline before treatment with abiraterone or enzalutamide (n = 18), the median AR-V7/AR-FL ratio was 21.0%.36 In men with detectable AR-V7 at baseline before treatment with taxane chemotherapies (n = 17), the median AR-V7/AR-FL ratio was 22.9%.50 These ratio values are higher than the 1%–2% ratio reported in previous studies of CRPC tumors collected before abiraterone and enzalutamide were used in the clinic,25,41 and the 5%–6% ratio was reported in a recent RNA-Seq study of mCRPC patients for whom the treatment history was largely not known.45 Among the 18 AR-V7-positive men evaluated in the abiraterone/enzalutamide study,36 16 patients had ≥1 follow-up sample collected during the course of treatment for further analysis. All 16 samples remained AR-V7-positive during treatment with enzalutamide or abiraterone, and the AR-V7/AR-FL ratio was not further increased. Among the 42 patients who were negative for AR-V7 at baseline, but had ≥1 additional follow-up sample for evaluation, six patients (four on enzalutamide and two on abiraterone) subsequently “converted” to AR-V7-positive during the course of treatment with enzalutamide or abiraterone. Clinical outcomes of patients who “converted” from AR-V7-negative to AR-V7-positive were intermediate between those who remained AR-V7-negative during treatment and those who were AR-V7-positive at baseline.36 Among the 17 patients with positive AR-V7 at baseline before treatment with taxane chemotherapies,50 12 patients had ≥1 follow-up sample collected during the course of treatment for further analysis. Among these 12 patients, 7 (58%) converted to AR-V7-positive to negative status during treatment with chemotherapy.50 These results suggest that the predictive values of AR-V7 may be specific to AR-targeting therapies, and a transition in AR-V7 status may re-sensitize such patients to further AR-directed therapies.53
CLINICAL IMPLICATIONS
There are currently six treatment options available to mCRPC patients.54 In addition to the AR-targeting abiraterone and enzalutamide, docetaxel55 and cabazitaxel56 (two taxane-based chemotherapies), sipuleucel-T57 (immunotherapy), and radium-22358 (radiopharmaceutical) have all resulted in survival improvement for men with mCRPC. Although multiple prognostic markers have been identified,59 no biomarker is available for treatment selection, making it impossible to select for or against particular therapies. Therefore, there is an unmet need to develop treatment selection markers to predict or indicate therapeutic benefit before mCRPC therapies are given. In addition, because mCRPC patients are often treated sequentially, serial measurements of a selection marker may help determine optimal sequencing of the various therapies, either as single agent or in combination with others. Serial testing of a biomarker will mandate a noninvasive approach since it is not practical to perform serial metastatic biopsies, an invasive procedure, in routine clinical practice. In a recent study,53 we have shown the feasibility of conducting serial AR-V7 measurements (n = 70) in the setting of patients (n = 14) with mCRPC undergoing multiple sequential therapies (total of 37 therapies). Finally, clinical trials seeking to develop new agents for prostate cancer or to expand the indications of approved therapies may benefit from a biomarker-driven or biomarker-stratified design, in which biomarker testing results are used for patient recruitment and/or randomization. Although our findings from pilot prospective studies suggest an unprecedented opportunity to develop AR-V7 as a treatment selection marker in both standard-of-care and clinical trial settings, more studies of larger sample size are needed to further evaluate the clinical utility. Many such trials are currently ongoing.
CONCLUSIONS
Recent studies focusing on the detection of AR-V7 and its association with treatment outcome have generated promising data supporting further development of AR-V7 as a treatment selection marker in contemporary treatment settings for mCRPC patients. These studies are possible following the discovery that AR-V7 can be reliably measured in CTCs from mCRPC patients. However, we acknowledge that current data are still considered preliminary mainly due to small sample sizes, and the potential utility may be limited by the requirement for detectable CTCs. Expanded, cross-institutional studies designed to further validate AR-V7 as a treatment selection marker is ongoing, and future studies using specimens other than CTCs, including metastatic biopsies or liquid biopsies (e.g., whole-blood RNA), may be explored. Going forward, it remains critical to consider a testing platform that is compatible with noninvasive serial sampling due to the need to conduct successive testing in mCRPC patients undergoing sequential treatments. Finally, many other molecular alterations in addition to AR-V7 may also have the potential to mediate drug resistance. It is possible to improve treatment outcome prediction by combining the different markers.
COMPETING INTERESTS
The author has served as a paid consultant/advisor for Astellas, Gilead, and Sanofi; has received research funding to his institution from Orion, Mirati, Astellas, Sanofi, and Gilead; and is a co-inventor of relevant technologies that have been licensed by his institution to A&G Pharmaceutical and Tokai Pharmaceuticals. Relevant financial relationships have been disclosed, reviewed, and approved by the Johns Hopkins University in accordance with its conflict of interest policies.
ACKNOWLEDGMENTS
The author wishes to thank all collaborators and investigators who contributed to the knowledge and insight in the topic covered in this short review. The author's laboratory is currently funded by a Prostate Cancer Foundation grant, an NIH grant R01 CA185297, and US Department of Defense Prostate Cancer Research Program grants W81XWH-13-2-0093 and W81XWH-15-2-0050.
REFERENCES
- 1.Huggins C, Hodges CV. Studies on prostatic cancer. I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. J Urol 2002. 1941;167:948–51. [PubMed] [Google Scholar]
- 2.Eisenberger MA, Blumenstein BA, Crawford ED, Miller G, McLeod DG, et al. Bilateral orchiectomy with or without flutamide for metastatic prostate cancer. N Engl J Med. 1998;339:1036–42. doi: 10.1056/NEJM199810083391504. [DOI] [PubMed] [Google Scholar]
- 3.Reiner WG, Scott WW, Eggleston JC, Walsh PC. Long term survival after hormonal therapy for stage D prostatic cancer. J Urol. 1979;122:183–4. [PubMed] [Google Scholar]
- 4.Bubley GJ, Carducci M, Dahut W, Dawson N, Daliani D, et al. Eligibility and response guidelines for phase II clinical trials in androgen independent prostate cancer: recommendations from the Prostate Specific Antigen Working Group. J Clin Oncol. 1999;17:3461–7. doi: 10.1200/JCO.1999.17.11.3461. [DOI] [PubMed] [Google Scholar]
- 5.Scher HI, Halabi S, Tannock I, Morris M, Sternberg CN, et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol. 2008;26:1148–59. doi: 10.1200/JCO.2007.12.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Attard G, Cooper CS, de Bono JS. Steroid hormone receptors in prostate cancer: a hard habit to break? Cancer Cell. 2009;16:458–62. doi: 10.1016/j.ccr.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 7.Chen Y, Sawyers CL, Scher HI. Targeting the androgen receptor pathway in prostate cancer. Curr Opin Pharmacol. 2008;8:440–8. doi: 10.1016/j.coph.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knudsen KE, Scher HI. Starving the addiction: new opportunities for durable suppression of AR signaling in prostate cancer. Clin Cancer Res. 2009;15:4792–8. doi: 10.1158/1078-0432.CCR-08-2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368:138–48. doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. 2014;371:1755–6. doi: 10.1056/NEJMoa1405095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–97. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 13.Antonarakis ES, Eisenberger MA. Is abiraterone acetate well tolerated and effective in the treatment of castration resistant prostate cancer? Nat Clin Pract Oncol. 2009;6:12–3. doi: 10.1038/ncponc1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Attard G, Reid AH, A’Hern R, Parker C, Oommen NB, et al. Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration resistant prostate cancer. J Clin Oncol. 2009;27:3742–8. doi: 10.1200/JCO.2008.20.0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reid AH, Attard G, Danila DC, Oommen NB, Olmos D, et al. Significant and sustained antitumor activity in post docetaxel, castration resistant prostate cancer with the CYP17 inhibitor abiraterone acetate. J Clin Oncol. 2010;28:1489–95. doi: 10.1200/JCO.2009.24.6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scher HI, Beer TM, Higano CS, Anand A, Taplin ME, et al. Antitumour activity of MDV3100 in castration resistant prostate cancer: a phase 1 2 study. Lancet. 2010;375:1437–46. doi: 10.1016/S0140-6736(10)60172-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, et al. Development of a second generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–90. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Antonarakis ES, Eisenberger MA. Expanding treatment options for metastatic prostate cancer. N Engl J Med. 2011;364:2055–8. doi: 10.1056/NEJMe1102758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Omlin A, Pezaro C, Mukherji D, Mulick Cassidy A, Sandhu S, et al. Improved survival in a cohort of trial participants with metastatic castration resistant prostate cancer demonstrates the need for updated prognostic nomograms. Eur Urol. 2013;64:300–6. doi: 10.1016/j.eururo.2012.12.029. [DOI] [PubMed] [Google Scholar]
- 20.Nakazawa M, Antonarakis ES, Luo J. Androgen receptor splice variants in the era of enzalutamide and abiraterone. Horm Cancer. 2014;5:265–73. doi: 10.1007/s12672-014-0190-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ryan CJ, Molina A, Griffin T. Abiraterone in metastatic prostate cancer. N Engl J Med. 2013;368:1458–9. doi: 10.1056/NEJMc1301594. [DOI] [PubMed] [Google Scholar]
- 22.Ferraldeschi R, Welti J, Luo J, Attard G, de Bono JS. Targeting the androgen receptor pathway in castration resistant prostate cancer: progresses and prospects. Oncogene. 2014;34:1745–57. doi: 10.1038/onc.2014.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ware KE, Garcia Blanco MA, Armstrong AJ, Dehm SM. Biologic and clinical significance of androgen receptor variants in castration resistant prostate cancer. Endocr Relat Cancer. 2014;21:T87–103. doi: 10.1530/ERC-13-0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, et al. Distinct transcriptional programs mediated by the ligand dependent full length androgen receptor and its splice variants in castration resistant prostate cancer. Cancer Res. 2012;72:3457–62. doi: 10.1158/0008-5472.CAN-11-3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, et al. Ligand independent androgen receptor variants derived from splicing of cryptic exons signify hormone refractory prostate cancer. Cancer Res. 2009;69:16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu R, Isaacs WB, Luo J. A snapshot of the expression signature of androgen receptor splicing variants and their distinctive transcriptional activities. Prostate. 2011;71:1656–67. doi: 10.1002/pros.21382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu C, Luo J. Decoding the androgen receptor splice variants. Transl Androl Urol. 2013;2:178–86. doi: 10.3978/j.issn.2223-4683.2013.09.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Plymate SR, Luo J. Androgen Responsive Genes in Prostate Cancer. New York, USA: Springer; 2013. The expression signature of androgen receptor splice variants and their distinctive transcriptional activities in castration resistant prostate cancer; pp. 201–13. [Google Scholar]
- 29.Hu R, Denmeade SR, Luo J. Molecular processes leading to aberrant androgen receptor signaling and castration resistance in prostate cancer. Expert Rev Endocrinol Metab. 2010;5:753–64. doi: 10.1586/eem.10.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jenster G, van der Korput JA, Trapman J, Brinkmann AO. Functional domains of the human androgen receptor. J Steroid Biochem Mol Biol. 1992;41:671–5. doi: 10.1016/0960-0760(92)90402-5. [DOI] [PubMed] [Google Scholar]
- 31.Guo Z, Yang X, Sun F, Jiang R, Linn DE, et al. A novel androgen receptor splice variant is up regulated during prostate cancer progression and promotes androgen depletion resistant growth. Cancer Res. 2009;69:2305–13. doi: 10.1158/0008-5472.CAN-08-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, et al. Androgen receptor splice variants mediate enzalutamide resistance in castration resistant prostate cancer cell lines. Cancer Res. 2013;73:483–9. doi: 10.1158/0008-5472.CAN-12-3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan SC, Selth LA, Li Y, Nyquist MD, Miao L, et al. Targeting chromatin binding regulation of constitutively active AR variants to overcome prostate cancer resistance to endocrine based therapies. Nucleic Acids Res. 2015;43:5880–97. doi: 10.1093/nar/gkv262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu Z, Chen S, Sowalsky AG, Voznesensky OS, Mostaghel EA, et al. Rapid induction of androgen receptor splice variants by androgen deprivation in prostate cancer. Clin Cancer Res. 2014;20:1590–600. doi: 10.1158/1078-0432.CCR-13-1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hornberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, et al. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration resistance and short survival. PLoS One. 2011;6:e19059. doi: 10.1371/journal.pone.0019059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, et al. AR V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371:1028–38. doi: 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Antonarakis ES, Luo J. Prostate cancer: AR splice variant dimerization clinical implications. Nat Rev Urol. 2015;12:431–3. doi: 10.1038/nrurol.2015.184. [DOI] [PubMed] [Google Scholar]
- 38.Liang M, Adisetiyo H, Liu X, Liu R, Gill P, et al. Identification of androgen receptor splice variants in the pten deficient murine prostate cancer model. PLoS One. 2015;10:e0131232. doi: 10.1371/journal.pone.0131232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120:2715–30. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu D, Zhan Y, Qi Y, Cao B, Bai S, et al. Androgen receptor splice variants dimerize to transactivate target genes. Cancer Res. 2015;75:3663–71. doi: 10.1158/0008-5472.CAN-15-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, et al. Constitutively active androgen receptor splice variants expressed in castration resistant prostate cancer require full length androgen receptor. Proc Natl Acad Sci U S A. 2010;107:16759–65. doi: 10.1073/pnas.1012443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang L, Lin C, Jin C, Yang JC, Tanasa B, et al. lncRNA dependent mechanisms of androgen receptor regulated gene activation programs. Nature. 2013;500:598–602. doi: 10.1038/nature12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tepper CG, Boucher DL, Ryan PE, Ma AH, Xia L, et al. Characterization of a novel androgen receptor mutation in a relapsed CWR22 prostate cancer xenograft and cell line. Cancer Res. 2002;62:6606–14. [PubMed] [Google Scholar]
- 44.Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell. 2015;163:1011–25. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–28. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Welti J, Rodrigues DN, Sharp A, Sun S, Lorente D, et al. Analytical validation and clinical qualification of a new immunohistochemical assay for androgen receptor splice variant-7 protein expression in metastatic castration-resistant prostate cancer. Eur Urol. 2016 doi: 10.1016/j.eururo.2016.03.049. Doi: 10.1016/j.eururo.2016.03.049. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pan Q, Saltzman AL, Kim YK, Misquitta C, Shai O, et al. Quantitative microarray profiling provides evidence against widespread coupling of alternative splicing with nonsense mediated mRNA decay to control gene expression. Genes Dev. 2006;20:153–8. doi: 10.1101/gad.1382806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang X, Morrissey C, Sun S, Ketchandji M, Nelson PS, et al. Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PLoS One. 2011;6:e27970. doi: 10.1371/journal.pone.0027970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alix Panabieres C, Pantel K. Circulating tumor cells: liquid biopsy of cancer. Clin Chem. 2013;59:110–8. doi: 10.1373/clinchem.2012.194258. [DOI] [PubMed] [Google Scholar]
- 50.Antonarakis ES, Lu C, Luber B, Wang H, Chen Y, et al. Androgen receptor splice variant 7 and efficacy of taxane chemotherapy in patients with metastatic castration resistant prostate cancer. JAMA Oncol. 2015;1:582–91. doi: 10.1001/jamaoncol.2015.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Onstenk W, Sieuwerts AM, Kraan J, Van M, Nieuweboer AJ, et al. Efficacy of cabazitaxel in castration resistant prostate cancer is independent of the presence of AR V7 in circulating tumor cells. Eur Urol. 2015;68:939–45. doi: 10.1016/j.eururo.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 52.Steinestel J, Luedeke M, Arndt A, Schnoeller TJ, Lennerz JK, et al. Detecting predictive androgen receptor modifications in circulating prostate cancer cells. Oncotarget. 2015 doi: 10.18632/oncotarget.3925. Doi: 10.18632/oncotarget.3925. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakazawa M, Lu C, Chen Y, Paller CJ, Carducci MA, et al. Serial blood based analysis of AR V7 in men with advanced prostate cancer. Ann Oncol. 2015;26:1859–65. doi: 10.1093/annonc/mdv282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Basch E, Loblaw DA, Oliver TK, Carducci M, Chen RC, et al. Systemic therapy in men with metastatic castration resistant prostate cancer: American Society of Clinical Oncology and Cancer Care Ontario clinical practice guideline. J Clin Oncol. 2014;32:3436–48. doi: 10.1200/JCO.2013.54.8404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502–12. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 56.de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels JP, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration resistant prostate cancer progressing after docetaxel treatment: a randomised open label trial. Lancet. 2010;376:1147–54. doi: 10.1016/S0140-6736(10)61389-X. [DOI] [PubMed] [Google Scholar]
- 57.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, et al. Sipuleucel T immunotherapy for castration resistant prostate cancer. N Engl J Med. 2010;363:411–22. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 58.Parker C, Nilsson S, Heinrich D, Helle SI, O’sullivan JM, et al. Alpha emitter radium 223 and survival in metastatic prostate cancer. N Engl J Med. 2013;369:213–23. doi: 10.1056/NEJMoa1213755. [DOI] [PubMed] [Google Scholar]
- 59.Armstrong AJ, Eisenberger MA, Halabi S, Oudard S, Nanus DM, et al. Biomarkers in the management and treatment of men with metastatic castration resistant prostate cancer. Eur Urol. 2012;61:549–59. doi: 10.1016/j.eururo.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]