

Abstract
In spite of the development of new treatments for late stage prostate cancer, significant challenges persist to match individuals with effective targeted therapies. Genomic classification using high-throughput sequencing technologies has the potential to achieve this goal and make precision medicine a reality in the management of men with castrate-resistant prostate cancer. This chapter reviews some of the most recent studies that have resulted in significant progress in determining the landscape of somatic genomic alterations in this cohort and, more importantly, have provided clinically actionable information that could guide treatment decisions. This chapter reviews the current understanding of common alterations such as alterations of the androgen receptor and PTEN pathway, as well as ETS gene fusions and the growing importance of PARP inhibition. It also reviews recent studies that characterize the evolution to neuroendocrine tumors, which is becoming an increasingly important clinical problem. Finally, this chapter reviews recent innovative studies that characterize the compelling evolutionary history of lethal prostate cancer evidenced by polyclonal seeding and interclonal cooperation between metastasis and the importance of tumor clone dynamics measured serially in response to treatment. The genomic landscape of late stage prostate cancer is becoming better defined, and the prospect for assigning clinically actionable data to inform rationale treatment for individuals with this disease is becoming a reality.
Keywords: advanced prostate cancer, genomic alterations, lethal prostate cancer, precision medicine, targeted therapy, whole-genome sequencing
INTRODUCTION
Prostate cancer is the leading cause of cancer death in men worldwide. Men who develop metastasis typically receive androgen deprivation therapy (ADT) as their central therapy. ADT has been shown to result in a median survival of approximately 6 years.1 Recently, two large clinical trials have demonstrated that the addition of docetaxel chemotherapy to ADT significantly prolonged survival.2,3 Unfortunately, virtually all men will develop resistance to primary ADT, a state known as metastatic castrate-resistant prostate cancer (mCRPC). Over the past 5 years, several “second-generation” ADT drugs including abiraterone and enzalutamide have been developed, which have demonstrated significant clinical benefit and prolongation of survival in men with mCRPC.4,5 Unfortunately, some men fail to respond to these drugs and of those that respond all will eventually progress due to development of resistance and die from their disease. Clearly, many challenges persist in the management of men with mCRPC, and the most important is the challenge of matching patients with targeted therapies. High-throughput sequencing technologies have accelerated the molecular characterization of prostate cancer, which has led to the development of precision medicine for therapeutic decision making for men with advanced prostate cancer.6 This chapter will examine the current data on genetic alterations in advanced prostate cancer, the potential therapeutic targets of these alterations, the tumor clone dynamics of lethal prostate cancer metastases, and the challenges in the future for translational genomics in prostate cancer.
The genomic landscape of primary prostate cancer has been the focus of considerable research. Investigations of DNA and RNA genomic information in primary prostate tumors have led to the development of three FDA-approved and Medicare-covered tests.7 These tests mainly provide prognostic information to identify men at risk for recurrence and development of metastases. Other studies have identified recurrent somatic mutations, copy number alterations, and DNA rearrangements in primary prostate cancer, which have also demonstrated prognostic information but minimal therapeutic impact.8,9 The genomic landscape of metastatic prostate cancer is under considerable investigation; however, to date, there have been no FDA-approved assays developed to predict prognosis or response to treatment. Until recently, most investigators relied on autopsy samples or preclinical models; genomic characterization of fresh biopsy samples from living mCRPC individuals was limited due to challenges in obtaining adequate tumor tissue, particularly from bone metastasis. To meet these challenges, an international consortium of eight academic centers cooperated to develop a clinical sequencing infrastructure to conduct prospective whole-exome and transcriptome sequencing of bone or soft tissue tumor biopsies from a cohort of 150 mCRPC affected individuals receiving various treatments, including abiraterone, enzalutamide, and docetaxel.10 This large coordinated effort, which will be designated as the “International Consortium” in this chapter, was designed to determine the landscape of somatic genomic alterations in this cohort, assess differences between primary and metastatic prostate cancer, and discover potential clinically actionable information that could impact treatment decisions. In this study, 89% of affected individuals were found to harbor a clinically actionable aberration (65% of cases even when alterations of the androgen receptor were not considered). These will be explored in more detail later in this chapter. Figure 1 depicts a graphical abstract of the study.
Figure 1.
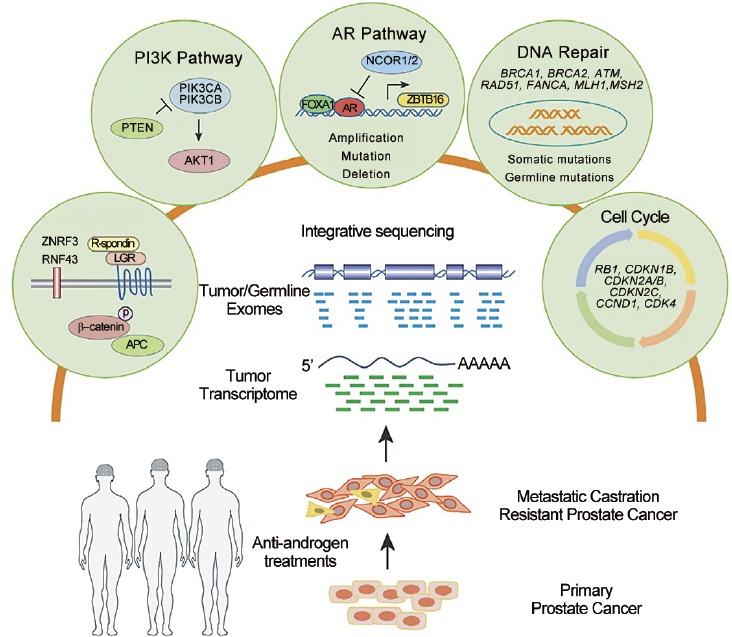
This graphical abstract outlines the multi-institutional integrative clinical sequencing analysis of samples from mCRPC individuals and depicts five pathways that harbor clinically actionable genomic alterations.10
ETS GENE FUSIONS
The most common chromosomal rearrangements in prostate cancer involve the 5’ untranslated region of the androgen-regulated gene TMPRSS2 and members of the ETS transcription family, ERG, or ETV1.11 Studies have confirmed ETS-related gene fusions in approximately 50% of primary prostate tumors. In the International Consortium study, recurrent ETS fusions were observed in metastatic tissues in 56% of patients, of which the majority was fused to ERG.10
Currently, there are no therapies in development that directly target this gene fusion. There are promising approaches, including synthetic lethality treatments and inhibition of specific protein-protein interactions of ETS transcriptional factors that are emerging. More specifically, there are preclinical data that provide a rationale for targeting components of the transcription factor complex associated with ETS through poly(ADP-ribose) polymerase (PARP) or DNA protein kinase inhibition.12 Therefore, knowledge of ETS gene fusion status, although not directly actionable, may allow therapy targeted to associated transcription factors that involve DNA repair pathways.
PARP AND DNA REPAIR PATHWAY
There is evidence that various ETS fusion transcription factors (predominantly ERG) interact in a DNA-independent manner with PARP1 and the catalytic subunit of DNA protein kinase (DNA-PK). PARP1 and DNA-PKcs function as coregulators of ERG transcriptional activity and ERG recruits PARP1 and DNA-PK to specific genomic loci during transcription.13 PARP1 and DNA-PK are also required for ERG-mediated cell invasion, intravasation, and metastasis. It was hypothesized that inhibition of PARP1 would inhibit ETS-positive prostate cancer growth; this was tested in xenograft models and demonstrated significant reduction in tumor growth in ETS-positive but not in ETS-negative xenografts.14 In addition, it was shown that the alkylating agent temozolomide potentiated the effect of a PARP inhibitor in several xenograft models.15 It has also been shown that ETS transcription factors drive DNA double-strand break formation. As a result, ETS-positive cancers may be susceptible to accumulation of DNA damage following inhibition of the DNA-repairing enzyme PARP1. This was demonstrated when the PARP inhibitor Olaparib was found to result in an increase in total levels of DNA damage when used to treat PC3-ERG cells or PC-3 cells with BRCA2 knockdown. These data demonstrate that similar to BRCA1/2-deficient tumors, ETS-positive, but not ETS-negative, prostate cancer models are susceptible to PARP inhibition through increased incidence of DNA double-strand breaks.14 Taken together, the existing data support that ETS-positive tumors are expected to respond with a higher probability of PARP inhibition than ETS-negative tumors, potentially making ETS status an important predictive biomarker. Further, adding PARP inhibition to other known prostate cancer treatments, such as abiraterone acetate or chemotherapy, may potentiate this effect. In fact, there is currently an ongoing clinical trial that stratifies mCRPC affected individuals based on ETS status assessed from tumor tissue from a metastatic site and then tests abiraterone acetate with or without the PARP1 inhibitor Veliparib (NCT01576172 – M Hussain PI). In this important Stand Up to Cancer study, valuable clinical information will be obtained to determine the role of ETS status and PARP inhibition as well as the effect of adding a PARP inhibitor to an effective treatment for mCRPC. In addition, use of a PARP inhibitor in unselected mCRPC individuals demonstrated that those achieving clinical benefit harbored biallelic BRCA2 loss.16
The International Consortium reported that integrative analysis of both somatic and pathogenic germline alterations in BRCA2 identified 12.7% of cases with loss of BRCA2, of which 90% exhibited biallelic loss.10 Pathogenic germline BRCA2 mutations were seen in 5.3% individuals with a subsequent somatic event that resulted in biallelic loss. This is a higher frequency than previously reported in primary prostate cancer. This group expanded the focus to other DNA repair/recombination genes and identified alterations in 22.7% of cases. These included biallelic loss of ATM, as well as BRCA1, CDK12, FANCA, RAD51B, and RAD51C. They concluded that if aberrations in BRCA1, BRCA2, and ATM all confer enhanced sensitivity to PARP inhibitors, 19.3% of mCRPC individuals would be predicted to benefit from this therapy.
There are emerging data that the DNA repair kinase DNA-PK functions as a selective modulator of transcriptional networks inducing cell migration, invasion, and metastases. Goodwin et al. recently characterized this repair kinase and identified its ability to promote metastases that it is upregulated in highly aggressive tumors and it predicts for poor survival.17 More importantly, these emerging data nominate DNA-PK as a promising therapeutic target.
ANDROGEN RECEPTOR (AR)
It has been known, since 1940, that hormones regulate prostate cancer, and it is now known that androgen signaling remains important despite castrate levels of testosterone. This occurs via various mechanisms, some of which include mutation or amplification of AR, presence of intratumoral testosterone production, and the presence of splice variants of the AR receptor.18,19,20,21 This knowledge has fueled the development of next-generation drugs such as abiraterone that blocks CYP17 used for androgen synthesis in the adrenal glands, testes, and within prostate tumors, and enzalutamide that competitively blocks AR, AR/ligand translocation into the nucleus, and binding to AR transcriptional elements in the nucleus. These drugs have demonstrated significant clinical benefit in individuals with mCRPC with improved overall and progression-free survival. Figure 2 depicts the AR pathway.
Figure 2.
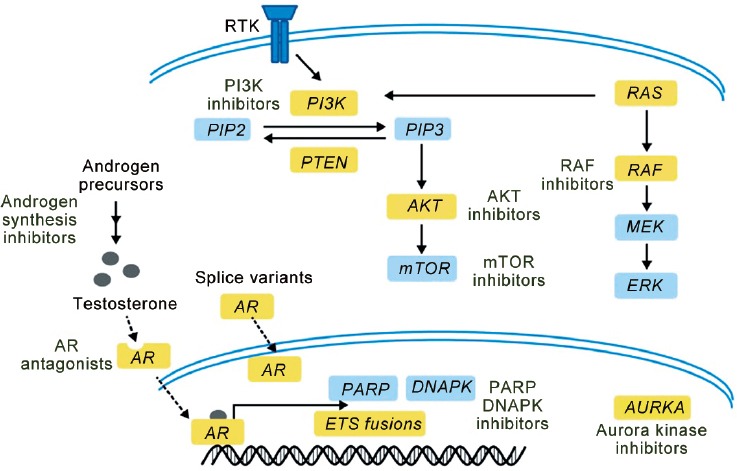
Pathway-guided treatment in prostate cancer. This diagram highlights pathways for targeting in prostate cancer.6
Current data suggest that more than 50% of mCRPC tumors display AR amplification or mutation.22 In addition, alternatively multiple spliced isoforms of AR that result in constitutively active androgen signaling have been described.21,23 The International Consortium found that in aggregate, 71.3% of cases harbored AR pathway aberrations, the majority of which were direct alterations affecting AR through amplification and mutation.10 In addition, alterations in AR pathway members were observed, including alterations in FOXA1 as a transcription factor, NCOR1/2 as negative regulators of AR, SPOP as a putative AR transcriptional regulator, and ZBTB16 that may negatively regulate AR.24,25,26
Currently, determination of AR alterations to inform treatment decisions remains investigational and is studied in various clinical research trials. Much attention has been paid to the relationship between AR splice variants and resistance to abiraterone and enzalutamide, and this important area is discussed elsewhere in this review. The presence of AR copy number alterations (CNAs) in cell-free DNA collected before treatment with abiraterone showed that median progression-free survival (PFS) and overall survival (OS) of patients with AR gene gain were significantly lower than nongained cases.27 In addition, patients with CYP17A1 gene gain had a lower PFS and OS than nongained cases. Others have also reported that AR copy number gain is associated with resistance to treatment with second-generation antiandrogens. Carreira et al. sequenced serial plasma and tissue samples from 16 ERG-positive patients with mCRPC treated with abiraterone or enzalutamide and showed a temporal association between clinical progression on treatment with emergence of AR mutations.28 Interestingly, AR mutations activated by glucocorticoids were found in about 20% of patients progressing on abiraterone and prednisolone or dexamethasone. These data support the use of AR alterations to predict resistance to AR-targeted therapy. There are less data available that inform treatment based on targeting other members of the AR pathway. Yang and Yu29 have reported on the role of FOXA1 in mCRPC tumors and are studying the effects of inhibitors of downstream TGF-β to render these tumors hormone-sensitive again and improve response to AR-targeted therapy.
PTEN AND P13K PATHWAY
The PTEN gene is a tumor suppressor that negatively regulates the PI3K-AKT-mTOR pathway. Thus, either loss or inactivation of PTEN genes leads to PI3K pathway activation. Somatic loss of PTEN occurs in multiple cancer types and provides a mechanistic rationale for PI3K pathway inhibitors for treatment.30 Figure 2 depicts the PTEN/PI3K/mTOR pathway. Genomic evidence of PTEN loss in prostate cancer through point mutation, deletion, or rearrangement has been observed in at least 50% of mCRPC cases.23,31 Epigenetic or posttranslational events may also result in functional PTEN inactivation. Studies have also demonstrated a relationship between ERG fusions and PTEN loss.32
The International Consortium reported that the PI3K pathway was commonly altered in mCRPC tumors, with somatic alterations in 49% of cases.10 This included biallelic loss of PTEN, as well as hotspot mutations, amplifications, and activating fusions in PIK3CA with resultant overexpression and activating mutations in AKT1. AKT is a serine/threonine protein kinase and is a component of the PI3K pathway. Boormans et al. observed mutations of AKT in 1.4% of 188 prostate cancer specimens.33 The International Consortium observed AKT mutations in 1.3% of cases. Mutations in another member of the PI3K catalytic subunit, PIK3CA and PIK3CB were also observed in 5.3% and 6.3% of cases, respectively.
PTEN loss and PI3K and AKT pathway alterations provide potential targets for treatment using inhibitors of the mTOR pathway. Marques et al.34 studied the AKT inhibitor AZD5363 and the PI3K inhibitor AZD8186 in combination with castration in PTEN-negative and PTEN-positive prostate cancer xenografts and observed significant growth inhibition of the PTEN-negative xenografts. Armstrong et al.35 studied BKM120 (buparlisib), an oral pan-class I PI3K inhibitor, in heavily pretreated men with mCRPC. Although there was no observed improvement in PFS over historic control data, this study did not select for PTEN loss. Vaishampayan et al.36 studied everolimus, an oral mTOR inhibitor, in combination with carboplatin in unselected mCRPC patients. There was minimal activity observed, but two patients with prolonged responses showed negative immunohistochemical staining for AKT. Other mTOR inhibitors, including ridaforolimus and temsirolimus, are tested in mCRPC individuals. With the ability to select for mCRPC individuals with alterations of the PI3K-mTOR pathway via tissue biopsy or cell-free DNA or CTCs, treatment with agents that inhibit this pathway will hopefully show benefit.
NEUROENDOCRINE TUMORS
Neuroendocrine prostate cancer (NEPC) is an aggressive subtype of prostate cancer that can arise de novo but much more commonly arises after treatment with hormonal therapy.37 It frequently metastasizes to visceral organs, responds briefly to chemotherapy, and patients typically survive <1 year. Histologically, NEPC differs from prostate cancer by the presence of small, round, and blue neuroendocrine cells which do not express androgen receptor or secrete PSA, but usually express neuroendocrine markers such as chromogranin A, synaptophysin, and neuron-specific enolase.38 The TMPRSS2-ERG gene fusion has been reported in approximately 50% of NEPC cases and suggests that NEPC is clonally derived from prostate cancer.39 The development of NEPC appears to be increasing as patients with mCRPC are receiving second-generation AR-targeted therapies such as abiraterone and enzalutamide. Poor molecular characterization of NEPC accounts for the lack of disease-specific therapies.
Beltran et al.40 studied 45 NEPC tumors using next-generation RNA sequencing and oligonucleotide arrays. Immunohistochemical analysis revealed the presence of the TMPRSS2-ERG gene fusion in 44% of cases, but NEPC foci lacked both ERG and AR expression. NGS analysis confirmed that NEPC showed low expression of known androgen-regulated genes and high expression of neuroendocrine-associated genes. Significant overexpression and gene amplification of Aurora kinase A (AURKA) were observed. AURKA is a serine/threonine kinase involved in mitotic spindle formation, centrosome separation, and the G2-M transition during the cell cycle and has oncogenic properties. Transfection of AURKA into a benign prostate cell line was shown to induce neuroendocrine differentiation as expressed by neuroendocrine markers SYP and NSE that could be suppressed by knockdown of the gene. They demonstrated that an MYCN-transfected LNCaP cell line and a cell line obtained from a patient with NEPC showed enhanced in vitro sensitivity to the Aurora kinase inhibitor PHA-739358 compared with control LNCaP cells. The inhibitor also resulted in significant tumor shrinkage in two NEPC xenografts.
This group also observed MYCN overexpression and amplification in NEPC tumors. Of interest, concurrent amplification of AURKA was observed in nearly all MYCN-amplified cases and experiments demonstrated costabilization between the two genes. Interaction of these two genes has also been demonstrated in neuroblastoma, another high aggressive neuroendocrine tumor. MYC has also been observed to occur with PI3K activation. Thus, targeting MYC and AURKA is an active area of clinical research with several ongoing trials utilizing AURKA inhibitors.
More recently, Small et al.41 reported characterization of NEPC in patients with mCRPC resistant to abiraterone or enzalutamide. These preliminary results from the Stand Up to Cancer West Coast Dream Team showed that pathology from metastatic tumor samples identified classic small cell histology in 12% of cases. Of interest, a distinct histology intermediate to small cell cancer and adenocarcinoma was observed in 27% of cases. They concluded that the development of NEPC in mCRPC resistant to second-generation AR-targeted therapies is far more common than previously appreciated and results in poor survival. They performed RNAseq and expression analysis to develop a 50- gene expression signature that should provide insight into the biology and treatment of NEPC.
Individuals with mCRPC with a clinically defined aggressive variant (AVPCa) have tumors that share clinical features with small cell cancer. Aparicio et al.42 performed immunostaining and subjected DNA to OncoScan evaluation for copy number alterations (CNAs) in these tumors. 67% stained negative for RB1 and 32% for AR. Negative AR staining was positively correlated with RB1, NKX3-1, and AURKA. The most common CNA were 8q24 amplification (MYC), PTEN loss, and RB1 loss. This study demonstrates that progression to the aggressive phenotype may require multiple genetic alterations.
CELL-CYCLE PATHWAY
Retinoblastoma (RB1) is a tumor suppressor gene with somatic alterations in multiple cancers. RB1 represses cell cycle activity via transcription of genes involved in DNA synthesis and mitosis. There are various mechanisms to somatically inactivate RB1, which occur through genomic deletion in prostate cancer and have been observed in 20%–60% of cancers.43 The International Consortium observed RB1 loss in 21% of cases. They observed other cell cycle genes in mCRPC including focal amplifications involving CCND1 in 9% of cases as well as less common (<5%) events in CDKN2A/B, CDKN1B, and CDK4.10 These cell cycle alterations may result in enhanced response to CDK4 inhibitors and preclinical models of mCRPC predict activity in prostate cancer. Palmbos et al.44 studied PD0332991 (palbociclib), a novel specific inhibitor of CDK4, in RB-positive mCRPC individuals. In addition, indirect targeting of RB1 inactivation is explored through MDM2 and HDAC inhibitors.45
RAS-RAF PATHWAY
Genomic studies of prostate cancer have demonstrated canonical BRAF and KRAS mutations in approximately 1%–2% of cases.23,46 Mutations of BRAF result in constitutive activation of RAF kinase. In addition to the successful application of RAF and MEK inhibitors in BRAF-mutant melanoma, other inhibitors are in development for other BRAF-mutant cancers. Activating gene fusions involving BRAF and KRAS has been observed in 1%–2% of prostate cancer cases. These gene rearrangements result in the expression and constitutive activity of an RAF kinase that is sensitive to RAF inhibitors. Although relatively rare, these RAS-RAF pathway alterations represent potential treatment targets in mCRPC individuals. Figure 2 depicts the RAF pathway.
WNT PATHWAY
The International Consortium reported that 27/150 (18%) cases harbored alterations in the Wnt signaling pathway.10 This has not been previously reported. Activating mutations were seen in CTNNB1, but alterations in APC were also observed, which have not been previously described in mCRPC individuals. Through integrative analysis, alterations in other Wnt signaling genes, including RNF43 and ZNRF3, were identified.47 R-spondin fusions involving RSP02 associated with RSP02 overexpression were also observed. Overall, 6% of affected individuals with aberrations in RNF43, ZNRF3, or RSP02 were identified. Individuals with these aberrations would be predicted to respond to porcupine inhibitors.48
EVOLUTIONARY HISTORY OF METASTASIS
It is unclear if a single clone metastasizes and remains dominant over the course of tumor evolution or whether multiple foci with different genomic patterns at diagnosis metastasize. Liu et al.49 performed copy number analysis on 94 separate cancer sites from 30 men who died of prostate cancer and concluded that copy number analysis data confirmed the monoclonal origin of lethal metastatic prostate cancer. More recently, the concept of polyclonal seeding has been introduced and studied. Gundem et al.50 used whole-genome sequencing to characterize multiple metastases arising in prostate tumors in ten patients. They grouped clonal and subclonal mutations within each sample. By plotting the cancer cell fractions of mutations from pairs of samples, they determined the clonal relationship between the constituent subclones and found evidence for polyclonal seeding of metastasis. They observed that most tumor sites harbored clonal mutations indicative of truncal mutations but also harbored multiple subclonal mutations. They concluded that metastasis usually occurs in the form of spreading among distant sites, rather than as separate waves of invasion directly from the primary tumor. They found that lesions affecting tumor suppressor genes usually occurred as single events whereas mutations in genes involved in AR signaling commonly involved multiple, convergent events in different metastases. These observations support the “seed and soil” hypothesis in which rare subclones develop metastatic potential within the primary tumor. Over time, clonal diversification occurs as a result of constraining necessity to bypass ADT. These important observations underscore the complexity of genetic alterations within metastases and the existence of multiple clonal and subclonal alterations that require careful analysis using whole-genome sequencing.
TUMOR CLONE DYNAMICS AND SERIAL TESTING
The aforementioned somatic genetic alterations typically originate from a single biopsy sample at a single point in time in a patient with mCRPC. Individuals with mCRPC undergo sequential treatments over the course of their disease including castration therapy as well as second-generation AR-targeted drugs such as abiraterone and enzalutamide and frequently chemotherapy. The effect of various treatments over time on clonal dynamics and genomic alterations is not well understood. To better understand these tumor clone dynamics, Carreira et al.28 sequenced serial plasma and tumor samples from 16 ERG-positive patients. They identified multiple independent clones in metastatic disease that are differentially represented in tissue and circulation. They also showed in several patients that clonality was not conserved over time and observed continuously changing patterns of relative frequencies of common deletions in sequential tumor and plasma samples. In some cases, dominant deletions became subclonal or undetectable with different treatments and then reemerged at a dominant frequency at a later time point. One example was the emergence of AR mutations activated by glucocorticoids in about 20% of patients progressing on abiraterone and prednisolone or dexamethasone, exemplifying the importance of sequential monitoring to identify agents that may be driving tumor progression. Overall, resistant clones showed complex dynamic heterogeneity suggesting distinct mechanisms of resistance at different sites that changed based on treatment selection pressure. This study underscores the complexity of tumor clone dynamics and genomic alterations in various tumor sites over time in advanced prostate cancer. Serial assessment of genetic alterations in metastatic tumor sites in response to changing treatments poses challenges due to the frequent presence of tumor in bone. It is anticipated that use of circulating cell-free DNA or CTCs will allow frequent serial assessments over time without the need for tissue biopsies.51
CLINICAL TRANSLATION OF GENETIC INFORMATION
Translation of genomic data for clinical application is ongoing, with availability of clinical trials targeting androgen signaling, the PI3K/mTOR/AKT pathway, ETS fusions and PARP, DNA-PKcs activity, MYC amplification, and others. Figure 3 outlines a path to genomics-driven treatment in which a fresh tumor biopsy is done to determine molecular stratification for inclusion into a clinical trial. Enrichment for the more common mutations, such as androgen signaling, ETS fusions, and PI3K pathway activation, may allow enrollment into more traditional trials with random assignment between groups whereas rare or private molecular disease subsets will require studies based on these specific alterations. For all patients, repeat biopsies, typically at the time of development of tumor progression, will be required to evaluate mechanisms of resistance and the development of new or changing subclones that can inform appropriate changes in treatment. To facilitate this process, multidisciplinary sequencing tumor boards have been established at some institutions to review genomic data and recommend clinically actionable treatments.
Figure 3.
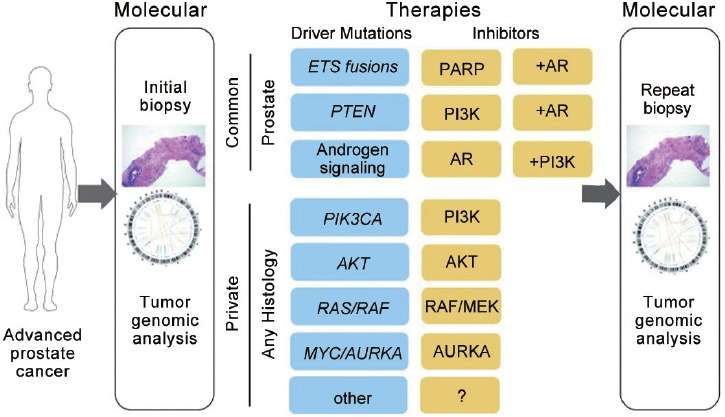
This diagram outlines a path to genomics-driven treatments in prostate cancer. Because of the prevalence of PI3K pathway activation, ETS rearrangements, and androgen signaling, genomic enrichment of patients for these common disease subsets may follow traditional trial structures including combination treatments. For rare or private molecular disease subsets, these patients may be better suited for studies based on the specific molecular aberration. For all treatments, repeat biopsy will be valuable for evaluating mechanisms of resistance and evolution of clonal and subclonal genomic alterations.6
SUMMARY AND CHALLENGES
The application of genomic data in mCRPC individuals to inform rationale treatment is becoming a reality. Now, there is a much greater understanding of tumor clone dynamics and the evolution of lethal metastatic disease. The International Consortium has published important genomic data that show that the majority of mCRPC individuals harbors clinically actionable molecular alterations. Clinical trials based on genomic data are now available but more are in development. Nonetheless, many important challenges remain. The process of obtaining sufficient viable tumor from osseous sites remains a significant challenge although some investigators have reported a relatively high success rate in this area. Genomic analysis of cell-free DNA and circulating tumor cells (liquid biopsy) is a very attractive alternative to sequential biopsies. There are challenges with this technology in that independent clones may be differentially represented in tissue and circulation. Current high-throughput sequencing technologies have limitations in that they do not adequately account for the influence of the tumor microenvironment or the effect of tumor epigenetics. In addition, the presence of tumor heterogeneity remains a significant challenge. Some of these topics are addressed elsewhere in this review. Despite these challenges and limitations, the future of integrated clinical genomics in advanced prostate cancer appears bright. The molecular characterization of prostate cancer demonstrates significant complexities but through the efforts of many investigators around the world, our understanding of these complex alterations is becoming much greater. Ultimately, our goal is to harness our understanding of genomics to identify actionable targets and develop novel treatments for men with advanced prostate cancer to improve and extend their lives.
COMPETING INTERESTS
None.
REFERENCES
- 1.Hussain M, Tangen CM, Berry DL, Higano CS, Crawford ED, et al. Intermittent versus continuous androgen deprivation in prostate cancer. N Engl J Med. 2013;368:1314–25. doi: 10.1056/NEJMoa1212299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sweeney CJ, Chen YH, Carducci M, Liu G, Jarrard DF, et al. Chemohormonal therapy in metastatic hormone-sensitive prostate cancer. N Engl J Med. 2015;373:737–46. doi: 10.1056/NEJMoa1503747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.James ND, Spears MR, Clarke NW, Dearnaley DP, de Bono JS, et al. Survival with newly diagnosed metastatic prostate cancer in the “docetaxel era”: data from 917 patients in the control arm of the STAMPEDE trial. Eur Urol. 2015;67:1028–38. doi: 10.1016/j.eururo.2014.09.032. [DOI] [PubMed] [Google Scholar]
- 4.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. 2014;371:424–33. doi: 10.1056/NEJMoa1405095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roychowdhury S, Chinnaiyan AM. Advancing precision medicine for prostate cancer through genomics. J Clin Oncol. 2013;31:1866–73. doi: 10.1200/JCO.2012.45.3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bostrom PJ, Bjartell AS, Catto JW, Eggener SE, Lilja H, et al. Genomic predictors of outcomes in prostate cancer. Eur Urol. 2015;6167:1–12. doi: 10.1016/j.eururo.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 8.Hieronymus H, Schultz N, Gopalan A, Carver BS, Chang MT, et al. Copy number alteration burden predicts prostate cancer relapse. Proc Natl Acad Sci U S A. 2014;111:11139–44. doi: 10.1073/pnas.1411446111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu W, Xie CC, Thomas CY, Kim ST, Lindberg J, et al. Genetic markers associated with early cancer-specific mortality following prostatectomy. Cancer. 2013;119:2405–12. doi: 10.1002/cncr.27954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–28. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rubin MA, Maher CA, Chinnaiyan AM. Common gene rearrangements in prostate cancer. J Clin Oncol. 2011;29:3659–68. doi: 10.1200/JCO.2011.35.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yap TA, Shahneen KS, Carden CP, de Bono JS. Poly (ADP-ribose) polymerase (PARP) inhibitors: exploiting a synthetic lethal strategy in the clinic. CA Cancer J Clin. 2011;61:31–49. doi: 10.3322/caac.20095. [DOI] [PubMed] [Google Scholar]
- 13.Brenner JC, Ateeq B, Yong L, Yocum AK, Cao Q, et al. Mechanistic rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell. 2011;19:664–78. doi: 10.1016/j.ccr.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li ZG, Mathew P, Yang J, Starbuck MW, Zurita AJ. Androgen receptor-negative human prostate cancer cells induce osteogenesis in mice through FGF9-mediated mechanisms. J Clin Invest. 2008;118:2697–710. doi: 10.1172/JCI33093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palma JP, Wang YC, Rodriguez LE, Montgomery D, Ellis PA, et al. ABT-888 confers broad in vivo activity in combination with temozolomide in diverse tumors. Clin Cancer Res. 2009;15:7277–90. doi: 10.1158/1078-0432.CCR-09-1245. [DOI] [PubMed] [Google Scholar]
- 16.Mateo J, Hall E, Sandhu S, Omlin A, Miranda S, et al. Antitumor activity of the PARP inhibitor olaparib in unselected sporadic castration-resistant prostate cancer in the TOPARP trial. Ann Oncol. 2014;25:1–41. [Google Scholar]
- 17.Goodwin JF, Kothari V, Drake JM, Zhao S, Dylgjeri E, et al. DNA-PKcs-mediated transcriptional regulation drives prostate cancer progression and metastasis. Cancer Cell. 2015;28:97–113. doi: 10.1016/j.ccell.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner ME, et al. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. 1995;332:1393–8. doi: 10.1056/NEJM199505253322101. [DOI] [PubMed] [Google Scholar]
- 19.Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Kainanen R, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9:401–6. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- 20.Ang JE, Olmos D, de Bono JS. CYP17 blockade by abiraterone: further evidence for frequent continued hormone-dependence in castrate-resistant prostate cancer. Br J Cancer. 2009;100:671–5. doi: 10.1038/sj.bjc.6604904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371:1028–38. doi: 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–43. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tomlins SA, Rhodes DR, Pemer S, Dhanasekeran SM, Mehra R, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–8. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 24.Geng C, He B, Xu L, Barbieri CE, Eedunuri VK, et al. Prostate cancer associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proc Natl Acad Sci U S A. 2013;110:6997–7002. doi: 10.1073/pnas.1304502110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44:685–9. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsieh CL, Botta G, Gao S, Li T, Van Allen EM, et al. PLZF, a tumor suppressor genetically lost in metastatic castration-resistant prostate cancer, is a mediator of resistance to androgen deprivation therapy. Cancer Res. 2015;75:1944–8. doi: 10.1158/0008-5472.CAN-14-3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salvi S, Casadio V, Conteduca V, Burgio SL, Menna C, et al. Circulating cell-free AR and CYP17A1 copy number variations may associate with outcome of metastatic castration-resistant prostate cancer patients treated with abiraterone. Br J Cancer. 2015;112:1717–24. doi: 10.1038/bjc.2015.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carreira S, Romanel A, Goodall J, Grist E, Ferraldeschi R, et al. Tumor clone dynamics in lethal prostate cancer. Sci Transl Med. 2014;6:254ra125. doi: 10.1126/scitranslmed.3009448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang YA, Yu J. Current perspectives on FOXA1 regulation of androgen receptor signaling and prostate cancer. Genes Dis. 2015;2:144–51. doi: 10.1016/j.gendis.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–50. doi: 10.1146/annurev.pathol.4.110807.092311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–20. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carver BS, Tran J, Gopalan A, Chen Z, Shaikh S, et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet. 2009;41:619–24. doi: 10.1038/ng.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boormans JL, Korsten H, Ziel-van der Made AC, van Leenders GL, Verhagan PC, et al. E17K substitution in AKT1 in prostate cancer. Br J Cancer. 2010;102:1491–4. doi: 10.1038/sj.bjc.6605673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marques RB, Ashraf A, de Ridder CM, Stuurman, D, Hoeben S, et al. High efficacy of combination therapy using PI3K/AKT inhibitors with androgen deprivation in prostate cancer preclinical models. Eur Urol. 2015;67:1177–85. doi: 10.1016/j.eururo.2014.08.053. [DOI] [PubMed] [Google Scholar]
- 35.Armstrong AJ, Halabi S, Healy P, Alumkal JJ, Yu EY, et al. Phase II trial of the P13 kinase inhibitor BKM120 with or without enzalutamide in men with metastatic castration resistant prostate cancer. J Clin Oncol. 2015;33:290. Suppl; Abstract 5025. [Google Scholar]
- 36.Vaishampayan U, Shevrin D, Stein M, Heilbrun L, Land S, et al. Phase II trial of carboplatin, everolimus, and prednisone in metastatic castration-resistant prostate cancer pretreated with docetaxel chemotherapy: a prostate cancer clinical trial consortium study. Urology. 2015;86:1206–11. doi: 10.1016/j.urology.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Palmgren JS, Karavadia SS, Wakefield MR. Unusual and under-appreciated: small cell carcinoma of the prostate. Semin Oncol. 2007;34:22–9. doi: 10.1053/j.seminoncol.2006.10.026. [DOI] [PubMed] [Google Scholar]
- 38.Wang W, Epstein JI. Small cell carcinoma of the prostate: a morphologic and immunohistochemical study of 95 cases. Am J Surg Pathol. 2008;32:65–71. doi: 10.1097/PAS.0b013e318058a96b. [DOI] [PubMed] [Google Scholar]
- 39.Lotan TL, Gupta NS, Wang W, Toubaji A, Haffner MC, et al. ERG gene rearrangements are common in prostate small cell carcinomas. Mod Pathol. 2011;24:820–8. doi: 10.1038/modpathol.2011.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beltran H, Rickman DS, Park K, Chae SS, Sboner A, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011;1:487–95. doi: 10.1158/2159-8290.CD-11-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Small EJ, Huang J, Youngren J, Sokolov A, Aggarwal RR, et al. Characterization of neuroendocrine prostate cancer (NEPC) in patients with metastatic castration resistant prostate cancer resistant to abiraterone or enzaluatmide: preliminary results from the SU2C/PCF/AACR West Coast Dream Team. J Clin Oncol. 2015;33:268. Suppl; Abstract 5003. [Google Scholar]
- 42.Aparicio A, Shen L, Tapia EL, Lu JF, Chen HC, et al. Molecular characterization of clinically defined aggressive variant prostate cancer in prospectively collected tissues and corresponding patient derived xenografts. J Clin Oncol. 2015;33:288. Suppl; Abstract 5055. [Google Scholar]
- 43.Sharma A, Yeow WS, Ertel A, Coleman I, Clegg N, et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J Clin Invest. 2010;120:4478–92. doi: 10.1172/JCI44239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Palmbos PL, Feng F, Tomlins SA, Kelly WK, Morgans AK, et al. A randomized phase II study of androgen deprivation therapy with or without PD0332991 in RB-positive metastatic hormone-sensitive prostate cancer. J Clin Oncol. 2015;33:295. doi: 10.1158/1078-0432.CCR-21-0024. Suppl; Abstract TPS5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomized phase 2 study. Lancet Oncol. 2015;16:25–35. doi: 10.1016/S1470-2045(14)71159-3. [DOI] [PubMed] [Google Scholar]
- 46.Palanisamy N, Ateeq B, Kalyana-Sundaram S, Pflueger D, Ramnarayanan M, et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer, and melanoma. Nat Med. 2010;16:793–8. doi: 10.1038/nm.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014;159:176–87. doi: 10.1016/j.cell.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu J, Pan S, Hsieh MH, Ng N, Sun F, et al. Targeted Wnt-driven cancer through the inhibition of porcupine by LGK974. Proc Natl Acad Sci U S A. 2013;110:20224–9. doi: 10.1073/pnas.1314239110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu W, Laitinen S, Khan S, Vihinen M, Kowalski J, et al. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat Med. 2009;15:559–65. doi: 10.1038/nm.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JM, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520:353–7. doi: 10.1038/nature14347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frenel JS, Carreira S, Goodall J, Roda D, Perez-Lopez R, et al. Serial next generation sequencing of circulating cell free DNA evaluating tumour clone response to molecularly targeted drug administration. Clin Cancer Res. 2015;21:4586–96. doi: 10.1158/1078-0432.CCR-15-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]